Pediatric Churg Strauss syndrome is a rare disorder that is extremely rare during childhood. A total of 63 cases of pediatric Churg Strauss syndrome have been reported in the world.
The aim of this paper is to describe the 64th case of pediatric Churg Strauss syndrome and the first case of Churg Strauss syndrome in Iraq. The patient illness was misdiagnosed as tuberculosis and the use of anti-tuberculosis medication was associated with life threatening complications and the development of serious corneal opacity because of delay in receiving the appropriate therapy.
It is an unnecessary and unfortunate experience for the patient to receive management that is very inappropriate in the presence of a therapeutic intervention that can induce remission of the disease.
This case illustrates the difficulty and importance of the awareness to reach a diagnosis in a rare syndrome or disorder for which there is an effective therapy.
pediatric, churg-strauss syndrome, misdiagnosis
Pulmonary eosinophilia including eosinophilic pneumonia is characterized by significant systemic and pulmonary manifestations such as fever, sweating at night, cough, difficulty breathing, weight loss, blood eosinophilia, peripheral infiltrates on chest radiograph, and a good response to corticosteroid therapy [1,2].
Pulmonary eosinophilia which can be caused by Churg Strauss syndrome is rare in children and can be misdiagnosed by pediatricians [1,2].
Chronic eosinophilic pneumonia was first described by Carrington in 1969.Tuberculosis was the initial diagnosis in most of Carrington cases, However, the patients' condition deteriorated on chemotherapy [2].
Churg Strauss syndrome was first described by Jacob Churg and Lotte Strauss in 1951. They called the condition "allergic granulomatosis." The syndrome is associated with the development of autoimmune systemic necrotizing vasculitis with inflammation of small and medium-sized blood vessels. It has highly variable presentation and course. The condition was sometimes called eosinophilic granulomatosis with polyangiitis [1].Pediatric Churg Strauss syndrome is a rare disorder that is extremely rare during childhood. A total of 63 cases of pediatric Churg Strauss syndrome have been reported in the world [3-7].
The aim of this paper is to describe the 64th case of pediatric Churg Strauss syndrome and the first case of Churg Strauss syndrome in Iraq.
A six-year old girl with convergent squint (abducent nerve palsy) and corneal opacity on the right eye (Figure 1) was seen early during November, 2016, because of evidence of recent hepatitis B infection which developed following the receive of several blood transfusions.

Figure 1. The girl had convergent squint (abducent nerve palsy) and corneal opacity on the right eye
Table 1 shows the results of tests for hepatitis B virus which were performed on the 20th of October 2016. HBs, antigen HBe antigen, HBc antigen were all positive despite they were negative several weeks earlier.
Table 1. The results of tests for hepatitis B virus (October, 20, 2016)
Serum HBs antigen
Chromatographic immunoassay for the qualitative detection of hepatitis B virus surface antigen. |
Positive |
Serum HBe antibody
Chromatographic immunoassay for the qualitative detection of hepatitis B virus envelope antibody |
Negative |
Serum HBc antibody
Chromatographic immunoassay for the qualitative detection of hepatitis B virus core antibody |
Positive |
Serum HBe antigen
Chromatographic immunoassay for the qualitative detection of hepatitis B virus envelope antigen. |
Positive |
Serum HBs antibody
Chromatographic immunoassay for the qualitative detection of hepatitis B virus surface antibody |
Negative |
Liver enzymes were shown to be elevated on earlier on the eighth of October while the serum albumin was normal. SGOT was 119 iu/L (Normally less than 50), SGPT was 121 iu/L (Normally less than 5), and serum albumin was 4.5g/L. The child had history of cough and wheezing more than one year earlier.
Her illness was first started at the age of five years with the development of keratitis in association with respiratory symptoms. She had fever, night sweats, cough and dyspnea with pulmonary infiltrates on chest radiographs. She also had hepatomegaly. Marked eosinophilia was demonstrated on blood counts.
