Abstract
Research on the human microbiome has revolutionized our approach to metabolic and inflammatory disorders in the past decade. Obesity and type 2 diabetes (T2DM) were among the first diseases where characteristic intestinal flora changes could be associated with specific biochemical consequences that are causally related to disease-development.
The diabetes disease spectrum appears to be a specific area of microbiome-associated disease where the metabolic and inflammatory factors “converge” which may develop in part independently from each other resulting in the “Janus-face” of this disease. On the one hand the link between glycemic control and the risk for diabetes complications is well established, however even suitably controlled blood glucose levels do not prevent diabetes complications in all cases. From this point of view neuropathy is one of the most under-diagnosed and most hardly reversible complications. As part of the growing prevalence of T2DM, it is one of the priority areas of morbidity and quality of life for patients. Despite this fact, neither screening of neuropathy nor secondary prevention is reassuringly addressed and resolved on the public health level. Microbiome research may contribute to understanding the link between lifestyle factors and specific risk factors in developing the wide range of diabetes complications.
We intend to outline novel microbiome associated putative mechanisms and their relation to the lifestyle risk factors of T2DM patients. This may provide a solid help foundation for lifestyle interventions, one of the most evident, yet most difficult and underutilized opportunities in the management of these patients.
Keywords
microbiome, neuropathy, type 2 diabetes mellitus, propionate sensing, gut barrier, Guillain-Barre syndrome
Abbreviations
BBE - Bickerstaff Encephalitis
GBS - Guillain-Barré Syndrome
HCMV - Human Cytomegalovirus
HHV-5 - Human Herpes Virus 5
HMP - Human Microbiome Project
IR - Insulin Resistance
IRS - Insulin Receptor Substrate
LPS - Lipopolysaccharide
Mtorc1 - Mammalian Target of Rapamycin Complex 1
NASH - Nonalcoholic Steatohepatitis
PAMP - Pathogen-Associated Molecular Pattern
PRR - Pattern Recognition Receptor
SCFA - Short Chain Fatty Acid
TLR - Toll-Like Receptor
T2DM - Type 2 Diabetes Mellitus
UC - Ulcerative Colitis
The human microbiome
The human microbiome is a complex ecosystem, the importance of which has become widely known in the last decade. Bacterial flora colonizing 7 body parts of healthy volunteers was identified in connection with the Human Microbiome Project (HMP) using next-generation sequencing [1]. The microbiota is a set of bacterial species that colonize the human body, and the name microbiome refers to a set of bacterial genes that can be uniquely defined for each person. This makes the deviations in the background of each disease group from the healthy average statistically reproducible and exactly comparable and in the future, it may also have diagnostic significance for the individual in terms of disease risk or pathogenesis and may serve as an important step to the new era of personalized medicine [2].
Early research on the human microbiome revealed that the vast majority of bacteria colonizing the human body (about 80%) lives in the colon and is significant in terms of weight, corresponding to a total weight of approximately 2kg in an 80kg person. Regarding its total gene pool, it exceeds about 150 times the human gene pool [3].
The bacteria constituting the human microbiome have a significant effect on digestion, their metabolic products are absorbed, and they provide about 40-60% of the fatty acids circulating in the blood [4]. They play a role in the absorption of vitamins; trace elements and microelements and they also help to produce them and make them usable by the human body [5]. In terms of digestion, absorption and nutrient supply, their role is very similar to that of bacteria sitting on the hair roots of plants, which live in close symbiosis and commensalism with the host [6]. The equilibrium of their biodiversity and distribution is a good indicator of the environmental damage to which the host is / was exposed, and its cumulative effect can be traced in their gene pool measured at any given time [7].
The Western-type lifestyle, dietary habits, and mass-production agriculture-goods all had a critical effect on the composition of the human microbiome, and one the first significant result of HMP was the causative association of obesity and T2DM with the injured microbiome [8]. Since then, its pathomechanism and its relationship to insulin resistance have been mapped: it has been shown that due to the so-called propionate sensing phenomenon, bacterial short-chain fatty acids in skeletal muscle led to the activation of mTORC1 through the activation of an intracellular signaling pathway, which inhibits insulin receptor substrate phosphorylation [9].
Inhibition of the insulin signal thus plays an important role in the development of insulin resistance (IR), and together with compensatory hyperinsulinemia simultaneously causes increased glucose uptake and ketone body accumulation in adipocytes [10]. However, the falling blood glucose curve characteristic for hyperinsulinemia slows down thinking, leads to tension and encourages increased sugar intake. As the evolutionary interactions between the gut flora in the past hundreds of thousands of years of human evolution, it is not surprising that biochemical mechanisms have evolved that bacteria can “use” to “let the host know” what kind of food is optimal for their living conditions [11]. In a healthy state of symbiosis this is a fine-tuning regulator enabling a long-term adaptation in terms of the balance of the available nutrients and the human and bacterial ecosystems. Industrialized nutrition, however and in particular the consumption of high-energy processed foods have made nutrient intake more and more efficient in terms of time and energy intake. At the same time, it has created strong evolutionary pressure on bacteria that may significantly reduce the diversity of the intestinal flora during a human lifespan and subsequently evolve the microbiome pattern characteristic of obesity and T2DM [12].
