Excess fructose associates with increased production of reactive oxygen species (ROS) that inhibits enzymes such as aconitase, which should affect mitochondrial metabolism. Yet there is a lack of studies investigating the impact of excess fructose on mitochondrial metabolic pathways and substrate utilization. We evaluated the impact of excess fructose on hepatocyte mitochondrial enzymes; citrate synthase (CS), aconitase and glutamate-oxaloacetate transaminase (GOT). Also, high-resolution using complex I-linked substrates [pyruvate+malate (PM), glutamate+malate (GM) and PGM]. Fructose decreased the activities of aconitase and GOT by 35% and 47% respectively. Respiration at Leak, OXPHOS and ETS states were reduced with GM but not PM or PGM. Thus, excess fructose inhibits GOT activity, reduced mitochondrial leak respiration, OXPHOS and ETS capacity. These changes were observed with complex-1 linked respiration (substrates GM). Therefore, fructose impairs mitochondrial respiration and substrate utilization via the enzyme GOT.
fructose, mitochondrial enzymes, mitochondrial respiration, complex I-linked substrates
There are increasing health concerns about the excess consumption of fructose in the form of high fructose corn syrup in modern diets [1]. Upon consumption, approximately 75% of fructose is metabolised by hepatocytes via fructolysis for glycogenesisA, lactate production and triglyceride synthesis [2]. The first step in this pathway is the phosphorylation of fructose, to fructose 1-phosphate by the high affinity ketohexokinase (fructokinase) enzyme. Because this reaction has a low Michaelis constant, and is not controlled allosterically or hormonally, plasma fructose is rapidly cleared and adenosine triphosphate (ATP) inside the hepatocyte is rapidly depleted [3]. Depletion of ATP stimulates adenosine monophosphate deaminase that activates the purine degradation pathway, leading to uric acid production [4,5]. Excess uric acid increases mitochondrial reactive oxygen species (ROS) production in hepatocytes [6,7] which induces defects in a number of ROS-sensitive mitochondrial enzymes including aconitase [8]. Excessive ROS production [9] and reduced aconitase activity [10] is known to affect mitochondrial metabolism. Yet there is a lack of studies providing extended investigation into the impact of excess fructose on mitochondrial metabolic pathways and substrate utilization. Such investigations is important, as the data generated could build upon previous research, and contribute to the body of knowledge on the adverse metabolic effects of excess fructose. We hypothesise that fructose impairs mitochondrial metabolic pathways and substrate utilization in hepatocytes. Therefore, the aim of this study was to investigate the effects of excess fructose on mitochondrial metabolic pathways and substrate utilization.
Cell culture
The HepG2 cell line was obtained from the Council for Scientific and Industrial Research (CSIR) (Stellenbosch, South Africa). Cell lines were initially propagated in 25 cm2 flasks (Bibby Sterilin, Stone, Staffordshire, UK), at 37 °C in 5 mL DMEM (Gibco BRL, Inchinnan, Scotland) containing 10% (v/v) fetal bovine serum, 20 mM HEPES, 10 mM NaHCO3, 100 μg mL-1 penicillin G, and 100 μg mL-1 streptomycin sulfate (Whittaker Bioproducts, Walkersville, MD, USA) at pH 7.5. Cells were incubated for 24 h to permit attachment and grown to semi-confluence and passaged 1:3 every 4-5 days. One group of cells received normal growth medium (control group, DMEM for 72 hours), while the second group were exposed to growth medium supplemented with excess fructose (15 mM, 72 hours) as previously reported [6].
Enzyme activity measurements
Sample preparation: HepG2 cells were homogenised in extraction buffer (0.1 M Tris-HCl; 15 mM Tricarballylic acid; pH 7.8) and incubated on ice for 20 min. The homogenate was centrifuged at 10 000 rpm for 20 min and the supernatant was used as a sample for further experimentation. Subsequent enzyme assays were performed according to modified protocols previously described by Wang et al. [11].
