stomatocytosis, spherocytosis, protein 4.2
Hereditary spherocytosis (HS) is a type of inherited hemolytic anemia that is characterized by chronic, non-immune hemolysis with spherocytes on peripheral smear, high mean corpuscular hemoglobin concentration (MCHC) and increased osmotic fragility. The most common mode of inheritance is autosomal dominant. Ankyrin-1 deficiency is the most frequent cause of HS in USA and Europe. In contrast, deficiency of protein 4.2 secondary to autosomal recessive mutation in EPB42 gene is the most common cause of HS in Japan and is a very rare occurrence in Caucasian population. Moreover, heterozygous mutation in the gene (EPB42) encoding for protein 4.2 has never been shown to cause symptomatic hemolytic anemia [1]. Here we present a case of spherocytosis in a patient with parenteral nutrition-associated liver disease (PNALD) who was found to have a heterozygous novel mutation in the erythrocyte protein 4.2 gene. The patient was also found to have a remarkable stomatocytosis. We speculate that the concomitant occurrence of stomatocytosis and spherocytosis in this patient is a rare phenomenon as result of a combination of his genetic mutation and acquired risk factors for development of symptomatic hemolytic anemia. Post splenectomy thromboembolic complications of hereditary stomatocytosis have been described earlier [2]. Interestingly, our patient developed portal veinous thrombosis without splenectomy.
A 13-month-old European American male with history of gastroschisis, jejunal atresia, short gut syndrome following bowel resection and parenteral nutrition-associated liver disease (PNALD) was referred to our clinic for severe anemia. Family history was not suggestive of any hemolytic anemia, gallstones, splenectomy or multiple blood transfusions. At the time of presentation, he had failure to thrive along with mild icterus and hepatosplenomegaly. Laboratory tests revealed a hemoglobin of 8.6 gm/dL6.2 g/dL, with a reticulocyte count of 8.8 %, MCV of 83.5 fL, MCHC: 32.9 g/dL) with undetectable haptoglobin and elevated LDH (530 IU/L) indicating hemolysis. Patient had both direct and indirect hyperbilirubinemia. Peak total bilirubin was 8.0 gm/dL with a direct of 5.7 gm/dL. Blood smear was significant for remarkable stomatocytosis and occasional spherocytes at thirteen months of age. Over the next six months, anemia worsened, reaching a nadir of 6.2 gm/dL, necessitating blood transfusion. During the same time, stomatocytosis became more prominent along with increase in MCV (peak MCV- 115 fL), and a drop in MCHC with a nadir of 25.9 g/dL. He also developed progressive thrombocytopenia (platelet count was in the range of 72,000- 102,000/µL). Eosin-5-maleimide screening test for hereditary spherocytosis was normal. His blood group was O-ve and direct antibody test (DAT) was negative. RBC folate and serum Vitamin B12 were within normal level. Next generation sequencing of 10 genes associated with red blood cell membrane defects (ANK1, EPB41, EPB42, PIEZO1, RHAG, SLC2A1, SLC4A1, SPTA1, SPTB and XK) showed a novel heterozygous mutation in EPB42 gene (c.826C > T; p.R276W) resulting in substitution of tryptophan for arginine at amino acid 276 in RBC membrane protein 4.2. Ektacytometry confirmed increased osmotic fragility (Omin 169.5), and decrease EImax (0.548), consistent with mild hereditary spherocytosis (Figure 1 and Figure 2). Through the use of mutation prediction software, this amino acid substitution is predicted to be "deleterious" and "probably damaging". Immunoblotting in the RBC ghosts demonstrated presence of at least 50% protein 4.2 indicating that the other EPB42 allele is normal (Figure 3). Other relevant lab tests for hemolytic anemia including glucose- 6- phosphate dehydrogenase, hemoglobin electrophoresis and pyruvate kinase screen were within normal limit. Later, patient developed massive hepatosplenomegaly with progressive worsening of direct and indirect hyperbilirubinemia with transamnitis (peak ALT of 237 IU/L and AST of 613 IU/L). Patient was treated with iron on couple of occasions for borderline MCV, however it was not shown to significantly improve anemia. Transfusions were also given as needed for anemia (7 gm/dL or he is symptomatic. Splenectomy was not performed due to contradicting indications for surgery in patients with stomatocytosis. At the age of 23 months, an ultrasound of abdomen revealed nonocclusive thrombus within the main portal vein, with sluggish (5 cm/sec) antegrade flow along with a small amount of ascites, a small gall stone, hepatosteatosis and massive hepatosplenomegaly. Clinical, laboratory and imaging findings were consistent with signs of liver failure. At the last follow up, patient was awaiting a liver transplant. Lab tests on parental blood samples could not be done since the patient’s father was untraceable and the patient was later lost to follow up due to difficult social situations.