- From insulin resistance to diabetes
A well-known phenomenon in medicine is that several pathogenic factors may go down one common “ultimate final” mechanism of action resulting in the characteristic of the symptomatology. A typical example is diarrhea, the ultimate common pathway of which is the pathogenesis of osmotically active intestinal contents accumulating in the colon, which prevents the reabsorption of water [13]. There can be a number of reasons why osmotically active intestinal contents enter the colon, starting with, for example, the paralysis of Na/K ATPase induced by a bacterial toxin, which forms an ion-enriched chymus [14], consumption of indigestible sugar containing food (eg lactose in case of lactose sensitivity [15]), or nutrients entering the colon after incomplete absorption from the small intestine due to accelerated motility or indigestion [16].
To the best of our knowledge, one of the important causes of insulin resistance is the overproduction or decreased use of short-chain fatty acids (SCFAs), especially propionate, produced by certain members of the intestinal flora. This is typically due to a dysbiotic intestinal flora caused by a shift in the proportion of bacterial families responsible for the overproduction of propionate. As a result, bacterial metabolism of the ingested food typically results in propionate overproduction, which causes mTORC1 activation through the previously mentioned propionate sensing mechanism described in 2018, and this leads to the inhibition of the intracellular signaling pathway from the insulin receptor [17].
As a clinician understanding its role in the development of insulin resistance and subsequently T2DM is extremely important, as the treatment planning involves lifestyle changes in terms of diet, exercise, and circadian rhythm, sleeping profiles of the patients [18].
First and foremost, it is critically important to understand the development of Type 2 diabetes trait and the causal relationship with obesity. The first step is propionate overproduction in the gut. The most important final common route seems here the development of the pathogenic propionate/butyrate metabolite levels in the gut, as the uniqueness of the intestinal flora makes it difficult to predict the ability of the 70% individual [19] pattern to produce “summa” propionate [20]. Understanding this is of great importance, as an insulin-resistant phenotype can develop also without clinical insulin resistance in case of high energy consumption. Typical examples are professional athletes [21]. In their case, neither obesity nor clinical insulin resistance are associated with the altered microbiome pattern, and as long as the increased propionate production is accompanied by constant, high-intensity muscle work associated with the shifted intestinal flora pattern, no clinical symptoms will appear. Once the propionate sensing mechanism “turns on,” however, the first symptoms are deterioration in skeletal muscle performance, rising insulin levels following IR, and disruption of skeletal muscle carbohydrate metabolism [22].
It is important to see that the components of the shifted flora which are dominant in propionate production create a predominance of propionate as long as the overgrowth persists. Thus, dietary intervention should be conscious and consistent for 3-6 months during and even after clinical symptoms, in order to maintain a diet capable of modifying the intestinal flora and to achieve a “low-energy-harvest” metabolic trait. At the same time, induction of heat production by adipose tissue and the intermittent skeletal muscle training are the most effective for maximum propionate consumption.
The ketogenic shift of “cardio” physical exertion and its maintenance of lower intensity over a long period of time was a misconception about activating adipose tissue metabolism. The idea was that the release of excess body weight is caused by the ketogenic shift of skeletal muscle work, which after a while “turns on” and is as a consequence able to mobilize the stored fat. However, indeed in case of the insulin-resistant phenotype, high insulin levels push blood sugar, even derived from a protein-dominant diet, towards SCFA storage and together with the daily sedentary intellectual work leads to reactive hypoglycemia and difficulty concentrating following post-diet insulin peaks. This, through increased coffee and snack consumption, on the one hand maintains blood sugar levels and current SCFA storage, and on the other hand the amount of caffeine consumed during the day by reducing sleep depth and quality reduces the amount of nutrients stored in nerve cells (Nisse particles, etc). The latter in turn further exposes the patient’s mental activity to prompt meals, causing constant snacking.
Thus, in the activation of skeletal muscles and adipose tissues, the intermittent load is much more efficient, which enables the body to adapt much better and to mobilize skeletal muscle energy reserves at the current peak load. Such maximum load/rest cycles have a proven effectiveness in increasing muscle mass and more effective propionate "burning" per time units. In terms of the adipose tissue, however, its role in heat management should be emphasized from an evolutionary point of view. Because of the limited time available for daily movement, restoring cold tolerance / excess heat production is of great importance. There are few mitochondria in the degenerated adipose tissue of an obese individuals [23]. It was previously a misconception that the brown adipose tissue of newborns, which contains countless mitochondria, and loss with aging turning into white adipose tissue typical of adults is normal. This is indeed a reversible, degenerative phenomenon. Swimming is an excellent method for reversing this, as with the slower muscle work the need for heat production of adipose tissue is more intense due to the lower heat production of skeletal muscles and the insulating effect of adipose tissue on a large surface due to high water conductivity does not really reduce this need. Gradual induction of all these elements: significant metabolic activity, intermittent load and continuous heat release can be induced simultaneously by metabolic activation of skeletal muscle and adipose tissue with little risk of joint damage [24].
