Atherosclerosis is a progressive inflammatory arterial disease triggered by interplay between immune and vascular cells. Development of atherosclerotic plaque requires endothelial retention of low-density lipids and its oxidation and immune cell recruitment into the arterial wall. However, recently against the existing dogma of this inside-out signalling, perivascular adipose tissue (PVAT) has been emerged as central regulator of out-side in signalling to modulate vascular tone and pathophysiology of atherosclerosis. Furthermore, under atherogenic conditions, PVAT exhibits remarkable plasticity and contributes to cardiovascular disease. However, understanding of regulatory mechanism by which PVAT plasticity modulates vascular functions particularly atherosclerosis, remains unexplored. This brief review is focused on understanding the in-depth roles of PVAT in atherosclerotic plaque progression, with the special focus on PVAT plasticity and its therapeutic implication for treatment of atherosclerosis.
atherosclerosis, PVAT, inflammation, adipose tissue
Atherosclerosis is a chronic inflammatory disease characterized by endothelial dysfunction, lipid deposition and inflammatory infiltration [1]. Endothelial dysfunction/injury caused by high shear stress promotes initiation of atherosclerosis, followed by subsequent adhesion of circulating inflammatory cells to the dysfunctional endothelium. These processes are further aggravated by accumulation of cholesterol in the arterial wall and lead to atherosclerosis [2]. One of the identified features of endothelial cells in response to local inflammation is dedifferentiation and switching of endothelial-to-mesenchymal transition which may be involved in the loss of endothelial phenotypes towards unspecialized mesenchymal-like cells that could undergo redifferentiation into mesodermal cell types, including adipocytes, in response to local inflammation [3]. This process can be triggered by various inflammatory cytokines and TGF-β which are produced in inflamed adipose tissue [4]. Adipose tissues play a significant role in atherosclerosis progression and are broadly classified as white adipose tissue (WAT) and brown adipose tissue (BAT). Initial studies have supported the hypothesis that dysfunctional WAT might be positively associated with atherosclerosis development, whereas activation of BAT may protect against atherosclerosis development. A third type beige adipose tissue (BeAT), has also been identified which is characterized by high expression of the brown adipocyte marker uncoupling protein-1 (UCP-1) [5].
Based on anatomical location, most recent addition to the family of adipose tissues is Perivascular adipose tissue (PVAT) which surrounds the blood vessels. It shares characteristics to both BAT and WAT and is an active component of the blood vessel walls and is reported to be involved in vascular homeostasis [6]. It has a very important function in providing structural support to the vessels and has been found to be removed from the vessels regularly highlighting the pro- contractile function of the PVAT [7]. The roles of PVAT in atherosclerosis development have been extensively studied by various researchers [8-11] and there is growing evidence that suggests that phenotypes of PVAT differ on anatomical location, morphology and differentiation, as well as in cell composition [12]. Furthermore, PVAT releases number of vasoactive factors that triggers both endocrine and paracrine signalling effects on the vascular wall, influencing its structure and function [13]. In atherosclerosis, PVAT increases in volume and becomes dysfunctional with altered cellular composition and molecular characteristics. Various investigators have demonstrated that the presence of PVAT in healthy tissue is ‘anti-contractile [14]. PVAT inflammation results in invasion of leukocytes into the perivascular adipose layer in response to injury of vessel wall [6]. PVAT exhibits both a deleterious and a protective role depending on the anatomical location. The PVAT mediated molecular and cellular responses involve extensive inflammation that is very peculiar and different from classical visceral or subcutaneous adipose tissue or from adventitia, emphasizing the unique structural and functional features of this adipose tissue compartment [15]. The dysfunctionality of PVAT is characterized by its associated inflammatory character, oxidative stress, decreased production of vaso‐protective adipocyte‐derived relaxing factors (ADRF) and increased production of paracrine factors such as resistin, leptin, cytokines (IL‐6 and TNF‐α) and chemokines [RANTES (CCL5) and MCP‐1 (CCL2) [15]. These adipocyte‐derived factors initiate inflammation and are instrumental in inflammatory cell infiltration that includes T cells, macrophages, dendritic cells, B cells and NK cells [16] ultimately leading to formation of atherosclerotic lesion.
Endothelium dysfunction – inside- out and PVAT mediated outside –in signaling during atherosclerosis progression
As per outside –in theory during atherosclerosis process, PVAT is found to be involved in instigating a new paradigm where the process of developing atherosclerotic plaque is initiated at the outside of the vessel wall and progressed towards the intima involving adventitia in forming new blood vessels [17].
