Cells may die from accidental cell death (ACD) or regulated cell death (RCD). ACD is a biologically uncontrolled process, whereas RCD involves a carefully programmed ritual of a tidy death. Under physiological conditions programmed cell death (PCD) and regulated cell death (RCD) are synonymous. ACD occurs from exposure to severe toxicological injury or higher degrees of hypoxia (a process often called necrosis). RCD involves tightly structured signaling cascades and molecularly defined effector mechanisms in response to various lethal stimuli (a process often called apoptosis). A growing number of novel non-apoptotic forms of RCD have been identified and are increasingly being implicated in various human pathologies. Programmed apoptosis consists of apoptosis, as well as anoikis. Multiple mechanisms and phenotypes compose programmed non‑apoptotic cell death, including vacuole‑presenting cell death (autophagy, entosis, methuosis and paraptosis), mitochondrial‑dependent cell death (mitoptosis and parthanatos), iron‑dependent cell death (ferroptosis), immune‑reactive cell death (pyroptosis and NETosis), as well as other types, such as necroptosis. The in-depth comprehension of each of these lethal subroutines and their intercellular consequences may uncover novel therapeutic targets for the avoidance of pathogenic cell loss. In the present review article, the hallmarks of different cell death modes are systematically described. At the end, we proceed to discuss how different forms of RCD and mitochondrial dysfunction are involved in the pathogenesis of Covid-19 and can be therapeutic targets.
regulated cell death, apoptosis, necrosis, inflammation, mitochondrial dysfunction, Covid-19
Careful observation with the light microscope in the 1950s and 1960s demonstrated the significance of physiological cell death in the process of development. In early 1970s, a process of cell death characterized by a stereotyped set of structural changes was also observed in very wide of physiological circumstances: negative selection in the immune system, cytotoxic T cell killing, atrophy induced in hormones and others stimuli, the growth and regression of tumours, and tissue development after exposure to teratogens [1]. The fact that the distinctive, stereotyped structural changes characterizing cell death in these circumstances were identical to those found in cell death during normal development raised the possibility that a “programmed” death pathway, similar to that in development, might be available in adult tissues and adopted in response to a variety of lethal stimuli. To emphasize the uniformity of the process suggested by these observations, and to highlight its potential significance as the counterpoise to mitosis in cell member homeostasis, this type of death was called apoptosis by John Kerr, Andrew Wyllie, and Alastair Currie in 1972 [2].
In apoptosis, structurally, the dying cell loses contact with its neighbours, undergoes a dramatic process of bubbling, blebbing and shrinkage, and disintegrates into a cluster of membrane-bounded fragments (Figure 1). Inside, there lie compacted organelles, for the most part structurally normal, but there is prominent and characteristic chromatin condensation that starts focally under the nuclear membranes and extends around and eventually throughout the whole nucleus. Apoptosis cells in tissues are swiftly recognized and phagocytosed by their neighbours, or by specialized phagocytes, in whose phagosomes they are competently destroyed within a few hours (Figure 2). Hence tissues can shrink to half their original cell number in a day or two, with little disturbance in structure, no inflammation process, and a few accumulating dead cells.