In summary, from a practical point of view, the level of propionate circulating in the blood and reaching the tissues has a significant influence on the development of IR. Measuring propionate levels is expensive and difficult to carry out in daily clinical practice, but an increase in induced insulin levels provides a good approximation [25]. The primary therapeutic goal is to reduce propionate levels, which is possible through both increased use and reduced production, but in the long run, reducing the proportion of propionate-producing flora is unavoidable with moderate physical activity. The use of mTORC1 inhibitors, such as metformin, is an additional factor that may temporarily reverse the inhibition of IRS phosphorylation even at higher propionate levels, but it has no protective effect in terms of other complications of T2DM associated with the diseased microbiome and PAMPs. [26] (Figure 1).
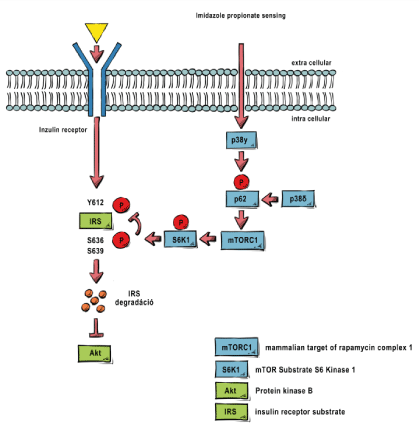
Figure 1: Propionate sensing mechanism.
Inflammation and the barrier
In addition to the metabolic effects of the intestinal flora, further important elements of the diabetes disease spectrum are inflammation and target organ complications [27]. These are closely correlated with disease progression, but to our best knowledge, they are not causally related to changes in blood glucose levels. This is crucial in the prevention of the complications of diabetes, as an important but not sufficient condition for their avoidance is the metabolic balance achieved with medication and with insulin treatment [28].
If we look at the clinical syndrome “on a skyscraper view”, this explains why these lesions are not specific to diabetes: micro- and macroangiopathy or neuropathy do not occur in diabetes only, their various spectra can also develop in an idiopathic manner, and the onset and severity can change individually [29,30].
Most evidence for the microbiome theory of complications comes from research on non-alcoholic fatty liver and steatohepatitis (NASH). Today, about 30% of liver failures requiring transplantation arise from this disease group in the U.S., so its public health significance is growing worldwide with a sharp increase together with the incidence of diabetes spectrum [31].
It is not fatty degeneration but the inflammation that lies in the center of progression of NASH [32]. The sinusoid circulation of the liver ensures that metabolites, toxins and inflammatory metabolites entering the portal venous blood from the intestine do not enter the systemic circulation directly [33].
The inflammatory effects of the bacterial cell wall components have long been established. Since the introduction of infusion therapy, a fever phenomenon commonly occurred, as in the old hospital infusion plants, due to the bacterial infection of the filling line despite pasteurization. Even though there was no living organism in the infusion solution, the biochemically lipopolysaccharide-type prokaryotic cell wall components still had a fever-causing effect [34]. This was a well-known phenomenon long before the discovery of innate immunity and its mechanisms, and these fever-causing bacterial particles were called bacterial “endotoxins” [35]. A characteristic clinical manifestation of endotoxins is the pathogenesis of acute pancreatitis, where the severity of the disease is closely related to the intestinal bacterial endotoxin translocation [36]. Given the pathogenesis, it is not surprising that prophylactic antibiotic treatment did not improve prognosis, however, restoring the integrity of the intestinal barrier by early jejunal feeding has taken a dramatic turn in the prognostic endpoints even in the case of the most severe forms of the disease [37].
Since then, the evolutionarily highly conserved biochemical signalling mechanisms of innate immunity have been described [38]. It has become clear that the biochemical differences of prokaryotic / eukaryotic cell constituents are key factors in activation and structurally highly heterogeneous molecules are also able to activate the defense mechanism. Accordingly, today they are no longer described as endotoxins or LPS, but are noted by an acronym “pathogen-associated-molecular-pattern” (PAMP) [39].
In the center of the pathogenesis of NASH there are PAMP structures entering the portal vein from the gut, which are able to activate innate immunity in the sinusoid circulation (via circulating tissue histiocytes - antigen-presenting Kupffer cells) and induce local inflammatory response [40]. The sign of this inflammation are elevated transaminase levels, the extent of which correlates with the severity of the inflammation [41]. The sinusoid circulation thus corresponds to a “filter system” for protection against intestinal bacterial infections through the portal venous system. However, this defense system is sensitive to PAMP-type bacterial antigens that enter the portal and then the sinusoid circulation through the damaged barrier. The level of PAMP concentration and the duration of inflammation together determine what local destruction will result from the phenomenon [42]. Overall, the prognosis is not different from other chronic inflammatory structural changes in the liver, which are also characteristic of chronic viral hepatitis: fibrosis followed by risk of progression to hepatocellular carcinoma [43].