Many studies and observations have contributed to the outside-to-inside signalling paradigm, in which changes in the adventitia and/or Pericoronary adipose tissue (PCAT) could also induce alterations in the blood vessel wall thus, having a deleterious influence on coronary artery disease development and progression. The role of periadventitial endotoxin, MCP-1, IL-1β, or oxidized LDL elicited inflammatory cell influx into the arterial wall, coronary vasospasm, or intimal lesions, suggesting the significant role of these bioactive molecules from the pericoronary tissues in altering arterial homeostasis [18-21]. Endothelial dysfunction is reportedly characterized by loss of nitric oxide (NO) and production of vasoprotective substances, such as prostacyclin (PGI2) and enhanced production of vascular damaging and pathologically activating molecules such as ROS, endothelin and thromboxane [22]. Vasoprotective agents including NO have potent anti‐inflammatory properties, which are triggered through inhibitory effects on adhesion molecule and chemokine expression. Loss of vasoprotective agents is further augmented by release of chemokines such as RANTES, CCL2 and CXCL10 [21,23], which can induce leukocyte migration or activation. Elevated expression of adhesion molecules like ICAM‐1 (intracellular adhesion molecule) and VCAM‐1 expression, on the vascular endothelium, establishes the link between endothelium dysfunction and inflammation [24]. When this dysfunction occurs in microvessels and vasa vasorum of PVAT, it will lead to the development of perivascular infiltration. Another important factor of endothelium dysfunctioning is oxidative stress characterized by the overproduction of superoxide anion and hydrogen peroxide. It results in rapid scavenging of NO in the blood vessel wall and leads to activation of redox‐sensitive genes within the endothelium, VSMCs and adventitia. VSMCs are a significant source of chemokines and cytokines, such as CCL2, CCL7, CCL20, CXCL1, CX3CL1, CXCL5 and IL‐6, IL‐23a and IL‐1β [25]. Numerous pro‐inflammatory genes including cytokines and chemokines as well as adhesion molecules are redox sensitive, establishing a link between vascular oxidative stress and inflammatory processes [26]. Similarly, chemokine receptors, such as CCR2, CCR5 and CXCR4, are also up‐regulated by oxygen radicals [27,28]. Increased expression of key chemokines in the vascular wall are observed at the early stages of atherosclerosis and these can be essential for the induction of perivascular inflammation. Ide et al. reported that CXCL10 (IP‐10) enhanced the expression of RAS components in endothelial cells [23], making it almost a prototypical ‘bidirectional’ role of released cytokine in atherosclerosis, through which the vessel wall can regulate inflammation and inflammatory cells. Endothelial dysfunction can also reduce endothelial nitric oxide synthase (eNOS) expression, further reducing NO production and bioavailability, with ensuing adverse consequences on vasodilation, and significantly increase peripheral resistance of blood vessels. Recent studies have shown that PVAT expresses eNOS [29], and removal of PVAT reduces basal NO production in small arteries from healthy individuals, suggesting that PVAT contributes to vascular NO production [8]. It has also been demonstrated that PVAT aggravate the macrophage infiltration in the perivascular area of the aorta and resulting in increased production of inflammatory cytokines ultimately leading to vascular inflammation and increased atherosclerotic lesions in the aortic wall [30]. Consistently, insulin receptor-deficient Apoe-/- mice showed severe atherosclerosis and increased expression of TNF-α and leptin while there was decreased adiponectin in adipose tissues, including thoracic PVAT [8]. It is known that adiponectin normalizes endothelial function through a mechanism involving increased eNOS phosphorylation and controls blood pressure through an endothelial-dependent mechanism [31]. Thus, endothelial dysfunction and vascular oxidative stress may initiate and execute PVAT inflammation evoked by key risk factors for atherosclerosis. All these observations indicate a bidirectional relationship between the vascular endothelium and PVAT mediated inflammation in the adventitial layer ultimately leading to migration of VSMCs and phenotypic switching followed by neovascularization. These recurrent alterations form the vulnerable plaques and leads the progression of atherosclerosis as depicted in Figure 1.
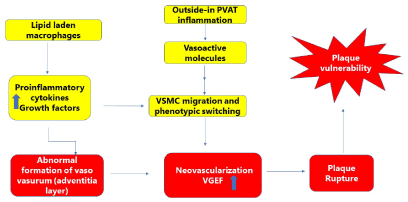
Figure 1. Crucial PVAT trigger in flipping Inside-out and Outside-in signaling in the pathogenesis of atherosclerosis. Lipid laden macrophages mediated inside-out signaling triggers vascular inflammation via release of proinflammatory cytokines and various growth factors affect the vessel wall and promote atherosclerotic lesion formation. However, in outside – in theory, PVAT mediated inflammation in the adventitial layer leads to vascular smooth muscle cell migration and phenotypic switching followed by neovascularization. These recurrent alterations form the vulnerable plaques and leads the progression of atherosclerosis
Chronology of atherosclerosis: PVAT mediated cellular and molecular events
Atherosclerotic plaques within the arterial walls are mediated in the form of lesions within the arterial wall and are mediated through interplay between a plethora of cellular mechanisms and interactions between endothelial cells (ECs), smooth muscle cells (SMCs) and inflammatory cells. Numerous reports suggested that high fat diet initiates lipid accumulation and leads to chronic endothelial injury, which further leads to increased permeability of endothelium caused by factors cytokines including IFN-α, CCL2, IL-6, TNF-α, IFN-γ, CCL3, CCL5, CXCL2 are released from adipose tissues to the circulation [4,15]. Sequential events, further result in promoting adhesions of circulating immune cells (monocytes) to its surface before infiltration into intima [32]. Evidently, the lipid laden macrophages (foam cells) triggers the secretion of pro-inflammatory cytokines and growth factors that can signal vascular smooth muscle cell (VSMC) within the medial layer to migrate to the intima and proliferate [33]. Apart from bidirectional cross talk between endothelial dysfunction and PVAT of the blood vessels, there are certain novel mechanisms where structure like the adventitia via PVAT have been recognized as active participants in the progression of atherosclerotic plaques through interaction with cellular players (including monocytes/macrophages, VSMCs, lymphocytes) of atherosclerosis [8].
