Instability of CAG triplet repeat encoding poly-glutamine (polyQ) stretch in the androgen receptor (AR) gene has been implicated as a putative mechanism in spinal bulbar muscular atrophy (SBMA) or Kennedy’s disease. Although the underlying mechanisms are not completely understood, suggested pathological pathways of SBMA involve the formation of AR nuclear and cytoplasmic aggregates. Here we discuss the role of polyQ chain length extension in the pathophysiology of SBMA and the potential therapeutic targets.
Androgen receptor, spinal bulbar muscular atrophy, Neurodegenerative disease, aggregation
Instability of repetitive DNA sequences within the genome is associated with a number of human diseases [1]. The largest group of triplet repeat diseases remains those in which the expansion resides in the gene coding sequences and comprises expression of expanded poly-glutamine (polyQ) tracts [2-4]. Among such DNA repeats, expansion of trinucleotide repeats is recognized as a major cause of neurodegenerative diseases [5,6]. For example, instability of CAG triplet repeat encoding polyQ stretches in the target gene has been implicated as a putative mechanism in inherited neurodegenerative diseases such as Huntington’s and Kennedy’s or Spinal bulbar muscular atrophy (SBMA). Although the target proteins associated with these phenotypes are quite distinct, the mutated forms share an expanded CAG gene product as a mediator of protein misfolding/aggregation [7,8]. The SBMA is an X-linked progressive motor neuron disease, without affecting other neurons [9,10]. The clinical features of SBMA correlate with a loss of lower motor neurons in the brainstem and spinal cord, and with marked myopathic and neurogenic changes in skeletal muscle. Numerous studies have shown that that androgen receptor (AR) gene (Figure 1) with expanded CAG codon repeats exhibits selective neurological phenotypes and neurodegeneration and causes spinal and SBMA or Kennedy’s disease [11-14]. This pathogenic repeat expansion in the AR gene leads to loss of AR function and nuclear aggregation in motor neurons.
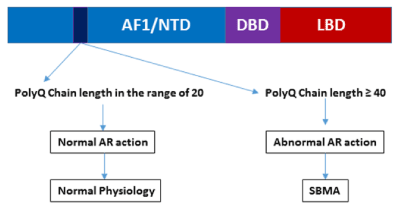
Figure 1. A diagram showing possible physiological and pathological scenarios due to altered polyQ chain length in the AR.
It has been reported that initiation of SBMA pathogenesis is closely related to the formation of androgen-dependent intracellular AR aggregates due to production of aberrant conformational changes in the AR protein [11,15,16]. These conformational changes could be linked to the release of associated chaperone protein complexes and/or to the AR nuclear translocation. In fact, nuclear localization appears to be associated with AR-polyQ neurotoxicity as cytoplasmic retention of AR-polyQ has been correlated with a decrease in its toxicity [17,18]. Normal individuals have ~20 repeats, while SBMA patients have ~40 or more CAG repeats. Extended polyQ chain length induces the formation of inclusion bodies in the affected neurons, suggesting that proteolytic cleavage may be responsible for enhanced toxicity of the AR gene products [10,11]. Interestingly, the AR-polyQ becomes toxic only after its activation by endogenous androgen ligands, possibly because of aberrant androgen-induced conformational changes of the AR-polyQ, which generate misfolded species [16]. Certain AR coregulatory proteins are also reported to be sequestered into the nuclear inclusions in SBMA supports the notion that AR transcriptional dysregulation may be a potential pathological mechanism leading to SBMA. Due to critical role of these coregulatory proteins including coactivators in the AR-mediated gene regulation, any dysregulation of AR:coactivator binding may perturb normal AR physiology leading to disease conditions [19,20].