Figure 1. Apoptotic event
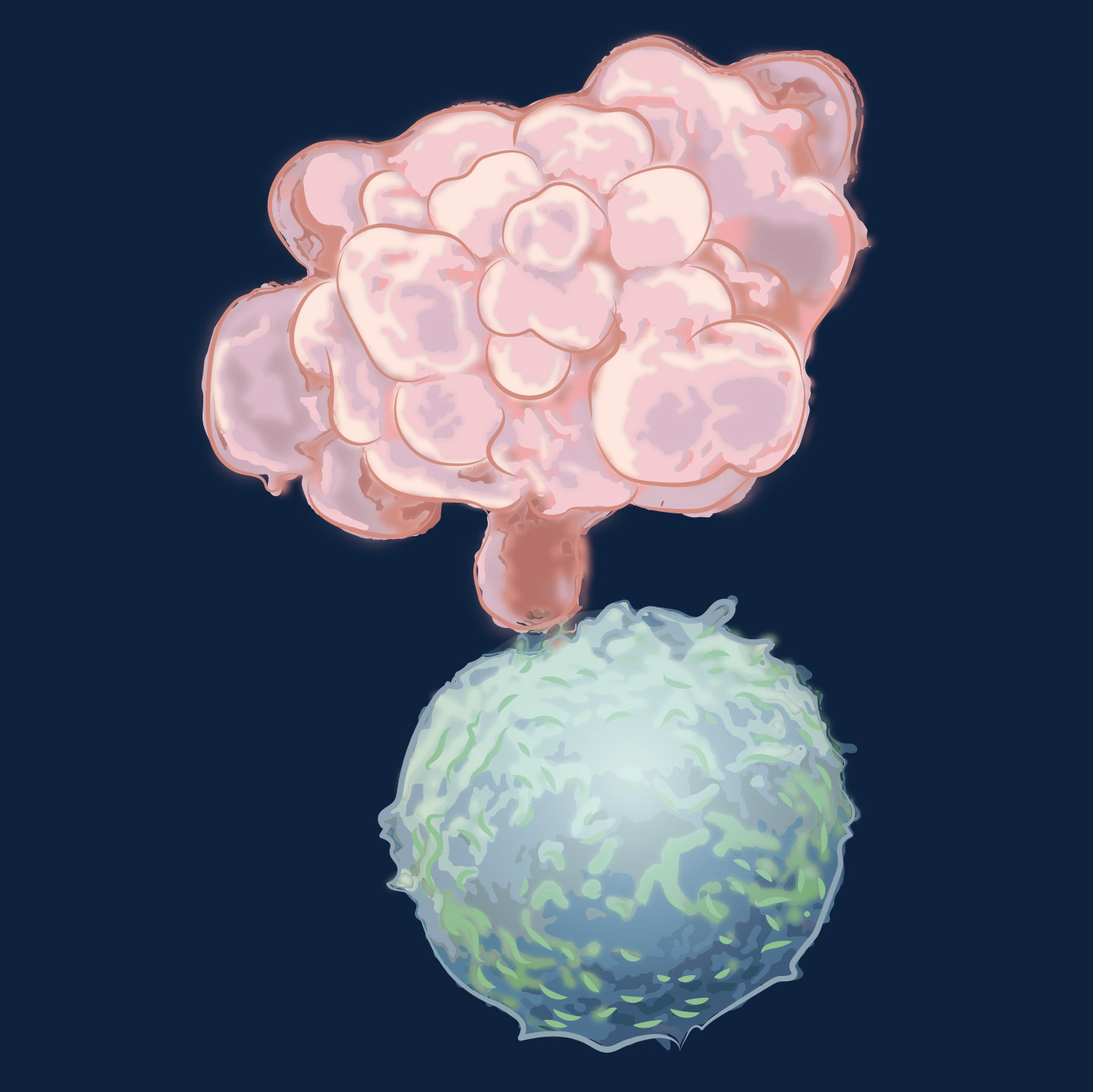
A cancer cell (pink) undergoes apoptosis after receiving a death signal from a killer T lymphocyte (blue). The dying cell has shrunk and its cytoplasm has developed cluster of grape-like forms (blebs). It will soon disintegrate into membrane-bound fragments.