Alcohol-induced liver damage has also been shed new light by this new pathogenetic theory. Alcohol can dose-effectively increase the permeability of the intestinal barrier. The exact biochemical details are still unknown, but it is probably due to its hydrophobic properties by dissolving intestinal (partly bacterial) mucus, which plays a key role in the barrier formation. The consumption of even 1 dl of wine increases the amount of PAMP entering the portal circulation by about 30%. Its individual variance is known, which is related to the genetic variability of barrier structures (e.g. tight junction) [44].
Ulcerative Colitis is on the one end of this disease spectrum, characterized by impaired barrier function and an inflammatory response to bacterial PAMPs arising from the large bowel, the greatest residue of bacteria in the human body [45]. In light of this, it is not surprising that primary sclerotic cholangitis with progressive autoimmune liver disease [46] and the concomitant sinusoidal inflammatory phenomenon, vanishing bile duct syndrome [47], formed a distinct group in terms of intestinal microbiome within ulcerative colitis patients. The fact that gluten-sensitive enteropathy shows 40% concordance in these families, which corresponds to the clinical syndrome of the genetic barrier defect affecting the small intestine, also sheds new light on pathogenesis [48]. Thus, the development of certain diseases within the same family is shaped by genetic predisposition and environmental effects. The inflammatory or barrier-damaging shift of the intestinal flora-forming species in the colon leads to UC syndrome [49], while small bowel abnormalities may shift the clinical appearance toward increased gluten load [50,51], and other factors toward celiac disease. The multifactorial pathogenesis is not yet known, but it is an intensively investigated field of gastroenterology [52].
Thus, circulating PAMP structures of intestinal origin play a central role in the development of systemic inflammation, and the nature of PAMP antigens (distribution and proportion of bacteria forming the intestinal flora) and their amount are of particular importance in their clinical manifestation [53]. The nature of PAMP antigens is primarily related to the state of the intestinal barrier [54]. Of course, PAMPs, which act as inflammatory “foci,” may not only be of intestinal origin, about 80% of the PAMPs originate from there. In addition, any infectious nodule, sepsis, can induce this process and the body’s “filter” system, depending on the foci, can react with local inflammation and with its characteristic clinical symptoms. Among the important filter systems where this pathomechanism may play a role are the kidney [55], the blood-brain barrier [56], ciliary body and the Bruch's membrane in the eye - the inflammatory filter defect of the latter is in the center of the pathogenesis of glaucoma and macular degeneration in the elderly [57].
These differ in the pathogenesis and clinical manifestation from classical immune complex disorders and also from the antigenic mimicry mechanism of rheumatic fever. Whereby analogous disease entities to the latter are common in relation to the intestinal microbiome, which will be discussed later [58]. In the intestinal microbiome the first such potentially pathogenic bacterium, Prevotella copri, was found to be responsible for up to 40% of cases of newly onset rheumatoid arthritis (NORA) [59]. In the case of Prevotella induced arthritis a number of hosts related, and environmental factors play together to induce a dominant IL17 producing inflammatory phenotype. It is the damaged gut barrier, however, that determines why some patients with high Prevotella abundance status do evolve arthritis and others do not [60]. Here again, the disease is multifactorial, in which Prevotella dysbiosis is a necessary but not sufficient factor to develop the disease [61] (Figure 2&3).
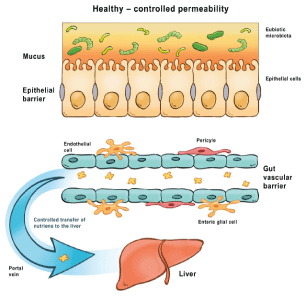
Figure 2: Pathogenesis of cirrhosis induced by pathogen-associated molecular patterns- Health controlled permeability.
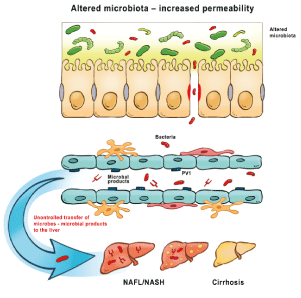
Figure 3: Pathogenesis of cirrhosis induced by pathogen-associated molecular patterns- Altred microbiota increased permeability.
Neuropathy and inflammation
Neuropathy is defined as a damage to the central or peripheral nervous system typically associated with inflammation that leads to target organ dysfunction. The clinical picture can be very diverse based on the type of nerve fibers damaged, the location of the injury, and its time course. Since the pathomechanism of neuropathies in the most clinically significant disease groups is not well understood, their classification is not precise. This is also highlighted by the fact that treating the cause of the most common forms of neuropathy is not resolved: such as neuropathy caused by diabetes [62], by old age [63], by alcohol [64] and drug-induced (iatrogenic) neuropathy [65]. Therefore, usually clinical descriptions prevail, such as sensory, motor, autonomic [66], mixed, mono- [67] and polyneuropathies [68] with connotation and with modest prognostic outlook.
Microbiome research can bring significant new insight into the pathogenesis of neuropathies. This approach is particularly important and useful, especially in the light of recent discoveries regarding the use of alcohol [69] and the pathogenesis of diabetes in association with the microbiome [70], as it provides a way to stop the progression, intervene directly and possibly provide optimal conditions for slow regeneration and possibly improve patients' quality of life until appropriate controlled randomized clinical trials are completed in the coming years and targeted therapeutic interventions will be available [71,72], especially in the case of the Guillain-Barré syndrome.