Citrate synthase: The following components were added in a cuvette: 473 µl of citrate synthase buffer (0.1 M Tris-HCl, 1.25 mM 5,5’-dithiobis-[2-nitrobenzoic acid] in deionized water, pH 8.0), 2 µl of sample and 25 µl of Oxaloacetate-Acetyl-CoA solution (50 mM oxaloacetic acid, 5 mM of Acetyl-CoA sodium salt in deionized water; pH 6.5) and was vortexed thoroughly. The absorbance was read in the spectrophotometer at 412 nm after setting a blank reading with pure buffer. The absorbance was used to calculate the specific enzyme activity (fmol.cell-1.min-1) [11].
Aconitase: A reaction mix (490 µl) was prepared and consisted of 0.5 mM NADP+ solution, 5 mM Aconitase Buffer (0.005 v/v chloroform, 10 mM sodium citrate and 160 mM triethanolamine in dH2O; pH 7.4), 0.5 mM MgCl2 and isocitrate dehydrogenase standard. Ten microliters of this sample were added into a cuvette and vortexed thoroughly. The mixture was incubated for 2-3 minutes at room temperature to equilibrate, and the absorbance was read in the spectrophotometer at 340 nm after blanking with buffer. The absorbance was used to calculate the specific enzyme activity (fmol.cell-1.min-1) [11].
Glutamate-oxaloacetate transaminase: The activity of glutamate-oxaloacetate transaminase was measured with a standard commercially available ELISA Kit (Sigma Aldrich, Kempton Park, and Johannesburg). Cells were homogenised in ice-cold GOT buffer, sonicated and centrifuged at 13 000 rpm for 10 min. The standards used ranged from a concentration of 0- 10 nmol well-1. The reaction mix [enzyme, substrate and a developer] (1 mL) and the supernatant (50 µL) were added into each well. The mixture was vortexed, the plate was incubated at 37 °C for 3 min and measurements (A450) were taken every 5 min for a total period of 30 min. The final GOT activity was calculated by using the formula and data were expressed as nmol.min-1 ml-1 , as previously reported [11].
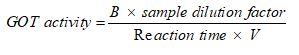
B=Amount (nmol) of glutamine generated between the initial and the final reading
Reaction time=Tfinal-Tinitial (min)
V=Sample volume added to each well.
Mitochondrial respiratory rates were measured at 37°C using the Oroboros 2K oxygraph (Oroboros® instruments, Innsbruck, Austria) [12,13]. Control and fructose-treated cells were trypsinized and centrifuged at 16 000 rpm for 3 min. Cells were counted, (100 000 cells per mL), centrifuged and the cell pellet suspended in 2 mL of DMEM (200 000 cells per 2 mL) containing of digitonin (10 mg/mL DMSO) for 15 min to induce permeabilisation. To assess whether permeabilisation did not damage the cells, cytochrome-c titration was included in the respiratory protocol. As described in literature [13], a spike in the oxygen flux signal, would signify that digitonin damaged mitochondrial integrity, in which case the experiments would be discontinued. Digitonin treated cells were washed with and re-suspended in respiration media (MIR05, 2 mL), and added into each oxygraph chamber. The chambers were hyper-oxygenated (from 200 to 450 μmol L-1) and the oxygen flux was allowed to stabilize for about 30 minutes. The oxygen flux (pmol.s-1.mL-1) was recorded using DatLab software (Oroboros® instruments, Innsbruck, Austria). The following complex I-linked substrate combinations were used to assess respiration at LEAK, OXPHOS and ETS states: a) 5 mM pyruvate+2 mM malate (PM) b) 10 mM glutamate+2 mM malate (GM) and c) 5 mM pyruvate+10 mM glutamate+2 mM malate (PGM) [12,13]. We chose PM substrate because it is metabolised via aconitase in the tricarboxylic acid cycle, whereas GM substrate bypasses this enzyme. Hence, we wanted to assess which substrate combination would be sensitive to tricarboxylic acid cycle enzyme defects, in addition, we used PGM since it is the commonly used substrate for complex-I linked respiration.