Tuberculosis was considered despite it cannot account for marked eosinophilia and despite that sputum exam for acid fast bacilli was negative. Hepatitis Anti-genes and anti-bodies for hepatitis B virus were negative during that time. The keratitis which was causing blurring of the cornea was considered to be viral and was treated by ophthalmologist accordingly. The corneal inflammation was treated mainly with acyclovir ophthalmic preparations. Anti-tuberculosis therapy was used for several weeks, but without any benefit.
She received triple anti-tuberculosis therapy (INH, rifampicin, and pyrazinamide). Anti-tuberculosis therapy was initiated on the second of November 2015. On the second of January 2016, pyrazinamide was stopped, and all anti-tuberculosis therapy was discontinued on the tenth of January 2016.
During January 2016, she developed pallor, jaundice, and lethargy. She was afebrile and had hepatosplenomegaly. Liver was palpable 10 cm below the subcostal margin, and the spleen was palpable 8 cm below thesubcostal margin. There was no lymphadenopathy, and the rest of physical examination was not significant.
Laboratory was performed on the eleventh of January 2016. Hemoglobin was 4.4 g/dl with normochromic normocytic red blood cells. There was also neutrophil leukocytosis with white cell count of 16.800/ml and neutrophil count was 67% while the lymphocyte count 30%. Platelet count was 380.000/ml and no abnormal cell was seen on blood film. She received blood transfusion daily for seven days.
A bone marrows specimen was examined on the 13th of January 2016. The bone marrow was hypercellular with myelopoisis hyperplasia. There was predominance of intermediate stage in giant forms. There was also erythropoesis hyperplasia with macro-normoblastic megaloblastic maturation. Marked erythroid hyperplasia megaloblastic maturation, and the myelopoisis/ erythropoesis ratio was 1:2 suggesting hemolytic anemia with feature of folate deficiency.
Laboratory tests on the 14th of January 2016 showed the followings:
- Hemoglobin was 5.1g/dl with normochromic red blood cells, and the presence of spherocytes, polychromatic macrocytes, and nucleated blood cells (2 per 100 blood cells).
- There was also neutrophil leukocytosis with white cell count of 19.400/ ml and neutrophil count was 62% while the lymphocyte count 24%.
- Reticulocyte count was 18% and the corrected reticulocyte count was 7%.
- Platelet count was adequate.
- Total bilirubin was 8.2 mg/dl with the indirect bilirubin 5.7mg/dl.
- SGPT was 38 iu/L and SGOT was 25 iu/ L.
- Test for G-6-PD was negative, but direct Coombs was positive.
On the 17th of January 2016 laboratory tests showed the followings:
- Hemoglobin was 6.5g/dl with normochromic red blood cells, and autoagglutination.
- Normal white cell count of and morphology; white blood cell count 4.600/ ml, neutrophil count was 68% while the lymphocyte count 22%.
- Platelet count was 167.000/ml.
- Total bilirubin was 8 mg/dl with the indirect bilirubin 5 mg/dl. SGPT was 47 iu/L and SGOT was 101 iu/ L.
- The ESR was very high 145 mm/hour, direct Coombs was strongly positive (3 +++). Serum LDH was also very high 1900 iu/L (Normal: 200-400 iu/L).
On the 19th of January, 2016 laboratory tests suggested cold autoimmune hemolytic anemia. Hemoglobin was 6.5g/dl; white blood cell count was 10.000/ml with 62 % neutrophils, 24 % lymphocytes, 7% monocytes, and 4% myelocytes.
Red blood cells were generally normochromic with slight anisopokilocytosis, and many macrocytes. There were two nucleated red blood cells per 100 with agglutination of red blood cells. The platelet count was within normal limits.
The urine was dark in color and was positive for urobilinogen and strongly positive for bile pigments (+++). Microscopic urine examination showed 1-3 red blood cells/HPF and 8-12 pus cells/HPF.