In PolyQ neurodegenerative disorders including SBMA, the mutant protein disrupts multiple downstream pathways, and toxicity likely results from the cumulative effects of altering a diverse array of cellular processes, suggesting that potential treatments targeting a single downstream pathway are likely to be incomplete and unsuccessful. In recent years, our understanding of the underlying mechanisms that appear to play a significant role in SBMA pathophysiology has increased dramatically, and several of these pathways and mechanisms have been investigated as possible therapeutic targets. Some of the therapeutic strategies that have been tested in SBMA include gene silencing, protein quality control and/or increased protein degradation, androgen deprivation, and modulation of AR activity and functions. Evidence from various studies that include both in vitro and in vivo models support a role for testosterone binding and nuclear translocation of the AR as the trigger for SBMA [21-27]. However, trails using antiandrogen therapy, commonly used in the treatment of advanced prostate cancer, yielded disappointing findings, despite highly promising animal studies. Other studies have shown AR:HSP70 complex and small molecules such as trehalose as a potential therapeutic target for SBMA [25,28]. Based on these studies, a potential therapeutic model can be proposed (Figure 2). Finally, it is also now clear that in addition to a greater understanding of the molecular mechanisms that underlie disease, the development of an effective disease modifying therapy for polyQ-mediated neurodegenerative diseases such as SBMA will require a coordinated research effort with diverse areas of scientific and clinical expertise as well as patient groups.
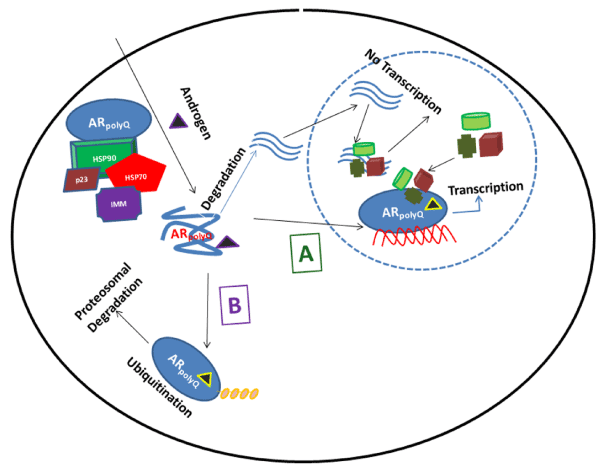
Figure 2. A therapeutic model of the regulation of the AR polyQ. The expanded AR polyQ tract allows transition into a distinct conformation that may cause toxicity as a monomer or it may self-associate to form toxic oligomers, which could assemble into larger aggregates leading to intracellular inclusions. The principal toxic effects of the aberrantly folded protein may include alterations in transcription, metabolism or impairment of the proteasome or stress response pathways. Under pathological conditions (due to polyQ chain length extension), the structure of natively-folded functional WT AR protein (AR WT) is compromised due to partial folding (AR polyQ) that could result in unwanted degradation. These polyQ containing peptide fragments enter the nucleus, sequester AR coregulatory proteins resulting into formation of intranuclear inclusions and thereby blocking the AR-mediated transcriptional regulation. The AR structure modulators (A) can convert (AR polyQ) back into (AR WT) conformation, and thereby restoring proper functions of protein by allowing it to translocate to nucleus where it binds to androgen response element (ARE) DNA sequences and interacts with critical coregulatory proteins (shown by different colors and shapes) leading to the AR target gene transcription. On the other hand chaperone-dependent ubiquitin ligases (B) may facilitate client protein ubiquitination and promote AR polyQ clearance by the proteasome. This strategy may alleviate polyglutamine toxicity by facilitating degradation of the mutant protein.