To induce LEAK respiration, one of the three substrate combinations was added to the chambers containing permeabilized cells [12,13]. Thereafter, 2.5 mM adenosine diphosphate (ADP) was added to stimulate oxidative phosphorylation (OXPHOS) capacity that was followed by addition of 10 μM Cytochrome-C to check mitochondrial outer membrane integrity [14]. Oligomycin (2.5 μM) was added to induce another LEAK state. Carbonyl cyanide m-chlorophenyl hydrazine (CCCP), a chemical uncoupler, was titrated in a 0.05 μM stepwise manner until the maximum capacity of the electron transfer system (ETS) capacity was reached. Rotenone (0.5 μM), an inhibitor of complex I, was added in order to determine residual oxygen consumption (ROX). Experiments were repeated using the three-substrate combinations at different occasions. Ascorbate and TMPD were titrated for measurement of complex-4 linked respiration/mitochondrial content. Oxygen fluxes at all respiratory states were normalised to the TMPD flux/ complex 4-oxygen flux in order to correct for variations in cell/mitochondrial content in each of the oxygraph chambers. Mitochondrial respiratory fluxes were expressed as pmolO2/s*ml/CIV, while control ratios and coupling efficiencies did not have an SI-unit [13].
Data are presented as the mean ± standard error of mean (SEM). Statistical comparison of the differences between groups were analysed for statistical significance using an unpaired T-test and Mann-Whitney U test performed with GraphPad Prism Software (version 5.00, GraphPad Software, San Diego, CA, USA). When comparing data, a difference with p<0.05 was considered significant.
Enzyme activity measurements
Figure one shows the activities of citrate synthase, aconitase and glutamate-oxaloacetate transaminase in fructose-treated and control HepG2 cells. Fructose had no influence on the activity of citrate synthase after 72-hour exposure (Figure 1A). In contrast, fructose significantly reduced (p<0.011) the activity of aconitase (Figure 1B) and glutamate-oxaloacetate transaminase (Figure 1C) by 35% and 47%, respectively, in comparison to untreated cells.
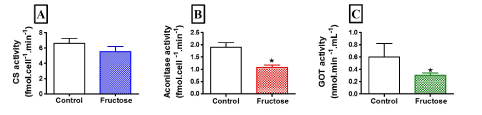
Figure 1. Comparison of activities for citrate synthase (A), aconitase (B) and glutamate-oxaloacetate transaminase (C) in control and fructose-treated HepG2 cells. Data are presented as means, and vertical bars indicate SEM (n=6 in each group). *P<0.011 by comparison with control cells
Mitochondrial respiration
Figure 2 compares LEAK respiration, and OXPHOS and ETS capacities when using a PM substrate combination in fructose-treated and control HepG2 cells. Fructose did not affect complex 4-oxygen flux, which was consistent throughout all experiments and therefore used as normalising factor for respiratory data (data not shown). Fructose had no significant effect on oxygen flux when using PM as shown by the representative oxygraph trace (Figure 2A, an uncorrected trace) and Figure 2B showing the mean oxygen flux at various respiratory states (normalised to complex 4-oxygen flux). Of note, fructose significantly reduced (p<0.028) oxygen flux in all respiratory states when GM was used as substrate (Figure 3A, an uncorrected trace and Figure 3B, normalised to complex 4-oxygen flux). Moreover, the combination of PGM substrates had no influence on oxygen flux (Figure 4A, an uncorrected trace and Figure 4B, normalised to complex 4-oxygen flux).