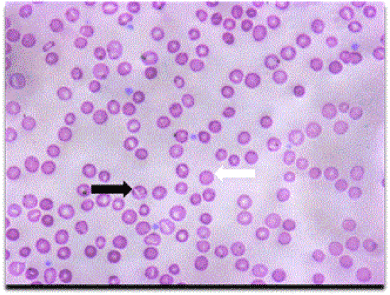
Figure 1. Patient’s peripheral smear showing typical numerous stomatocytes (black arrow) and spherocytes (white arrow).
Figure 2. Ektacytometry curve of the patient showing shift to the right of Omin and Hyper point.
Figure 3. Immunoblotting for protein 4.2 and GAPDH demonstrates decreased but not absent protein 4.2 in the patient’s RBC ghosts, compatible with heterozygous EPB42 mutation.
We present a unique case of concomitant occurrence of stomatocytosis and spherocytosis in a patient with heterozygous novel mutation in the erythrocyte protein 4.2 gene and PNALD. This case presented us with a diagnostic dilemma and proved to be challenging in making therapeutic decision regarding splenectomy since hereditary stomatocytosis is a contraindication due to the risk of life threatening deep veinous thrombosis [2].
RBC phenotype of our patient showed a progressive increase in MCV reaching > 110fL, with a decrease in MCV in the 25-30 g/dL range. Metabolic causes for macrocytosis were ruled out due to a normal serum vitamin B12 and RBC folate levels. These findings can be explained by impressive stomatocytosis. In hereditary stomatocytosis the red blood cells have an increased MCV especially the hereditary overhydrated stomatocytosis (HOS) variant where MCV is higher than 110 fL with decreased or normal MCHC [3].
Genetic testing with NGS showed a heterozygous novel mutation in EPB42 gene. The immunoblot showed a partial deficiency of protein 4.2 consistent with heterozygosity. Hereditary spherocytosis secondary to autosomal recessive mutations in EPB42 gene is characterized by stomatocytes and spherocytes on peripheral smear similar to our patient. However, heterozygous mutation in EPB42 gene has never been shown to cause of symptomatic spherocytic anemia [4,5]. Even though we were not able to conclusively prove a causal link between heterozygous mutation in protein 4.2 (EPB42) gene and symptomatic spherocytic anemia, we speculate that the severe anemia was a product of partial deficiency of protein 4.2 and other acquired factors like hypersplenism and lipid abnormalities of RBC membrane secondary to TPN/PNALD. Also, we did not find any genetic mutation consistent with hereditary stomatocytosis, a rare entity that has been recently linked to a mutation in FAM38A gene encoding for PIEZO1 protein or a deficiency of stomatin [3]. Hence, the impressive stomatocytosis could have been due to red cell membrane lipid abnormality secondary to TPN dependence as has been previously described in neonates receiving total parenteral nutrition [6]. Stomatocytosis has also been reported secondary to chronic liver disease as was the case in our patient. Certain drugs are known to be stomatocytogenic in vitro but our patient was not on any drugs that are known to cause stomatocytosis [7,8]. A decrease in stomatin protein, a red blood cell membrane protein that acts as a channel regulator, has also been implicated in HOS [9]. This specialized study of stomatin protein, along with cation content of RBCs and lipid profile of RBC membrane, would have been the next logical step in the evaluation of this patient. Unfortunately, further investigation to delve into the genetic versus acquired cause of the anemia was compromised due to the loss of patient to follow up and unavailability of parental blood samples for genetic testing.