On the 20th of January, cold agglutinin test was positive with a titer exceeding 1024.
On the 21st of January, total bilirubin was 26 mg/dl; SGPT was 245 iu/dl.
1-Blood transfusion twice; one with AB positive blood (her blood group), and one with O positive blood.
2-Prednisolone 2mg/kg/day for three days but was switched to parenteral methyl prednisolone 500 mg per square meter in two divided doses for three days because of the lack of benefit to oral prednisolone. Methyl prednisolone was started on the 20th of January, 2016.
3-Vitamin B 12, 1 mg ampoule on alternate days.
4-Folic acid 5mg daily.
5-Azithromycin.
On the 25th of January, 2016, she received blood transfusion. After three days, IgM and IgG antibody titers were found to be normal. The patient was discharged from hospital after stabilizing hemoglobin level, return of urine to normal and reduction in hepatosplenomegaly. On discharge, the patient was kept on
1-Prednisolone 2mg/Kg /day (30 mg daily in two divided doses).
2-Folic acid tablet 1 mg daily.
3-B12 1 mg ampoule weekly.
On the 9th of March, 2016, laboratory tests showed normal renal function tests and elevated liver enzymes. Serum creatinine was 18 μmol/L, blood urea was 3.2 mmol/L, SGOT was 60 iu/L (Normally less than 50), and SGPT was 177 iu/L (Normally less than 50).
On the 20th of March 2016, laboratory tests showed a total serum bilirubin of 1.6 mg/dl (Direct 1.2 mg/dl and indirect 1.4 mg/dl). Serum SGOT was 81 u/L and serum SGPT was 181u/L).
Chest radiographs (Figure 2) taken during March 2016 showed initially accentuation of the brocho-vascular markings, which progressed to diffuse reticulo-nodular shadowing suggestive of bronchopneumonia. Both costo-phrenic angles were clear.
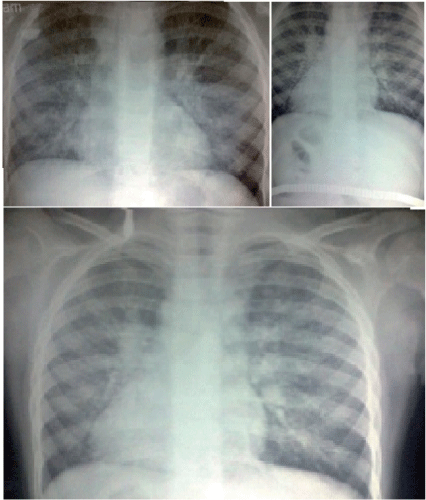
Figure 2. Chest radiographs taken during March, 2016 showed initially accentuation of the brocho-vascular markings, which progressed to diffuse reticulo-nodular shadowing suggestive of bronchopneumonia
The deterioration in the respiratory symptoms and chest radiographs occurred following discontinuation of steroids which was used to treat the hemolytic anemia caused by anti-tuberculosis medications.
On the 18th of June 2016, she developed fever, anorexia, cough, abdominal pain, and she had hepatosplenomegaly.
An abdominal ultrasound performed on the 19th of June 2016 showed mild hepatosplenomegaly. The liver was mildly enlarged with normal echo pattern and no focal lesion. The intra-hepatic biliary tree and hepatic veins were normal. The spleen was mildly enlarged (103 mm) with normal parenchyma and normal splenic veins. All other organs including gall bladder, pancreas, kidneys, and bladder were all normal.
General stool examination on the 19th of June showed few red blood cells and the stool was semi-solid with no other abnormality.
Urine test performed on the 27th of June was negative for bile pigments.