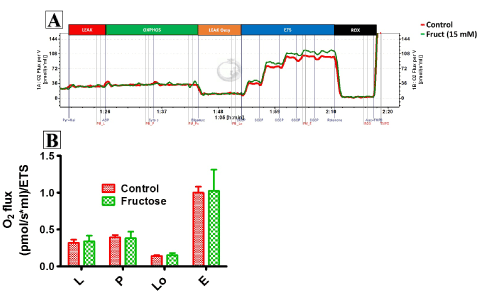
Figure 2. Representation of O2 consumption trace and (A) pulled data (B) of permeabilized control and fructose treated HepG2 cells (15 mM) using PM. The sequence of titration was 5 mM Pyruvate +2 mM Malate, 2.5 mM ADP, 10 µM Cytochrome C, 2.5 µM Oligomycin, steps of 0.05 µM CCCP, 0.05 µM Rotenone and 2 mM ascorbate +0.5 mM tetramethyl-p-phenylenediamine (TMPD). Data are presented as means, and vertical bars indicate SEM (n=7 in each group)
Figure 3. Representation of O2 consumption trace and (A) pulled data (B) of permeabilized control and fructose treated HepG2 cells using GM. The sequence of titration was similar to that of fig 2 except for leak substrates, 10 mM Glutamate +2 mM Malate. Data are presented as means, and vertical bars indicate SEM (n=7 in each group). *P<0.028 by comparison with control cells
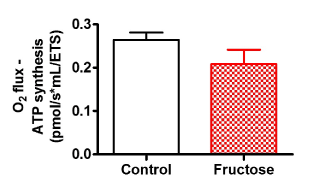
Figure 4. Representation of O2 consumption trace and (A) pulled data (B) of permeabilized control and fructose treated HepG2 cells using PGM. The sequence of titration was similar to that of fig 2 and 3 except for leak substrates, 5 mM Pyruvate +10 mM Glutamate +2 mM Malate. Data are presented as means, and vertical bars indicate SEM (n=7 in each group)
The present study investigated the effects of excess fructose on mitochondrial metabolic pathways and substrate utilization. We found that 72 hrs of 15 mM fructose exposure inhibited aconitase by 35% and GOT, by 47%, but had no influence on citrate synthase. Furthermore, fructose reduced oxygen flux in all the mitochondrial respiratory states (LEAK, OXPHOS and ETS capacity) measured when GM but not PM was supplied as substrates.
Previous studies demonstrated that fructose increases ROS production in hepatocytes that reduces mitochondrial aconitase activity [5,7]. These effects were accompanied by accumulated citrate content and increased triglyceride concentration [5,7]. Our study provides further insight into the effects of fructose on mitochondrial enzymes, as we show that fructose inhibited activities of aconitase and GOT. This effect on GOT activity is observed in our study, either because we implemented longer fructose exposure (72 hrs versus 48 hours) or due to difference in cell type (hepatocytes versus L6). As opposed to previous work [6], our study did not show a change in CS activity. This could be attributed to different levels of ROS between our hepatocytes and their L6 cells [6]. This could have affected the mitochondrial enzymes differently, and thus account for the reduced aconitase and GOT activity yet unaffected CS in our study.
However, we did not measure ROS in our study and therefore future experiments are required to investigate the impact of fructose on the various types of ROS in hepatocytes and to correlate this to the different effects on the relevant enzymes. Another possible reason for not observing any effect of fructose exposure on CS activity could be due to the type of in vitro model used in the study (L6 versus HepG2 cells). L6 myotubes are mouse cells, whereas HepG2 cells are human liver carcinoma cells that have a tendency to be resistant to a variety of stimuli [15,16], and they have an upregulated carbohydrate metabolism [17,18]. To this end, another study has shown that in a model of HepG2 mitochondrial dysfunction, CS activity remained unaltered despite the presence of a pathophysiologic stimulus [19]. The resistant nature of HepG2 cells could mean that they have CS enzymes that are more resistant to the impact of fructose, and the increased carbohydrate metabolism could be underlined by increased CS activity that is not inhibited by fructose. These reasons may provide some explanation as to why fructose reduced activities of aconitase and GOT but not CS in our study albeit subject to debate.