Role of cellular players in PVAT inflammation
Macrophages were the first immune cells identified in AT [34]. They are also the most abundant cell type in typical visceral and subcutaneous AT, representing more than 50% of all leukocytes [35]. The increase in the density of CD68+ macrophages and CD20+ B lymphocytes called periplaque PVAT leads to the instability of plaques by carrying the lipoproteins from the PVAT to the artery wall and may disrupt the vessel tone [16]. Infiltration of M2 type (anti-inflammatory) macrophages in PVAT and in adventitia during atherosclerosis [28] produces array of cytokines such as IL‐6, IFN‐γ and TNF‐α that change the vascular and PVAT cellular events through the release of adipokines [15]. Recent studies suggest significant infiltration of M2 macrophages in PVAT, which may regulate the release of adipokines. Macrophages in PVAT also regulates the release of mediators that modulate the expression of co‐stimulatory ligands and their functions related to T cell activation through antigen presentation. This activation further, can trigger the aggregation of other pro inflammatory cells like CD4+, CD8+, B cells and dendritic cells [36]. These PVAT mediated proinflammatory cytokines (IFN ‐γ, IL-17, etc) release in found to be associated with progression of atherosclerosis [8]. Another pathway suggested by Gaborit et al. in 2015 is the communication of PVAT with the atherosclerotic plaque through the infiltration of adipocytes in the adventitial layer [37]. The important function of this layer includes growth and repair of the vessel wall, and it also communicate between vascular endothelial cells and smooth muscle cells (SMCs) and their local tissue environment [38]. The adventitia is also critical in formation and regression of microvessels, nourish the media and intima. In response to injury and stress, fibroblasts from the adventitia proliferate, differentiate into myofibroblasts, and migrate to the intima [39]. During plaque development, it stimulates abnormal formation of vasa vasorum which can trigger neovascularization through the production of vascular endothelial growth factors (VEGF). These immature micro vessels are prone to rupture and may cause haemorrhage in plaques leading to plaque vulnerability. These neo vessels can also enhance monocytes recruitment and release of cytokines thus perpetuating inflammation. During PVAT inflammation, inflammatory mediators released from DCs stimulate T cells to produce pro-inflammatory cytokines by blocking the CD28/CD80/CD86 co-stimulatory axis between DCs and T cells [15]. However, the contributions of DCs and T cells in PVAT to atherosclerotic lesion formation remains largely unclear.
Proinflammatory cytokines and PVAT
During atherosclerosis, PVAT releases pro-inflammatory factors like leptin, adiponectin, resistin, visfatin, interleukins into circulation. Hypertrophic PVAT may release abundant free fatty acids into the perivascular area and blood vessel wall and trigger nuclear factor-κB (NF-κB), protein kinase C (PKC) and Toll-like receptors (TLRs) which in turn promote phosphorylation of insulin receptor substrate 1 (IRS-1) in PVAT and vascular cells reducing its ability to activate downstream PI3K/Akt signalling [8]. This could probably result in inhibition of eNOS synthesis and NO production, triggering changes in vascular modelling and accumulate oxidative stress, resulting in endothelial dysfunction. Parallelly, they also activate TLR2 and TLR4, which regulate the NF-κB signaling pathway in macrophages [40]. Modified lipids in PVAT adipocytes lead ROS mediated vascular inflammation. Evidence demonstrates a more complex two-way communication between the arterial and venous wall and their surrounding PVAT where signals originating from the vascular wall or lumen can affect PVAT phenotype, and signals from PVAT can influence structure and function of the vascular wall in a paracrine manner. Hence, it’s critical to study and decipher the dual role of PVAT in atherosclerotic plaque progression. PVAT triggers release of vasoactive substances to target VSMCs and regulate the pathological process of vascular remodelling [41]. Persistent inflammation during atherosclerosis triggers phenotypic switching of VSMCs, their proliferation and migration. These molecular modulations consecutively trigger dysfunctional vasculo-adipose balance and significantly alter the microenvironment, for once or recurrent alterations leading towards enhanced disease susceptibility [5].