Thus, it is didactically important to start with rare neuropathies with a revealed pathomechanism that can provide insight into potential detrimental factors and their natural clinical course, which can serve as a model for the study and treatment of causes of great public health importance.
First of all, Guillain-Barre syndrome needs to be mentioned: it is caused by an autoimmune inflammation trigerred by antibodies to PAMP structures that are cross-reactive with gangliosides during the diverse but in many cases induced immune response following intestinal infections through an antigen-mimic mechanism. The nature and severity of the clinical appearance (the subtype of GBS) depend on the infection causing it and the specificity of the antibodies produced. In terms of time course, it is an acute disease with rapid progression that occurs over a few days, typically affecting the motor innervation of the lower limb first and spreading proximally during its progression and also affecting the autonomic nervous system. Due to its extensive appearance, it is classified as a group of polyneuropathies. The most common cause is Campylobacter jejuni infection, which causes about 30% of all cases. An additional approximately 10% is caused by a variety of cytomegalovirus HCMV-HHV-5 [73]. Associations with GBS have been described in several other viral and bacterial infections, but association with vaccines is of more significant importance. The influenza vaccine should be highlighted, incidence of about 5-6 cases per 100 thousand inhabitants have been described. However, it has been shown that GBS can occur not only induced by the vaccine but also induced by the virus itself, so there is no doubt about the benefit of vaccination [74].
Glycolipids are involved in the development of innate immunity as important antigenic epitopes and are able to activate the TLR system, thus participating in the adhesion, initiation and activation of migration of inflammatory cells.
The clinical presentation of neuropathies did not correlate with TLR polymorphism and given the known course of GBS and the known pathological factors and antigenic mimicry mechanisms, most likely, most of these initiate neuronal damage through the antigen mimicry mechanism and based on the nature of the epitope will define its demyelinating or axonal form, its localization and specificity [75].
The classical acute forms of these diseases are associated with invasive intestinal infections, where the humoral immune response against bacterial cell wall epitopes also produce IgG antibodies instead of the adequate IgA type, locally effective immunoglobulin class. The regulation of this process and the defect of the regulation is one of the pivotal points in the mimicry formation of the antigen.
Autoimmunity develops when IgG antibodies raised against bacterial glycolipid cell wall epitopes cross-react with glycolipid groups on the neuronal membrane, primarily with ganglioside molecules: with ganglioside GM1, GD1a, GT1a and GQ1b groups [76]. For example, an antibody linked to the GQ1b molecule may be associated with the Miller Fisher subtype and related forms of GBS, including Bickerstaff encephalitis (BBE) [77].
The IgA / IgG “switch”, one of the keys to pathomechanism, has caused significant debates for decades. Genetic and HLA-dependent mechanism were investigated but not concluded before the discovery of the antigen presenting role of the intestinal microfold cells. It turned out that in the case of normal intestinal barrier function, bacterial antigens are typically presented through microfold cells, resulting in an IgA secretory immune response [78]. The significance is that it has no autoimmune potential through its local effect.
However, to the best of our knowledge, the development of an IgM / G antibody switch is not due to a genetic predisposition, but due to an alternative antigen presentation: in addition to impaired barrier function, bacterial antigen epitopes entering the interstitial space or, in the case of intestinal antigens, typically into the portal circulation, are able to induce a systemic, humoral immune response, which has an autoimmune potential known in a very broad spectrum. In neuropathies, for example, against the synovial membrane, the first major actor associated with the microbiome within the group of non-invasive intestinal bacteria was Prevotella copri. Also in this case, a non-invasive PAMP can provoke severe autoimmune arthritis by crossing the barrier. At the same time, when the barrier function is intact, Prevotella copri is an active member of the human intestinal flora in fiber breakdown and can typically enrich the intestinal flora in connection with raw vegan nutrition, without any pathological concomitant factors.
In both cases we are facing multifactorial pathogenesis centered on a glycolipid bacterial cell wall epitope, or other PAMP structure that crosses the intestinal or other barrier. The development of antigen mimicry and autoimmunity – in contrast to the invasive bacteria underlying GBS – analogous to invasive airway or urogenital pathogens known for 100 years, reactive arthritis includes antigenic epitopes of non-invasive bacteria. Accordingly, the course of the disease will not be “digital”: zero or one, but autoimmunity will be fluctuating and proportional to the amount of antigenic epitopes that always penetrate the barrier - which is also influenced by the slow and limited regeneration potential of these structures. The clinical picture is thus much more colorful, less homogeneous and in terms of time it has a long-time course.
Today, the role of more than 20 different glycolipid epitopes has been demonstrated in acute and chronic neuropathic syndromes. In particular, significant data is available regarding antibodies to acute axonal neuropathies and gangliosides GM1, GD1a, GM1b and GalNAc-GD1a, as well as antibodies against cranial, bulbar and sensory forms of GBS and gangliosides of GQ1b, GT1a, GD1b and GD3. The structure of these and the relevant bacterial PAMP structures, and Campylobacter jejuni are well documented and the correlation with neuronal attack points and their clinical manifestations have also been described. In parallel, the pathomechanism has been demonstrated in experimental models in vitro and in vivo [79].