Laboratory tests performed on the first of July, showed leukocytosis with absolute eosinophilia. White blood cell count was 14.000/ml with 48% neutrophils, 11% lymphocytes, and 39% eosinophils. Hemoglobin was 11.6g/dl and the red blood cells were normochromic normocytic with slight anisocytosis. No abnormal cells were seen on blood film. Reticulocyte count was 8%, and platelet count was 256.000/ml. The erythrocyte sedimentation rate was high at 99 mm/hour.
During July, 2016, she was experiencing high grade fever, poor appetite, cough, vomiting and severe abdominal pain.
On the eleventh of July, sputum specimen was negative for acid fact bacilli, and blood test with real time PCR for mycobacteria tuberculosis was also negative. Tuberculin skin test was also negative.
On the 12th of July, 2016 , A native and with intravenous contrast CT-scan was performed on the chest and abdomen (Figure 3).In the chest, there was diffuse reticulo-nodular shadowing with ground glass shadow. There was no lung mass or hilar lymphadenopathy or pleural effusion. In the chest, there was neither visceromegaly nor mass.
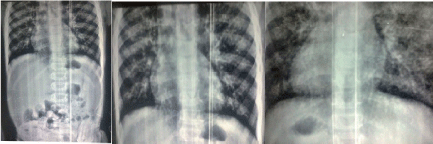
Figure 3. Chest contrast CT-scan the chest showed diffuse reticulo-nodular shadowing with ground glass shadow
The pancreas was enlarged in size and the enlargement was mostly of the head suggesting pancreatitis. Serum amylase level was high at 157 u/L (Normally less than 100 u/L).
Abdominal ultrasound examination performed on the 18th of July, 2016 also showed enlarged pancreas suggesting pancreatitis. The pancreas was enlarged in size in all parts with hypo-echoic texture and normal shape. The liver was also enlarged with normal texture.
On the 14th of July, 2016, laboratory tests showed the followings: Hemoglobin was 11.2 g/dl, white blood cell count 8.800/ ml, reticulocyte count was 9%, and platelet count was 236.000/ml. Blood urea was 5.2 mmol/l, and serum creatinine was 16 mmol/l. Liver function tests showed the following results: SGPT was 282 iu/l, SGOT was 371 iu/ l, and alkaline phosphatase was 492/l.
Laboratory tests performed on the 16th of July, showed the following results: White blood cell count was 6.300/ml with 30 % neutrophils, 13% lymphocytes, and 56 % eosinophils. Hemoglobin was 9.4g/dl. Reticulocyte count was 8.3%. The erythrocyte sedimentation rate was high at 100 mm/hour.
On the 16th of July, 2016, ultrasound of the abdomen was performed and showed small amount of fluid behind the child uterus. Ultrasound also showed increased size of the head of the pancreas to 4.3 cm and also the tail to 2.3 cm. The ultrasound report suggested the possibility of acute pancreatitis. Ultrasound of the abdomen was performed again on the 18th of July, 2016 and the pancreas was enlarged in all parts with hypo-echoic texture, and the possibility of pancreatitis was greatly increased.
On the 24th of July 2016, serum amylase was found to be elevated at 409 iu/L, but it declined to 108 iu/l on the 30th of July in association with disappearance of abdominal pain.
On the 25th of July, a specimen of bone marrow was found negative when tested with real-time PCR for mycobacteria tuberculosis.
However, the norm-cellular bone marrow specimen, which was taken on the 27th of July, showed prominent eosinophilia. There were active megakaryocytes with predominance of eosinophilic precursors, and no evidence of neoplastic cells.
On the 28th of July 2016, the treating physicians during that time decided to start two anti-tuberculosis agents, streptomycin and ethambutol as they considered them not associated with hepatitis or hemolytic anemia.
On the 30th of July, laboratory tests performed and showed the following findings:
Hemoglobin was 9.3g /dl, white blood cell count was 5500/ ml with 54% neutrophils and 34% lymphocytes.
Reticulocyte count was 2.5% and platelet count was 302.000/ml.