Figure 2. Apoptotic attack
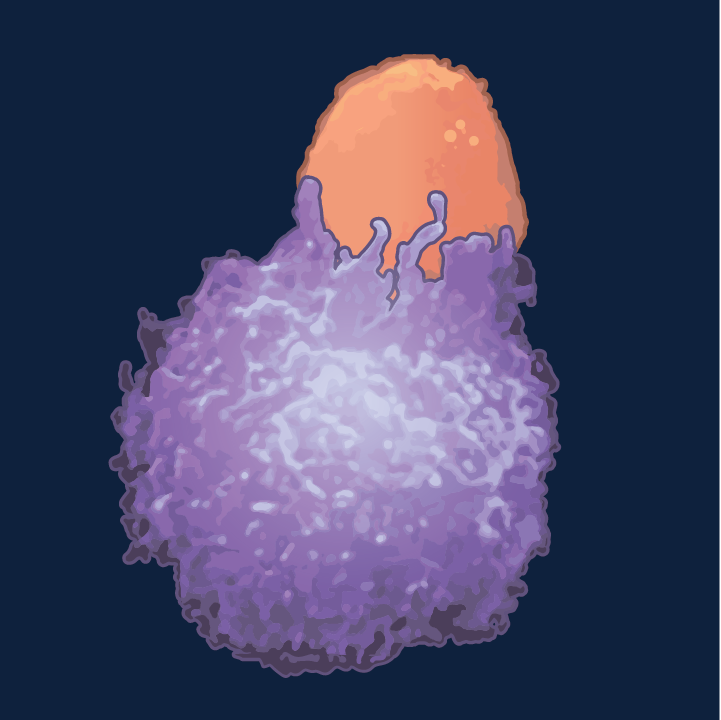
The pathway by which a killer T cell (orange) induces apoptosis in a cancer cell involves the expression of fas ligand, which binds to fas receptor on the target cell.
All of this is very different from the changes in cells exposed to severe injury (infection, toxins or physical injury) or hypoxia, wherein damage to energy-dependent membrane ion pumps leads to progressive cellular swelling (oncosis), a rupture (necrosis) and randomly degraded DNA, without severe chromatin condensation. When this occurs in tissues in vivo, there is usually an acute inflammatory reaction, apparently stimulated by neutrophils chemotactic factors originating from intracellular proteins lost from the necrotic cells (Figure 3). There has been a rapid expansion of apoptosis research in the last 30 years, and in addition to knowing that there are two major subtypes of apoptosis, new forms of non-apoptotic RCD have also been described. This monograph intends to concisely review the subject, highlighting the similarities and differences in the different RCD subtypes, as well as when various mechanisms act in series, in parallel or in "concert" to produce the RCD. In the end, the relationship of RCD and mitochondrial dysfunction in Covid-19 is explored.
Figure 3. Necrosis and apoptosis
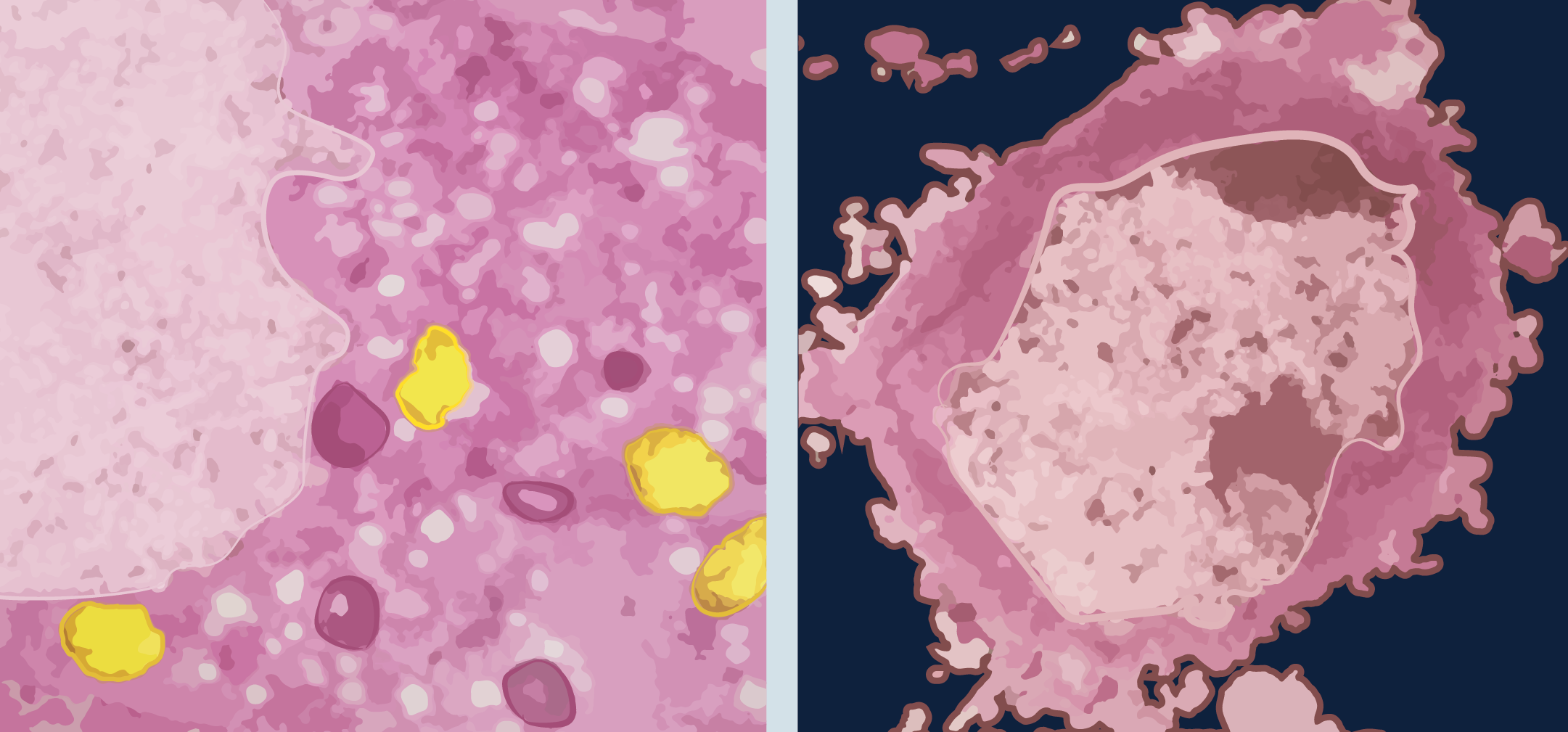
Cultured cerebellar granule cells showing morphological changes associated with necrosis (left) and apoptosis (right). The swelling necrotic cell has vacuolization in the cytoplasm and damage to the inner membrane of the mitochondria (yellow). The apoptotic cell shows chromatin condensation and blebbing.