Role of PVAT in vascular smooth muscle cells proliferation and migration
PVAT is found to release relaxing factors [42] and growth factors [43] which are involved in the proliferation of VSMCs. Study published by Takaoka et al. demonstrated that after implanting adipose tissue to surround the blood vessel affects neointimal formation following endovascular injury, indicating the important role of PVAT in VSMC growth and migration [44]. The adipocytes, stem cells, immune cells, and other components in PVAT, such as extracellular vesicles (EV), can produce and secrete PVAT-derived factors such as adipokines, cytokines and growth factors which regulate vascular remodelling. Mechanistically, TGF-β (transforming growth factor-β) signalling promotes differentiation of PVAT resident progenitors into VSMC since they express Tgfbr2 (transforming growth factor β receptor 2) and Anxa1 (annexin A1) [5]. Treatment of PVAT-derived progenitors with TGFβ promoted differentiation towards VSMC. The characteristics of those progenitors in PVAT and other signalling pathways which contribute to vascular remodelling remain unexplored. The effects of PVAT on intimal SMC infiltration have been investigated after transplantation of thoracic PVAT to wire-injured carotid arteries, where the presence of PVAT accelerated neointimal formation in an MCP-1 dependent manner [45,46]. Also, transplantation of PVAT derived from transgenic mice overexpressing adipose tissue-specific angiopoietin like 2 (ANGPTL2) accelerated neointimal hyperplasia induced by wire injury of the femoral artery, which was attenuated by transplantation of PVAT from Angptl2−/− mice [47]. Moreover, inflamed PVAT increases VSMC proliferation in a TGF-β dependent manner, suggesting that TGF-β secretion from PVAT can potentiate neointima formation [8]. Furthermore, It has also been shown that PVAT derived factors such as leptin, viafastin, TNF-α, IL-6 and IL-8, promote VSMC proliferation and migration [19]. As described earlier, PVAT has been argued to be both an instigator and a protective compensator in diseases at the levels of vascular remodelling and arterial plasticity. VSMCs are characterized by display of extensive plasticity and undergo phenotypic switching as response to injury to a proliferative state [48]. The same is observed during restenosis following angioplasty where VSMC proliferation and VSMC migration from media to intima are involved. The phenotypic switch is controlled through factors like visfatin released from PVAT, and may act as regulators for VSMC fate change [8]. The role of visfatin during diseased state, in proliferation of VSMC as well as inflammations is closely related to the genesis, development and rupture of atherosclerotic plaques [49]. It was even more interesting to note that the growth‐promoting effect of PVAT was significantly enhanced in aged rats and in high‐fat diet‐induced obese rats, suggesting the significant role of PVAT in atherosclerosis. PVAT derived growth factors, including thrombospondin-1, serpin-E1, TGF-β, PDGF-BB, VEGF (vascular endothelial growth factor), bFGF (basic fibroblast growth factor), PLGF (placental growth factor), HGF (hepatocyte growth factor), and ILGFBP-3 (insulin-like growth factor-binding protein-3), have well-known effects in stimulation of VSMC proliferation and migration [50] (Figure 2).
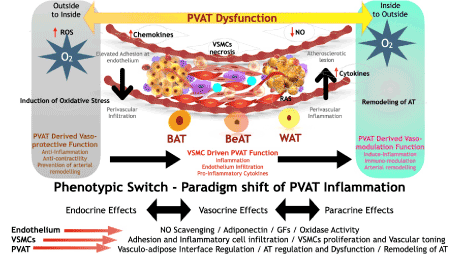
Figure 2. Paradigm shift of the regulatory role of PVAT in atherosclerosis. PVAT is pivotal to create a biomolecular balance with anti-inflammatory to pro inflammatory influences at the vasculo- adipose interface through the release of bioactive molecules like adipokines, adiponectins, growth factors and extra cellular vesicles
Role of PVAT in modulating cellular plasticity
Growing number of evidences suggested that the biological role of PVAT under physiological and pathological conditions is mediated by perivascular adipocytes surrounding human coronary arteries called pericoronary perivascular adipocytes. They exhibit adipogenic differentiation and elevated proinflammatory state leading to tremendous release of proinflammatory cytokine (monocyte chemoattractant peptide-1) [8] hence, contribute to increased inflammation of PVAT observed in atherosclerotic human blood vessels [19]. Adipocytes are also responsible for the release of anti-inflammatory molecules such as adiponectin. It has also been seen that inflammation of PVAT may be modified by environmental factors, such as high-fat diet and tobacco smoke, which are relevant to atherosclerosis. These findings suggested the diverse nature of the inflammatory phenotype of PVAT based on species, anatomic location, and environmental factors which vary depending on the stage of disease. PVAT is also found to be associated with VSMC’s plasticity in atherosclerosis depending on the release of inflammatory cytokines by PVAT. Contractile and anti- contractile characteristics can also have a very important role in the modulation of VSMC’s plasticity which may play significant role in stabilization /destabilization of the atherosclerotic plaques [51]. VSMCs plasticity in response to the changes in microenvironment acquire diverse range of structural and functional properties characterized by expression of a unique set of structural, contractile, and receptor