Total serum bilirubin was 0.9 mg/dl, SGPT was 116 was iu/l, and SGOT was 112 iu/l. Direct coombs test was negative, Urine examination showed normal findings, and was negative for bile pigments.
On the 10th of August, 2016, the treating physicians during that time decided to add INH in small dose and increasing it gradually while monitoring liver enzymes and reticulocyte count.
Treatment with prednisolone was also started. However, liver enzymes were found to be
elevated on the on the 27th of August. SGOT was 59 iu/L (Normally less than 50), and SGPT was 90 iu/L (Normally less than 50).
On the 27th of August, 2016, there was more elevation of liver enzymes. SGOT was 121 iu/L (Normally less than 50), and SGPT was 234 iu/L (Normally less than 50).
On the 19th of September 2016, laboratory tests were performed. The Hemoglobin was 14.1 mg and the reticulocyte count was 2%. Platelet count was 165.000/ml.
An ultrasound performed during October 2016 (Figure 4) showed mild acalculous cholecystitis and cholangitis. The gall bladder had normal size, but there was mild thickening of the mucosa, and no stone was present. There was also mild irregular dilatation (4-7 mm) of the common bile duct with mucosal thickening and without obvious distal obstruction. The liver had normal dimensions with coarse echo pattern and mild periportal fibrosis. The pancreas was normal in size and had normal echo pattern.
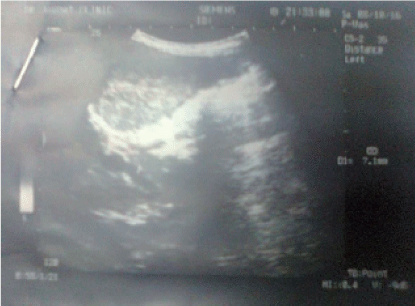
Figure 4. An ultrasound performed during October, 2016, showed mild acalculous cholecystitis and cholangitis
The diagnosis of the 64th case of pediatric Churg Strauss syndrome in the World and the first case of Churg Strauss syndrome in Iraq was made.
Table 2 shows the main features of the 64th case of Churg Strauss syndrome in the World.
Table 2. The main features of the 64th case of pediatric Churg Strauss syndrome in the World
1 |
Historty of asthmatic attacks. |
2 |
Unfixed pulmonary infiltrates with fever, night sweats, cough. |
3 |
Marked eosinophilia. |
4 |
Histological evidence of extra-vascular eosinophils: Bone marrow biopsy showing prominent eosinophilia. |
5 |
Mononeuropathy: Sixth cranial nerve palsy. |
6 |
Pancreatits presented with abdominal pain, and vomiting. The diagnosis of pancreatitis was based on high serum amylase and ultrasonographic findings of pancreatitis. |
7 |
Acalculous cholecystitis and cholangitis. |
8 |
Eye involvement: Keratitis. |
Hundreds of rare clinical syndromes are known to occur in humans. However, there is often inadequate professional knowledge, experience and awareness of their presentations and their most appropriate management, because of the small number of patients having each one of them. Diagnostic challenges and difficulties, diagnosis delay, and misdiagnoses, inappropriate or unsatisfactory management are well known to be associated with rare syndromes and disorders. It is expected that at least one third of rare syndromes and disorders are misdiagnosed more than once, or the diagnosis is delayed unnecessarily for a variety of reasons. Some rare syndromes often present with a symptom or symptoms of more common illnesses and are associated with variable presentations [1].
Diagnosis of a rare clinical syndrome may depend on whether the consulted physician has seen the rare condition before, and whether he /she has trained himself or herself to be aware of rare conditions and to consider them in the appropriate setting.
Some experts thought that it may take at least four years before a patient with a rare condition can receive the right diagnosis, generally after visiting and consulting several physicians [1].
In 2010, Twardowsky, et al. [8] from Brazil counted the reported cases of childhood Churg Strauss syndrome and found only 35 children and adolescent patients reported with the syndrome. Twardowsky, et al. reported the thirty-six pediatric case of Churg Strauss syndrome.