The role of PAMP translocation-induced innate immunity is also likely, its exact mechanism is less revealed, but its significance in the development of the syndrome accompanying neuropathy is well documented, including in neuropathic pain. Pattern recognition receptors here appear to play a prominent role in the development of pain and neurogenic inflammation following peripheral nerve damage. Most data are related to the TLR family, while the role of other PRRs is largely unknown [80].
Take home messages for the clinician
From all the above, modest, yet significant consequences can be drawn for the clinical practice: just like in the case of Guillain-Barré syndrome, there is hope for treating the cause and cessation of progression in diabetic peripheral neuropathies, which has a chronic long but progessive clinical course. historically.
The first step is the inhibition of abnormal bacterial PAMP-translocation. This is especially important in the light of the fact that some of the analgesics used in symptomatic treatment, non-steroidal anti-inflammatory drugs and socially acceptable levels of alcohol consumption significantly increase the permeability of the intestinal barrier.
In addition, certain foods and spices also increase the concentration of PAMPs measured in the portal circulation, e.g. chili, spicy food, high oxalate containing vegetables (raw spinach in salads, etc), emulsifier food additives (ones used in sauces and vegetable milk products).
Until potential bacterial antigenic epitopes are mapped and subjected to either vaccination with specific anti-idiotype or the antigen source itself is eliminated (eg through fecal transplantation) - patients can be provided with simple and inexpensive everyday lifestyle advices, and specific prebiotic and probiotic strategies can help as well by supporting the restoration of the barrier function.
References
- Human microbiome project consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. [Crossref]
- Yohe S, Thyagarajan B (2017) Review of clinical next-generation sequencing. Arch Pathol Lab Med 141: 1544-1557. [Crossref]
- Bermon S, Petriz B, Kajėnienė A, Prestes J, Castell L, et al. (2015) The microbiota: an exercise immunology perspective. Exerc Immunol Rev 21: 70-79. [Crossref]
- Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT (1987) Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 10: 1221-1227. [Crossref]
- Hill MJ (1997) Intestinal flora and endogenous vitamin synthesis. Eur J Cancer Prev 6: S43-S45. [Crossref]
- van der Heijden MG, Bardgett RD, van Straalen NM (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11: 296-310. [Crossref]
- Wu Z, Liu Q, Li Z, Cheng W, Sun J, et al. (2018) Environmental factors shaping the diversity of bacterial communities that promote rice production. BMC Microbiol 18: 51.
- Cani PD (2014) Metabolism in 2013: The gut microbiota manages host metabolism. Nat Rev Endocrinol 10: 74-76. [Crossref]
- Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, et al. (2018) Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 175: 947-961. [Crossref]
- Smith U (2002) Impaired ('diabetic') insulin signaling and action occur in fat cells long before glucose intolerance--is insulin resistance initiated in the adipose tissue? Int J Obes Relat Metab Disord 26: 897-904. [Crossref]
- Leitão-Gonçalves R, Carvalho-Santos Z, Francisco AP, Fioreze GT, Anjos M, et al. (2017) Commensal bacteria and essential amino acids control food choice behavior and reproduction. PLoS Biol 15: e2000862. [Crossref]
- Bibbò S, Ianiro G, Giorgio V, Scaldaferri F, Masucci L, et al. (2016) The role of diet on gut microbiota composition. Eur Rev Med Pharmacol Sci 20: 4742-4749. [Crossref]
- Field M (2003) Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest 111: 931-943. [Crossref]
- Marchelletta RR, Gareau MG, McCole DF, Okamoto S, Roel E, et al. (2013) Altered expression and localization of ion transporters contribute to diarrhea in mice with Salmonella-induced enteritis. Gastroenterology 145: 1358-1368. [Crossref]
- Misselwitz B, Butter M, Verbeke K, Fox MR (2019) Update on lactose malabsorption and intolerance: pathogenesis, diagnosis and clinical management. Gut 68: 2080-2091. [Crossref]
- Murray JA, Rubio-Tapia A (2012) Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol 26: 581-600. [Crossref]
- Molinaro A, Bel Lassen P, Henricsson M, Wu H, Adriouch S, et al. (2020) Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nature Commun 11: 5881. [Crossref]
- Kerrison G, Gillis RB, Jiwani SI, Alzahrani Q, Kok S, et al. (2017) The effectiveness of lifestyle adaptation for the prevention of prediabetes in adults: a systematic review. J Diabetes Res 2017: 8493145. [Crossref]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59-65. [Crossref]
- Zhang S, Wang H, Zhu MJ (2019) A sensitive GC/MS detection method for analyzing microbial metabolites short chain fatty acids in fecal and serum samples. Talanta 196: 249-254.