proteins and isoforms that can be correlated with differing patterns of gene expression [52]. Recent studies have identified signaling pathways and transcription factors (e.g., RhoA GTPase/ROCK, also known as Rho kinase, and serum response factor) that regulate the transcriptional activities of genes encoding proteins associated with the contractile apparatus. These types of changes can extend further during functional plasticity [53]. Anticontractile effects of PVAT could be regulated by uptake and metabolism of the vasoactive amines such as dopamine, norepinephrine, and serotonin in PVAT itself [54]. The PVAT’s anticontractile effects are also reported to be mediated by PVRF (PVAT-derived relaxing factors). Recent studies also demonstrated that PVAT secretes PVCF (PVAT-derived contracting factors), which induces blood vessel contraction. PVRF and PVCF influence local vascular tone through endothelium-dependent or endothelium-independent effects [5]. Endothelium-derived NO (nitric oxide), one of the most potent vascular dilators, stimulates guanylate cyclase in VSMCs, resulting in upregulated expression of GMP and related signalling cascades to induce blood vessel relaxation [55]. The anticontractile effects of PVAT on blood vessels might be mediated by PVAT-derived NO. Inhibition of NOS attenuated the anticontractile effects of PVAT. PVAT mediated release of vasoconstrictors helps in elicitation of contraction in mesenteric arteries suggesting PVAT induces contractile response. Therefore, it is possible that PVRF, such as NO, directly target VSMC to induce vessel relaxation. Numerous studies have also documented that PVAT controls vascular tone through potassium channels in VSMC. PVAT mediated release of PVRF such as H2S, Ang (1–7) and methyl palmitate, could induce vasodilation by opening of K+ channels on VSMC [56]. PVAT also releases PVCF such as calpastatin, Ang II, 5-HT, chemerin and norepinephrine which could induce vasoconstriction by, in some cases, still underdetermined mechanisms, albeit most likely mediated by their receptors on VSMC [8]. Thus, PVAT-derived bioactive factors collectively and coordinately regulate vascular tone by targeting VSMC.
Role of PVAT intervention in sustaining recurrent inflammation
Endothelial dysfunction is now well correlated with dysfunctional PVAT, which can be well characterized by the loss of protective functions of PVAT. PVAT-derived adiponectin inhibits plaque formation via a reduced vascular inflammatory response by secreting biologically active H2S (mechanism unknown), which improves endothelial function. PVAT reportedly has a protective role on vascular grafts, especially the no-touch saphenous vein, in patients undergoing coronary artery bypass providing mechanical protection [12]. Initially it was found to be related to aberrant adipokine profile, but it soon became evident that PVAT dysfunction is mobilized by inflammatory responses. Hence probably, perivascular adipocytes may de‐differentiate which were primarily lipid‐storing cells but become a metabolically active tissue that produces pro‐inflammatory adipokines and chemokines and become key factors of inflammation in atherosclerosis [8]. This is supported by the study where the loss of perilipin, which directly induces this change in PVAT phenotype, causes vascular dysfunction due to dedifferentiation of PVAT adipocyte and inflammatory cell infiltration [54]. Under normal physiological state, PVAT releases substances including beneficial adipocyte‐derived relaxing factor (ADRF), which has been shown to affect vasomotor tone and regulate important homeostatic blood vessel functions through endothelium-dependent or endothelium-independent mechanisms, including activating VSMC potassium channels [57]. However, under pathological conditions, PVAT becomes pro-atherogenic and secretes adipocytokines and chemokines which targets a variety of cell types including macrophages, ECs, T cells and VSMCs [8].
PVAT-derived adipocytokines such as adiponectin (APN) and IL-10 regulate VSMC migration and proliferation and help maintain the vascular tone through the control of endothelial vasodilator/constrictor release and other inflammatory cell migration [58]. However, in atherosclerotic conditions, homeostatic functions of PVAT become dysregulated and induces atherogenesis through secretion of adipocytokines such as leptin, visfatin, Tumour necrosis factor- α (TNF- α), interleukin-6 (IL-6) and monocyte chemoattractant protein (MCP-1) [59]. Alteration of endocrine functions of adipose tissue negatively affects the secretion of different adipocytokines under disease conditions. Adiponectin acts as a regulator of metabolic responses such as energy metabolism and insulin-sensitivity, and has pleiotropic effects during atherosclerosis [60]. The presence of high cholesterol and other inflammatory-inducing factors could probably induce the progression of disease due to decreased expression of adiponectin expression in adipose tissue during inflammation. Low-grade inflammation in atherosclerosis has been found to be associated with decreased plasma adiponectin, whereas advanced, chronic inflammation is associated with increased adiponectin levels. With available literature, it could be inferred the role of adiponectin regulation is complex in humans [61].