In 2012, Razenberg, et al. reviewed 50 cases of childhood Churg Strauss syndrome including their case and found that respiratory tract is most frequently involved with pulmonary infiltrates, asthma and sinusitis [3].
A total of 63 cases of pediatric Churg Strauss syndrome have been reported in the world [3-8].
In this paper, I am reporting the sixty fourth case and emphasizing the importance of the awareness to reach a diagnosis in a rare syndrome or disorder for which there is an effective therapy.
Logically, and in most instances, appropriate management is associated with making the correct diagnosis in time. Sometimes, inappropriate therapies result in unwanted effects and further complicate the presentation of the rare condition being treated.
Anyhow, it will continue to be difficult to diagnose a rare condition or disorder because it is impracticable for doctors to be familiar with a large number of rare conditions.
It is an unnecessary and unfortunate experience for the patient to receive management that is very inappropriate in the presence of a therapeutic intervention that can induce remission of the disease.
This case illustrates the difficulty and importance of the awareness to reach a diagnosis in a rare syndrome or disorder for which there is an effective therapy.
In 1990, Masi, et al. published criteria for the classification of Churg Strauss syndrome which were developed by the American College of Rheumatology by comparing 20 patients having Churg Strauss syndrome with 787 control patients with other forms of vasculitis. Masi, et al. considered the presence of four or more of six criteria yields a sensitivity of 85% and a specificity of 99.7%. The six criteria include [9]:
1-Asthma.
2-Eosinophilia greater than 10% on differential white blood cell count.
3-Mononeuropathy (including multiplex) or polyneuropathy.
4-Non-fixed pulmonary infiltrates on radiography.
5-Histological evidence of extra-vascular eosinophils.
6-Paranasal sinus abnormality.
The diagnosis of the 64th case of pediatric Churg Strauss syndrome in the World which is the first case of Churg Strauss syndrome in Iraq was based on the presence of five of the six diagnostic criteria used by the American College of Rheumatology [9].
The mononeuropathy in the first case of Churg Strauss syndrome in Iraq was presented as sixth cranial nerve palsy which was presented as convergent squint.
The association of Churg Strauss syndrome with cranial neuropathy is well recognized from Italy reported a case of Churg-Strauss syndrome characterized by the presence of multiple ophthalmological and neuroophthalmological lesions including mononeuritis of the fourth cranial nerve [1].
Vitali, et al. thought that the simultaneous occurrence of multiple ocular features in a patient with Churg-Strauss syndrome suggests that regional vasculitis may be the pathological mechanism underlying the multiple ophthalmological lesions in this disorder [1].
In 2005, Tsuda, et al. from Japan reported a 30-year-old man with Churg Strauss syndrome who had partial oculomotor nerve palsy with restrictions of elevation and adduction, and mydriasis was in the left eye [1].
Cranial magnetic resonance imaging showed an infarction lesion in the territory of the left superior median mesencephalic branch of the posterior cerebral artery. The case was the first case of oculomotor nerve palsy due to midbrain infarction associated with Churg-Strauss syndrome.
In 2008, Naitoh reported the fifth case of Churg-Strauss syndrome associated with oculomotor paralysis. Naitoh emphasized the rarity of cranial nerve paralysis in Churg-Strauss syndrome.
In 2012, Shimada, et al. reported the association of Churg Strauss syndrome with lower cranial neuropathy affecting the glossopharyngeal and vagal nerve palsy and presenting as dysarthria and dysphagia [1].
Ozaki, et al. (2012) from Japan reported a patient with Churg-Strauss syndrome who had eighth cranial nerve (Vestibulocochlear nerve) palsy which is extremely rare in Churg-Strauss syndrome [1].
In 2014, Byun, et al. from Korea described the occurrence of left facial and cochlear neuropathies which were detected in electro-diagnostic studies in a patient with Churg-Strauss syndrome [1].