- Emami M, Behforouz A, Jarahi L, Zarifian A, Rashidlamir A, et al. (2018) The risk of developing obesity, insulin resistance, and metabolic syndrome in former power-sports athletes - does sports career termination increase the risk. Indian J Endocrinol Metab 22: 515-519. [Crossref]
- Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, et al. (2010) Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5: e9085. [Crossref]
- Lee JH, Park A, Oh K, Lee SC, Kim WK, et al. (2019) The role of adipose tissue mitochondria: regulation of mitochondrial function for the treatment of metabolic diseases. Int J Mol Sci 20: 4924. [Crossref]
- Barbosa MA, Guerra-Sá R, De Castro UGM, de Lima WG, Dos Santos RAS, et al. (2018) Physical training improves thermogenesis and insulin pathway, and induces remodeling in white and brown adipose tissues. J Physiol Biochem 74: 441-454. [Crossref]
- Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, et al. (2019) Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet 51: 600-605. [Crossref]
- Koh A, Mannerås-Holm L, Yunn N, Nilsson PM, Ho Ryu S, et al. (2020) Microbial imidazole propionate affects responses to metformin through p38γ-dependent inhibitory AMPK phosphorylation. Cell Metab 32: 643-653.e4. [Crossref]
- Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP (2013) Diabetes mellitus and inflammation. Curr Diab Rep 13: 435-444. [Crossref]
- Chaturvedi N (2007) The burden of diabetes and its complications: trends and implications for intervention. Diabetes Res Clin Pract 76: S3-S12. [Crossref]
- Huysman E, Mathieu C (2009) Diabetes and peripheral vascular disease. Acta Chir Belg 109: 587-594. [Crossref]
- Zakin E, Abrams R, Simpson DM (2019) Diabetic neuropathy. Semin Neurol 39: 560-569. [Crossref]
- Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, et al. (2015) Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148: 547-555. [Crossref]
- Cobbina E, Akhlaghi F (2017) Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metabolism Rev 49: 197–211. [Crossref]
- Son G, Kremer M, Hines IN (2010) Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Pract 2010: 453563. [Crossref]
- Magalhães PO, Lopes AM, Mazzola PG, Rangel-Yagui C, Penna TC, et al. (2007) Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci 10: 388-404. [Crossref]
- Pfeiffer R (1892) Untersuchungen über das Choleragift. Zeitschrift für Hygiene und Infektionskrankheiten 11: 393–412.
- Ammori BJ, Leeder PC, King RF, Barclay GR, Martin IG, et al. (1999) Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg 3: 252-262. [Crossref]
- Windsor AC, Kanwar S, Li AG, Barnes E, Guthrie JA, et al. (1998) Compared with parenteral nutrition, enteral feeding attenuates the acute phase response and improves disease severity in acute pancreatitis. Gut 42: 431-435. [Crossref]
- De Arras L, Seng A, Lackford B, Keikhaee MR, Bowerman B, et al. (2013) An evolutionarily conserved innate immunity protein interaction network. J Biol Chem 288: 1967-1978. [Crossref]
- Ray A, Cot M, Puzo G, Gilleron M, Nigou J (2013) Bacterial cell wall macroamphiphiles: pathogen-/microbe-associated molecular patterns detected by mammalian innate immune system. Biochimie 95: 33-42. [Crossref]
- Bán O, Lisziewicz J, Nyúl D, Peták I, Tordai A, et al. (2019) A bélmikrobiom szerepe az alkoholos és nem alkoholos zsírmáj kialakulásában, progressziójában. Central Eur J Gastroenterolo Hepatol 5: 129-134.
- Wang L, Li J, Yang K, Zhang H, Wang Q, et al. (2020) Comparison and evaluation of non-invasive models in predicting liver inflammation and fibrosis of chronic hepatitis B virus-infected patients with high hepatitis B virus DNA and normal or mildly elevated alanine transaminase levels. Medicine 99: e20548. [Crossref]
- Son G, Kremer M, Hines IN (2010) Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Pract 2010: 453563. [Crossref]
- Alisi A, Carsetti R, Nobili V (2011) Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology 54: 1500-1502. [Crossref]
- Antón M, Rodríguez-González A, Ballesta A, González N, Del Pozo A, et al. (2018) Alcohol binge disrupts the rat intestinal barrier: the partial protective role of oleoylethanolamide. Br J Pharmacol 175: 4464-4479. [Crossref]
- Johansson ME, Gustafsson JK, Holmén-Larsson J, Jabbar KS, Xia L, et al. (2014) Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63: 281-291. [Crossref]
- Little R, Wine E, Kamath BM, Griffiths AM, Ricciuto A (2020) Gut microbiome in primary sclerosing cholangitis: A review. World J Gastroenterol 26: 2768-2780. [Crossref]
- Reau NS, Jensen DM (2008) Vanishing bile duct syndrome. Clin Liver Dis 12: 203-217. [Crossref]
- Shah A, Walker M, Burger D, Martin N, von Wulffen M, et al. (2019) Link between celiac disease and inflammatory bowel disease. J Clin Gastroenterol 53: 514-522. [Crossref]
- Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, et al. (2019) Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4: 293-305. [Crossref]