Antonopoulos et al. have demonstratedhow the reciprocal effects of systemic inflammation and brain natriuretic peptides (BNP) influence adiponectin expression in patients with heart failure using non-invasive imaging to establish that inflammation within PVAT and plaque are directly proportional [62]. This idea was further supported by a study published in Lancet in 2018 by same group utilising non-invasive imaging to establish correlation between PVAT and plaque inflammation. These studies raise further questions and probabilities that blocking inflammation in PVAT can also influence/alter plaque dynamics [63]. The homeostasis of anti-atherosclerosis adipokines and pro- atheromatous adipokines serum levels is perturbed in people with coronary disease and all these factors have potential as future biomarkers which can be useful during the follow up of the atheromatous plaque development (Figure 3).
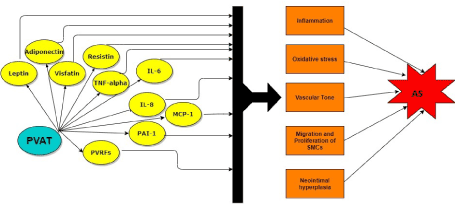
Figure 3. Role of PVAT in Atherosclerosis. PVAT plays a pivotal role in the development of atherosclerosis. Various factors like leptin, adiponectin, resistin, visfatin, interleukins, PVRFs etc are released by PVAT which mediate inflammation, oxidative stress, SMC migration and proliferation, neointimal hyperplasia and alter the vascular tone, consequently contributing to atherosclerosis (AS)
Location of adipose tissue with specific morphological distribution, composition, structure and its functionality have inherent role and these well correlated individual factors can serve as important factors in outside in theory elucidating the role of various pro inflammatory cytokines and signaling cascades in atheroprogression. PVAT, here in coordination with and progressive / recurrent inflammation becomes the pivotal checkpoint. The essential contribution of PVAT towards vascular homeostasis is crucial for biological designs as diagnostic and therapeutics for targeted restoration of the vasculo-adipose balance to prevent occurrence of acute cardiovascular events. Alterations in the paracrine control of PVAT in the regulation of arterial tone contributes to vascular dysfunction in obesity, hypertension, and cardiometabolic diseases. Perivascular relaxing factors, or perhaps their putative targets, might represent exciting new targets for the prevention and treatment of cardiovascular and metabolic diseases.
- Galkina E, Ley K (2009) Immune and inflammatory mechanisms of atherosclerosis (*). Annual Review of Immunology 27: 165-197.
- Mudau M, Genis A, Lochner A, Strijdom H (2012) Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovascular Journal of Africa 23: 222-231.
- Haynes BA, Yang LF, Huyck RW, Lehrer EJ, Turner JM, et al. (2019) Endothelial-to-mesenchymal transition in human adipose tissue vasculature alters the particulate secretome and induces endothelial dysfunction. Arteriosclerosis, Thrombosis, and Vascular Biology 39: 2168-2191.
- Makki K, Froguel P, Wolowczuk I (2013) Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN inflammation 2013: 139239.
- Chang L, Garcia-Barrio MT, Chen YE (2020) Perivascular adipose tissue regulates vascular function by targeting vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology 40: 1094-1109.
- Gollasch M (2017) Adipose-vascular coupling and potential therapeutics. Annual Review of Pharmacology and Toxicology 57: 417-436.
- Ramirez JG, O'Malley EJ, Ho WSV (2017) Pro-contractile effects of perivascular fat in health and disease. British Journal of Pharmacology 174: 3482-3495.
- Qi XY, Qu SL, Xiong WH, Rom O, Chang L, et al. (2018) Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword. Cardiovascular Diabetology 17: 134.
- Fitzgibbons TP, Czech MP (2014) Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: basic mechanisms and clinical associations. Journal of the American Heart Association 3: e000582.
- Kawahito H, Yamada H, Irie D, Kato T, Akakabe Y, et al. (2013) Periaortic adipose tissue-specific activation of the renin-angiotensin system contributes to atherosclerosis development in uninephrectomized apoE-/- mice. American Journal of Physiology Heart and Circulatory Physiology 305: H667-675.
- Faight E, Verdelis K, Ahearn JM, Shields KJ (2018) 3D MicroCT spatial and temporal characterization of thoracic aorta perivascular adipose tissue and plaque volumes in the ApoE-/- mouse model. Adipocyte 7: 156-165.
- Gil-Ortega M, Stucchi P, Guzmán-Ruiz R, Cano V, Arribas S, et al. (2010) Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology 151: 3299-3306.
- Gálvez-Prieto B, Somoza B, Gil-Ortega M, García-Prieto CF, de Las Heras AI, et al. (2012) Anticontractile effect of perivascular adipose tissue and leptin are reduced in hypertension. Frontiers in Pharmacology 3: 103.