In addition to the five of the six diagnostic features of Churg Strauss syndrome used by the American College of Rheumatology, the first case of the syndrome in Iraq also had gastrointestinal involvement including non-calculus cholecystitis and pancreatitis which are a well-recognized association in Churg Strauss syndrome.
Cojocaru M, et al. (2011) from Romania emphasized that the systemic autoimmune diseases including Churg Strauss syndrome can involve any part of the gastrointestinal tract, hepatobiliary system and pancreas [1].
The occurrence of non-calculus cholecystitis in Churg Strauss syndrome is well recognized. Imai, et al. from Japan emphasized that that acute acalculous cholecystitis can be the initial sign of a systemic vasculitis, specifically Churg Strauss syndrome [1].
In 2003, Nishie, et al. from Japan described a patient with acute cholecystitis and duodenitis associated with Churg-Strauss syndrome [10].
The patient of Nishie, et al. was a 36-year-old male, who had fever, abdominal pain, a transient infiltration in the left lower lung, and marked hypereosinophilia of 17,000/ml. Ultrasonographic and gastroendoscopic examinations revealed acute cholecystitis and duodenitis, respectively [10].
The patient also developed distally dominant mononeuritis multiplex, especially in the upper limbs. Ye L, et al. from China emphasized the rare occurrence of cholecystitis in Churg Strauss syndrome, reported a case and reviewed eleven cases of Churg Strauss syndrome -associated cholecystitis reported in the literature [11].
- Al-Mosawi AJ (2016) Pediatric Churg Strauss syndrome. (1st Edn) Saarbrücken, LAP Lambert Academic Publishing.
- Carrington CB, Addington WW, Goff AM, Madoff IM, Marks A, et al. (1969) Chronic eosinophilic pneumonia. N Engl J Med 280: 787-798. [Crossref]
- Razenberg FG, Heynens JW, Jan de Vries G, Duijts L, de Jongste JC, et al. (2012) Clinical presentation of Churg-Strauss syndrome in children: A 12-year-old-boy with ANCA-negative Churg-Strauss syndrome. Respir Med Case Rep 7: 4-7.
- Mutsaers ER, Witteveen R, van den Bosch-Ruis W, Kuijpers TW, van Houten MA, et al. (2013) A pseudoleukemic blood differentiation in a 13-year-old child: an extraordinary presentation of Churg-Strauss syndrome. Clin Rheumatol 32 Suppl 1: S7-S9.
- Gendelman S, Zeft A, Spalding SJ (2013) Childhood-onset eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome): a contemporary single-center cohort. J Rheumatol 40: 929-935.
- Albahri Z, Minxová L, LukeÅ¡ A, Mawiri AA, Å tefáÄková Å (2014) Churg-Strauss syndrome in childhood: a case report. J Child Neurol 29: NP98-100. [Crossref]
- Alyasin S, Khoshkhui M, Amin R (2015) Familial Churg-Strauss Syndrome in a Sister and Brother. Iran J Allergy Asthma Immunol 14: 338-340.
- Twardowsky AO, Paz JA, Pastorino AC, Jacob CM, Marques-Dias MJ, et al. (2010) Chorea in a child with Churg-Strauss syndrome. Acta Reumatol Port 35: 72-75. [Crossref]
- Masi AT, Hunder GG, Lie JT (1990) The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 33: 1094-100.
- Nishie M, Tomiyama M, Kamijo M, Kannari K, Tanosaki M, Baba M, et al. (2003) Acute cholecystitis and duodenitis associated with Churg-Strauss syndrome. Hepatogastroenterology 50: 998-1002. PMID: 12845966.
- Ye L, Lu X, Xue J (2016) Eosinophilic granulomatosis with polyangiitis complicated by cholecystitis: a case report and review of the literature. Clin Rheumatol 35: 259-263.