As described by King, et al. a combination of lab tests for HS increases the sensitivity of the diagnosis. Hence, we performed Ektacytometry in combination with gene testing and immunoblot test for protein 4.2 [10]. Ektacytometry testing for HS is characterized by decreased deformability index (DI), increased in osmotic fragility (increased Omin; osmolality at which 50% of red blood cells hemolyze) [1] as was seen in our patient. However, a similar ektacytometric curve may also be seen in HOS. However, even though OHS couldn’t be ruled out in our patient, it is an extremely rare clinical condition with only 20 cases reported so far [3,11]. Also, due to the 2 different phenotypes of RBCs present in our patient the ektacytometric curve was probably a reflection of both stomatocyte and spherocyte, making the interpretation of ektacytometric curve quite difficult. Additionally, heterozygous mutation in the protein RhAG, a glycoprotein associated with the Rh complex, is known to cause HOS. A complete lack of RhAG, or Rh null patients, may have a concurrent spherocytosis and stomatocytosis on smear but RhAG was found to be normal in our patient, thereby ruling out this diagnosis [11,12].
Another challenging aspect of this interesting case was the occurrence of refractory transfusion dependent anemia with massive splenomegaly. The case presented as a therapeutic challenge since a decision had to be taken about splenectomy for refractory hemolytic anemia with transfusion dependence and failure to thrive. Splenectomy is considered to be the treatment of choice for severe hemolytic anemia in HS patients [1]. However, hereditary stomatocytosis has been associated with hypercoagulable state and splenectomy is contraindicated due to considerable risk of life threatening deep veinous thrombosis, especially involving the portal vein [2]. Hence, splenectomy was not recommended. Unfortunately, our patient developed portal vein thrombosis despite avoiding splenectomy.
In summary, our case highlights a diagnostic dilemma about the relevance of a novel heterozygous mutation in EPB42 genes in causing symptomatic anemia in the setting of PNALD Further research would lead to a better understanding of the effects of TPN, chronic liver disease and lipid abnormality of the RBC membrane in causing hemolytic anemia. Finally, even though our case leaves many interesting unanswered questions, it alerts the physicians about the catastrophic consequences of hypercoagulability and splenectomy in patients with stomatocytosis. These findings have the potential to open new avenues to decipher links between RBC membrane defects and hypercoagulability.
No source of support needs to be acknowledged.
No funding was received from any institution, agency or any other source for this work.
- Perrotta S, Gallagher PG, Mohandas N (2008) Hereditary spherocytosis. Lancet 372: 1411-1426. [Crossref]
- [Crossref] Stewart GW, Amess JA, Eber SW, Kingswood C, Lane PA, et al. (1996) Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol 93: 303-310.
- [Crossref] Bruce LJ (2009) Hereditary stomatocytosis and cation-leaky red cells--recent developments. Blood Cells Mol Dis 42: 216-222.
- Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, et al. (2000) Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol 71: 118-135.
- Authors Kalfa TA, Connor JA, Begtrup AH (2016) EPB42-Related Hereditary Spherocytosis. [Crossref]
- Ross JD, Mendoza PE (1978) Acquired stomatocytosis in infants receiving parenteral fat emulsion. Pediatric Research 12: 473-473.
- Davidson RJ, How J, Lessels S (1977) Acquired stomatocytosis: its prevalence of significance in routine haematology. Scand J Haematol 19: 47-53. [Crossref]
2021 Copyright OAT. All rights reserv
- Wislöff F, Boman D (1979) Acquired stomatocytosis in alcoholic liver disease. Scand J Haematol 23: 43-50. [Crossref]
- Fricke B, Argent AC, Chetty MC, Pizzey AR, Turner EJ, et al. (2003) The "stomatin" gene and protein in overhydrated hereditary stomatocytosis. Blood 102: 2268-2277. [Crossref]
- King MJ, Garçon L, Hoyer JD, Iolascon A, Picard V, et al. (2015) International Council for Standardization in Haematology. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol 37: 304-325.
- Da Costa L, Galimand J, Fenneteau O, Mohandas N (2013) Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev 27: 167-78.
- Bruce LJ, Guizouarn H, Burton NM, Gabillat N, Poole J, et al. (2009) The monovalent cation leak in overhydrated stomatocytic red blood cells results from amino acid substitutions in the Rh-associated glycoprotein. Blood 113: 1350-1357.