- Zaborska KE, Wareing M, Edwards G, Austin C (2016) Loss of anti-contractile effect of perivascular adipose tissue in offspring of obese rats. International Journal of Obesity (2005) 40: 1205-1214.
- Nosalski R, Guzik TJ (2017) Perivascular adipose tissue inflammation in vascular disease. British Journal of Pharmacology 174: 3496-3513.
- Liu R, Nikolajczyk BS (2019) Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Frontiers in Immunology 10: 1587.
- Alkhalil M, Choudhury RP (2018) Current concepts in atherosclerosis. Indian Journal of Thoracic and Cardiovascular Surgery 34: 198-205.
- Miyata K, Shimokawa H, Kandabashi T, Higo T, Morishige K, et al. (2000) Rho-kinase is involved in macrophage-mediated formation of coronary vascular lesions in pigs in vivo. Arteriosclerosis, Thrombosis, and Vascular Biology 20: 2351-2358.
- Shimokawa H, Ito A, Fukumoto Y, Kadokami T, Nakaike R, et al. (1996) Chronic treatment with interleukin-1 beta induces coronary intimal lesions and vasospastic responses in pigs in vivo. The role of platelet-derived growth factor. The Journal of Clinical Investigation 97: 769-776.
- Prescott MF, McBride CK, Court M (1989) Development of intimal lesions after leukocyte migration into the vascular wall. The American Journal of Pathology 135: 835-846.
- Fernández-Alfonso MS, Gil-Ortega M, Aranguez I, Souza D, Dreifaldt M, et al. (2017) Role of PVAT in coronary atherosclerosis and vein graft patency: friend or foe? British Journal of Pharmacology 174: 3561-3572.
- Channon KM, Guzik TJ (2002) Mechanisms of superoxide production in human blood vessels: relationship to endothelial dysfunction, clinical and genetic risk factors. Journal of Physiology and Pharmacology: An Official Journal of the Polish Physiological Society 53: 515-524.
- Ide N, Hirase T, Nishimoto-Hazuku A, Ikeda Y, Node K (2008) Angiotensin II increases expression of IP-10 and the renin-angiotensin system in endothelial cells. Hypertension Research: Official Journal of the Japanese Society of Hypertension 31: 1257-1267.
- Weyl A, Vanscheidt W, Weiss JM, Peschen M, Schopf E, et al. (1996) Expression of the adhesion molecules ICAM-1, VCAM-1, and E-selectin and their ligands VLA-4 and LFA-1 in chronic venous leg ulcers. Journal of the American Academy of Dermatology 34: 418-423.
- Butcher MJ, Waseem TC, Galkina EV (2016) Smooth muscle cell-derived interleukin-17C plays an atherogenic role via the recruitment of proinflammatory interleukin-17A+ T cells to the aorta. Arteriosclerosis, Thrombosis, and Vascular Biology 36: 1496-1506.
- Shah D, Wanchu A, Bhatnagar A (2011) Interaction between oxidative stress and chemokines: possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology 216: 1010-1017.
- Zhang X, Chen X, Song H, Chen HZ, Rovin BH (2005) Activation of the Nrf2/antioxidant response pathway increases IL-8 expression. European Journal of Immunology 35: 3258-3267.
- Chan CT, Moore JP, Budzyn K, Guida E, Diep H, et al. (2012) Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt-treated mice. Hypertension (Dallas, Tex: 1979) 60: 1207-1212.
- Meziat C, Boulghobra D, Strock E, Battault S, Bornard I, et al. (2019) Exercise training restores eNOS activation in the perivascular adipose tissue of obese rats: Impact on vascular function. Nitric Oxide: Biology and Chemistry 86: 63-67.
- Stieber C, Malka K, Boucher JM, Liaw L (2019) human perivascular adipose tissue as a regulator of the vascular microenvironment and diseases of the coronary artery and aorta. Journal of Cardiology and Cardiovascular Sciences 3: 10-15.
- Quesada I, Cejas J, García R, Cannizzo B, Redondo A, et al. (2018) Vascular dysfunction elicited by a cross talk between periaortic adipose tissue and the vascular wall is reversed by pioglitazone. Cardiovascular Therapeutics 36: e12322.
- Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H (2014) Atherosclerosis: process, indicators, risk factors and new hopes. International Journal of Preventive Medicine 5: 927-946.
- Rudijanto A (2007) The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Medica Indonesiana 39: 86-93.
- Hirayama D, Iida T, Nakase H (2017) The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. International Journal of Molecular Sciences 19:
- Ferrante AW (2013) The immune cells in adipose tissue. Diabetes, Obesity & Metabolism 15 Suppl 3: 34-38.
- Shirai T, Hilhorst M, Harrison DG, Goronzy JJ, Weyand CM (2015) Macrophages in vascular inflammation--From atherosclerosis to vasculitis. Autoimmunity 48: 139-151.
- Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, et al. (2015) Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovascular Research 108: 62-73.
- Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, et al. (2001) Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circulation Research 89: 1111-1121.
- Stenmark KR, Nozik-Grayck E, Gerasimovskaya E, Anwar A, Li M, et al. (2011) The adventitia: Essential role in pulmonary vascular remodeling. Comprehensive Physiology 1: 141-161.
- Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, et al. (2003) Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR "homotolerance" versus "heterotolerance" on NF-kappa B signaling pathway components. Journal of Immunology (Baltimore, Md: 1950) 170: 508-519.
- Zhang YY, Shi YN, Zhu N, Zhao TJ, Yi-Jie Guo N, et al. (2020) PVAT targets VSMCs to regulate vascular remodeling: angel or demon. Journal of Drug Targeting: 1-38.
- Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, et al. (2002) Periadventitial fat releases a vascular relaxing factor. FASEB 16: 1057-1063.
- Barandier C, Montani JP, Yang Z (2005) Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: effects of aging and obesity. American Journal of Physiology Heart and Circulatory Physiology 289: H1807-1813.
- Takaoka M, Nagata D, Kihara S, Shimomura I, Kimura Y, et al. (2009) Periadventitial adipose tissue plays a critical role in vascular remodeling. Circulation Research 105: 906-911.
- Gu W, Nowak WN, Xie Y, Le Bras A, Hu Y, et al. (2019) Single-cell RNA-sequencing and metabolomics analyses reveal the contribution of perivascular adipose tissue stem cells to vascular remodeling. Arteriosclerosis, Thrombosis, and Vascular Biology 39: 2049-2066.
- Bailey-Downs LC, Tucsek Z, Toth P, Sosnowska D, Gautam T, et al. (2013) Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences 68: 780-792.
- Manka D, Chatterjee TK, Stoll LL, Basford JE, Konaniah ES, et al. (2014) Transplanted perivascular adipose tissue accelerates injury-induced neointimal hyperplasia: role of monocyte chemoattractant protein-1. Arteriosclerosis, Thrombosis, and Vascular Biology 34: 1723-1730.
- Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, et al. (2016) Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circulation Research 119: 1313-1323.
- Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. The New England Journal of Medicine 352: 1685-1695.
- Nicosia RF, Nicosia SV, Smith M (1994) Vascular endothelial growth factor, platelet-derived growth factor, and insulin-like growth factor-1 promote rat aortic angiogenesis in vitro. The American Journal of Pathology 145: 1023-1029.
- van Kuijk K, Kuppe C, Betsholtz C, Vanlandewijck M, Kramann R, et al. (2019) Heterogeneity and plasticity in healthy and atherosclerotic vasculature explored by single-cell sequencing. Cardiovascular Research 115: 1705-1715.
- Worth NF, Rolfe BE, Song J, Campbell GR (2001) Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motility and the Cytoskeleton 49: 130-145.
- Halayko AJ, Solway J (2001) Molecular mechanisms of phenotypic plasticity in smooth muscle cells. Journal of Applied Physiology (Bethesda, Md: 1985) 90: 358-368.
- Man AWC, Zhou Y, Xia N, Li H (2020) Perivascular adipose tissue as a target for antioxidant therapy for cardiovascular complications. Antioxidants (Basel, Switzerland) 9:
- Tsai EJ, Kass DA (2009) Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacology & Therapeutics 122: 216-238.
- Jackson WF (2018) KV channels and the regulation of vascular smooth muscle tone. Microcirculation (New York, NY: 1994) 25:
- Nava E, Llorens S (2019) The local regulation of vascular function: from an inside-outside to an outside-inside model. Frontiers in Physiology 10: 729.
- Miao C-Y, Li Z-Y (2012) The role of perivascular adipose tissue in vascular smooth muscle cell growth. British Journal of Pharmacology 165: 643-658.
- Grigoras A, Amalinei C, Balan RA, Giusca SE, Caruntu ID (2019) Perivascular adipose tissue in cardiovascular diseases-an update. Anatolian Journal of Cardiology 22: 219-231.
- Di Zazzo E, Polito R, Bartollino S, Nigro E, Porcile C, et al. (2019) Adiponectin as link factor between adipose tissue and cancer. International Journal of Molecular Sciences 20:
- Ouchi N, Walsh K (2007) Adiponectin as an anti-inflammatory factor. Clinica Chimica Acta 380: 24-30.
- Antonopoulos AS, Sanna F, Sabharwal N, Thomas S, Oikonomou EK, et al. (2017) Detecting human coronary inflammation by imaging perivascular fat. Science Translational Medicine 9.
- Oikonomou EK, Marwan M, Desai MY, Mancio J, Alashi A, et al. (2018) Non-invasive detection of coronary inflammation using computed tomography and prediction of residual cardiovascular risk (the CRISP CT study): a post-hoc analysis of prospective outcome data. Lancet (London, England) 392: 929-939.