- Cardoso-Silva D, Delbue D, Itzlinger A, Moerkens R, Withoff S, et al. (2019) Intestinal barrier function in gluten-related disorders. Nutrients 11: 2325. [Crossref]
- Hamilton I, Cobden I, Rothwell J, Axon AT (1982) Intestinal permeability in coeliac disease: the response to gluten withdrawal and single-dose gluten challenge. Gut 23: 202-210. [Crossref]
- Leccioli V, Oliveri M, Romeo M, Berretta M, Rossi P (2017) A new proposal for the pathogenic mechanism of non-coeliac/non-allergic gluten/wheat sensitivity: piecing together the puzzle of recent scientific evidence. Nutrients 9: 1203. [Crossref]
- Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124: 783-801. [Crossref]
- Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, et al. (2018) The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 15: 397-411. [Crossref]
- Gomez H, Ince C, De Backer D, Pickkers P, Payen D, et al. (2014) A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41: 3-11. [Crossref]
- Varatharaj A, Galea I (2017) The blood-brain barrier in systemic inflammation. Brain Behav Immun 60: 1-12. [Crossref]
- Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K (2016) Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci 73: 1765-1786. [Crossref]
- Negi S, Singh H, Mukhopadhyay A (2017) Gut bacterial peptides with autoimmunity potential as environmental trigger for late onset complex diseases: In-silico study. PLoS One 12: e0180518. [Crossref]
- Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, et al. (2019) Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis 78: 590-593. [Crossref]
- Mu Q, Kirby J, Reilly CM, Luo XM (2017) Leaky gut as a danger signal for autoimmune diseases. Front Immunol 8: 598. [Crossref]
- Horta-Baas G, Del Socorro Romero-Figueroa M, José Montiel-Jarquín A, Luisa Pizano-Zárate M, García-Mena J, et al. (2017) Intestinal dysbiosis and rheumatoid arthritis: a link between gut microbiota and the pathogenesis of rheumatoid arthritis. J Immunol Res 2017: 4835189. [Crossref]
- Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, et al. (2019) Diabetic neuropathy. Nat Rev Dis Primers 5: 41. [Crossref]
- Bouche P (2020) Neuropathy of the elderly. Rev Neurol 176: 733-738. [Crossref]
- Julian T, Glascow N, Syeed R, Zis P (2019) Alcohol-related peripheral neuropathy: a systematic review and meta-analysis. J Neurol 266: 2907-2919. [Crossref]
- Jones MR, Urits I, Wolf J, Corrigan D, Colburn L, et al. (2020) Drug-induced peripheral neuropathy: a narrative review. Curr Clin Pharmacol 15: 38-48. [Crossref]
- Schwartzlow C, Kazamel M (2019) Hereditary sensory and autonomic neuropathies: adding more to the classification. Curr Neurol Neurosci Rep 19: 52. [Crossref]
- Sanjaya A (2020) Meralgia paresthetica: finding an effective cure. Postgrad Med 132: 1-6. [Crossref]
- Siao P, Kaku M (2019) A clinician's approach to peripheral neuropathy. Semin Neurol 39: 519-530. [Crossref]
- Caslin B, Maguire C, Karmakar A, Mohler K, Wylie D, et al. (2019) Alcohol shifts gut microbial networks and ameliorates a murine model of neuroinflammation in a sex-specific pattern. Proc Natl Acad Sci U S A 116: 25808-25815. [Crossref]
- Sikalidis AK, Maykish A (2020) The gut microbiome and type 2 diabetes mellitus: discussing a complex relationship. Biomedicines 8: 8. [Crossref]
- Saxena A (2016) Probiotics as a potential alternative for relieving peripheral neuropathies: a case for Guillain-Barré syndrome. Front Microbiol 6: 1497. [Crossref]
- Brooks PT, Brakel KA, Bell JA, Bejcek CE, Gilpin T, et al. (2017) Transplanted human fecal microbiota enhanced Guillain Barré syndrome autoantibody responses after Campylobacter jejuni infection in C57BL/6 mice. Microbiome 92.
- Yuki N, Hartung HP (2012) Guillain-Barré syndrome. N Engl J Med 366: 2294-304. [Crossref]
- Lehmann HC, Hartung HP, Kieseier BC, Hughes RA (2010) Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis 10: 643-651. [Crossref]
- Willison HJ, Goodyear CS (2013) Glycolipid antigens and autoantibodies in autoimmune neuropathies. Trends Immunol 34: 453-459. [Crossref]
- Onodera M, Mori M, Koga M, Kamitsukasa I, Fukutake T, et al. (2002) Acute isolated bulbar palsy with anti-GT1a IgG antibody subsequent to Campylobacter jejuni enteritis. J Neurol Sci 205: 83-84. [Crossref]
- Shahrizaila N, Yuki N (2013) Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 84: 576-83. [Crossref]
- Rios D, Wood MB, Li J, Chassaing B, Gewirtz AT, et al. (2016) Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol 9: 907-916. [Crossref]
- Willison HJ, Yuki N (2002) Peripheral neuropathies and anti-glycolipid antibodies. Brain 125: 2591-2625. [Crossref]
- Ishikawa A, Miyake Y, Kobayashi K, Murata Y, Iizasa S, et al. (2019) Essential roles of C-type lectin Mincle in induction of neuropathic pain in mice. Sci Rep 9: 872. [Crossref]