Our study further demonstrated that fructose not only inhibited aconitase and GOT, but also metabolic pathways and substrate utilization. Due to the inhibitory effect of fructose on aconitase, we used PM and GM as substrate combinations in separate experiments. This was because PM involves aconitase while GM bypasses this enzyme during their metabolism in the TCA cycle. We wanted to tease out the substrate combination that would reflect impairment in respiration and we eventually used the combination of PGM as this is the commonly used substrate for accessing complex-I function. Fructose exposure had no influence on O2 flux in all respiratory states when using PM as a substrate (Figure 2). We explained these results based on Figure 5A; fructose-induced inhibition of aconitase prevented the conversion of citrate to 2-oxoglutarate hence causing accumulation of citrate inside the mitochondrial matrix. Due to this accumulation, citrate would then escape the mitochondria into the respiratory medium and respiration would continue as normal. The citrate that is expelled is used to make fatty acid and its escape reduces the efficiency of PM breakdown. Similar results of no effect were obtained with PGM substrate combination (Figure 4). Since PM and PGM had no effect on O2 flux this implies that these substrate combinations are not a suitable for assessing defects in mitochondrial enzymes.
Interestingly when using the GM substrate combination, complex 1-linked respiratory states were reduced in fructose-treated cells. These results could be explained based on (Figure 5B); fructose-induced inhibition of GOT would prevent the conversion of oxaloacetate to 2-oxoglutarate hence causing accumulation of oxaloacetate. Increased oxaloacetate would suppress malate dehydrogenase hence reducing NADP produced by this reaction. Other studies have reported that fructose reduces glutamate dehydrogenase activity [20,21] that could lead to a lesser conversion of glutamate to alpha-ketoglutarate and reduced mitochondrial respiration via the GM pathway. Therefore, it is likely that in our study, fructose reduced glutamate dehydrogenase in addition to GOT activity, hence resulting in reduced mitochondrial respiration with GM but not with PM and PGM. However, because we did not measure glutamate dehydrogenase activity in our study, we cannot provide a concrete analysis but suggest that the GOT impairment we observed is in part responsible for overall reduction of NADH/NAD+ levels that leads to reduced complex-I linked respiration with GM substrate. Although we did not measure NADH/NAD+ levels, previous studies have reported a link between fructose and reduced NADH/NAD+ levels [20].
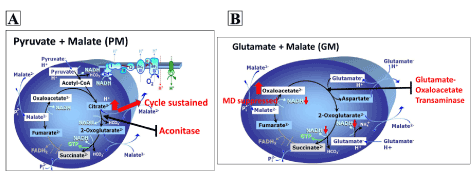
Figure 5. TCA cycle showing the involvement of aconitase when using PM (A) substrate and glutamate oxaloacetate transaminase when using GM (B) substrate (Modified from Gneiger [13])
In summary, novel findings of this study, is that in addition to aconitase, excess fructose inhibits GOT activity in hepatocytes. Furthermore, fructose reduced mitochondrial leak respiration, OXPHOS and ETS capacity, when we used the substrate combination GM. These changes were not seen with PM or PGM. Therefore, the adverse metabolic effects of excess fructose not only inhibit aconitase as previously reported, but also impairs mitochondrial respiration and substrate utilization via the enzyme GOT.
The authors declare that there are no conflicting interests.
EO conceived and designed the experiments, procured funding for the purchase of all the study/experimental expenses (reagents/materials/analysis tools), and contributed to data analysis/interpretation and writing of the manuscript. HPM performed the experiments and analysed the data, prepared draft manuscript and coordinated the completion of the manuscript, and undertook submission of the paper. GM contributed to the writing of the draft and final manuscript, contributed to data interpretation and overall presentation of the data within the manuscript and strategizing the layout, together with co-authors.