- Naiki H, Nagai Y (2009) Molecular pathogenesis of protein misfolding diseases: pathological molecular environments versus quality control systems against misfolded proteins. J Biochem 146: 751–756. [Crossref]
- Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75: 333-366. [Crossref]
- Bauer PO, Nukina N (2009) The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 110: 1737–1765. [Crossref]
- Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6: 11-22. [Crossref]
- Bates GP, Mangiarinim L, Daviesm SW (1998) Transgenic mice in the study of polyglutamine repeat expansion diseases. Brain Pathol 8: 699–714. [Crossref]
- Smith DL1, Portier R, Woodman B, Hockly E, Mahal A, et al. (2001) Inhibition of polyglutamine aggregation in R6/2 HD brain slices-complex dose-response profiles. Neurobiol Dis 8: 1017-1026. [Crossref]
- Cowin RM, Roscic A, Bui N, Graham D, Paganetti P, et al. (2012) Neuronal aggregates are associated with phenotypic onset in the R6/2 Huntington’s disease transgenic mouse. Behav Brain Res 229: 308–319. [Crossref]
- Simanainen U, Brogley M, Gao YR, Jimenez M, Harwood DT, et al. (2011) Length of the human androgen receptor glutamine tract determines androgen sensitivity in vivo. Mol Cell Endocrinol 342: 81–86. [Crossref]
- Kumar R, Atamna H, Zakharov MN, Bhasin S, Khan SH, et al. (2011) Role of the androgen receptor CAG repeat polymorphism in prostate cancer, and spinal and bulbar muscular atrophy. Life Sci 88: 565-571. [Crossref]
- Kumar (2012) Role of Androgen Receptor PolyQ Chain Elongation in Kennedy’s Disease and Use of Natural Osmolytes as Potential Therapeutic Targets. IUBMB Life 64: 879–884. [Crossref]
- Diamond MI, Robinson MR, Yamamoto KR (2000) Regulation of expanded polyglutamine protein aggregation and nuclear localization by the glucocorticoid receptor. Proc Natl Acad Sci USA 97: 657–661. [Crossref]
- Palazzolo I, Gliozzi A, Rusmini P, Sau D, Crippa V, et al. (2008) The role of the polyglutamine tract in androgen receptor. J Steroid Biochem Mol Biol 108: 245-253. [Crossref]
- Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, et al. (2010) Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron 67: 936–952. [Crossref]
- Rusmini P, Sau D, Crippa V, Palazzolo I, Simonini F, et al. (2007) Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol Aging 28: 1099–1111. [Crossref]
- Davies P, Watt K, Kelly SM, Clark C, Price NC, et al. (2008) Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J Mol Endocrinol 41: 301–314. [Crossref]
- Orafidiya FA, McEwan IJ (2015) Trinucleotide repeats and protein folding and disease: the perspective from studies with the androgen receptor. Future Sci OA 1: FSO47. [Crossref]
- Montie HL, Cho MS, Holder L, Liu Y, Tsvetkov AS, et al. (2009) Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 18: 1937–1950. [Crossref]
- Montie HL, Pestell RG, Merry DE (2011) SIRT1 modulates aggregation and toxicity through deacetylation of the androgen receptor in cell models of SBMA. J Neurosci 31: 17425–17436. [Crossref]
- Heinlein CA, Chang C (2002) Androgen receptor (AR) coregulators: an overview. Endocr Rev 23: 175-200. [Crossref]
- Poletti A, Negri-Cesi P, Martini L (2005) Reflections on the diseases linked to mutations of the androgen receptor. Endocrine 28: 243-262. [Crossref]
- Poletti A (2004) The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front Neuroendocrinol 25: 1-26. [Crossref]
- Vismara G, Simonini F, Onesto E, Bignamini M, Miceli V, et al. (2009) Androgens inhibit androgen receptor promoter activation in motor neurons. Neurobiol Dis 33: 395–404. [Crossref]
- Chevalier-Larsen ES, O'Brien CJ, Wang H, et al. (2004) Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci 24: 4778–4786. [Crossref]
- Katsuno M, Adachi H, Kume A, et al. (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35: 843–854. [Crossref]
- Giorgetti E, Rusmini P, Crippa V, et al. (2015) Synergic prodegradative activity of bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum Mol Genet 24: 64–75. [Crossref]
- Takeyama K, Ito S, Yamamoto A, et al. (2002) Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in drosophila. Neuron 35: 855–864. [Crossref]
- Nedelsky NB, Pennuto M, Smith RB, et al. (2010) Native functions of the androgen receptor are essential to pathogenesis in a drosophila model of spinobulbar muscular atrophy. Neuron 67: 936–952. [Crossref]
- Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, et al. (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9: 112-118. [Crossref]
2021 Copyright OAT. All rights reserv