The authors are grateful to Dr Maronel Steyn from CSIR, Stellenbosch, South Africa, for providing the HepG2 cell line that was used in the study. We also thank Dr Peter A. Hecker from Brigham Young University, Utah, USA for providing experimental protocols for citrate synthase and aconitase activity assays. National Research Foundation of South Africa, the funding body had no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
This study was funded by the National Research Foundation of South Africa. Funders did not have any role in the conception, data collection and writing of the manuscript.
- Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ (2002) Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr 76: 911-922. [Crossref]
- Feinman RD, Fine EJ (2013) Fructose in perspective. Nutr Metab (Lond) 10: 45. [Crossref]
- Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, et al. (2009) Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem 57: 763-774. [Crossref]
- Calvo RY, Araneta MRG, Kritz-Silverstein D, Laughlin GA, Barrett-Connor E (2014) Relation of serum uric acid to severity and progression of coronary artery calcium in postmenopausal white and Filipino women (from the Rancho Bernardo Study). Am J Cardiol 113: 1153-1158. [Crossref]
- Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, et al. (2012b) Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 7: e47948. [Crossref]
- Jaiswal N, Maurya CK, Arha D, Avisetti DR, Prathapan A, et al. (2015) Fructose induces mitochondrial dysfunction and triggers apoptosis in skeletal muscle cells by provoking oxidative stress. Apoptosis 20: 930-947. [Crossref]
- Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, et al. (2012a) Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress potential role in fructose-dependent and-independent fatty liver. J Biol Chem 287: 40732-40744. [Crossref]
- Gardner PR, Fridovich I (1991) Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J Biol Chem 266: 1478-1483. [Crossref]
- Schwall CT, Greenwood VL, Alder NN (2012) The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim Biophys Acta 1817: 1588-1596. [Crossref]
- Boquist, L, Ericsson, B, Lorentzon, R, Nelson L (1985) Alterations in mitochondrial aconitase activity and respiration, and in concentration of citrate in some organs of mice with experimental or genetic diabetes. FEBS Lett 183: 173-176. [Crossref]
- Wang H, Sreenivasan U, Gong DW, O'Connell KA, Dabkowski ER, et al. (2013) Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J Lipid Res 54: 953-965.
- Gnaiger E (2008) Polarographic oxygen sensors, the oxygraph and high-resolution respirometry to assess mitochondrial function. New York: John Wiley & Sons.
- Gnaiger E (2014) Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis. Innsbruck, Austria: OROBOROS Instruments.
- Jimenez-Lopez JM, Carrasco MP, Segovia JL, Marco C (2002) Resistance of HepG2 cells against the adverse effects of ethanol related to neutral lipid and phospholipid metabolism. Biochem Pharmacol 63: 1485-1490. [Crossref]
- Kuznetsov AV, Schneeberger S, Seiler R, Brandacher G, Mark W, et al. (2004) Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am J Physiol Heart Circ Physiol 286: H1633-H1641. [Crossref]
- Lee H, Uhm S, Shin JW, Jeon HM, Dongbang S, et al. (2015) HepG2 Cell Resistance against Camptothecin from a Lysosomal Drug Delivery. Chem Asian J 10: 2695-2700. [Crossref]
- Daniels VW, Smans K, Royaux I, Chypre M, Swinnen JV, et al. (2014) Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment. PLoS One 9: e106913. [Crossref]
- Pereira da Silva AP, El-Bacha T, Kyaw N, dos Santos RS, da-Silva WS, et al. (2009) Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem J 417: 717-726. [Crossref]
- Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Munoz-Yague T, Solis-Herruzo JA (2015) In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis Model Mech 8: 183-191. [Crossref]
- Han SJ, Choi SE, Yi SA, Jung JG, Jung IR, et al. (2016) Glutamate dehydrogenase activator BCH stimulating reductive amination prevents high fat/high fructose diet-induced steatohepatitis and hyperglycemia in C57BL/6J mice. Sci Rep 5: 37468. [Crossref]
- Spanaki C, Plaitakis A (2012) The role of glutamate dehydrogenase in mammalian ammonia metabolism. Neurotox Res 21: 117-127. [Crossref]