Apoptosis is a word made up of two Greek roots: "apo" and “ptosis” which means fall or detachment. An alternative meaning is "falling leaves" which describes the phenomenon of autumn leaves dying to make way for fresh new shoots in spring. It is a form of cell death in which a programmed sequence of events hermetically controlled leads to the elimination of cells without the release of dangerous substances into the surrounding environment. Apoptosis is crucial in the development and maintenance of cellular integrity and homeostasis by eliminating senescent, diseased, or unnecessary cells [3]. The loss of control of cell death and the dysregulation of apoptosis contributes to human diseases such as cancer, neurodegeneration (spinal muscular atrophy, Alzheimer´s and Parkinson´s diseases), autoimmune diseases, infectious diseases (bacteria and virus) and cardiovascular diseases, both in removal of cardiac myocytes in cases if hearth failure and in contributing to the extent of cerebral infarcts in stroke [4,5].
There are two types of apoptosis: extrinsic and intrinsic (mitochondrial) apoptosis (Figure 4). The extrinsic pathway of apoptosis starts from external stimuli to the cell. It is triggered by the oligomerization of transmembrane proteins from the superfamily of death receptors (DR) or when extracellular concentrations of specific factors reach a particular threshold, leading to the transduction of lethal signals or dependence receptors [6]. There are eight types of death receptors (DR1–DR8), which can be divided in two groups according to the adapter proteins used to propagate the signal [7]. Two well-defined DRs in extrinsic apoptosis are Fas cell surface death receptor (FAS; also known as CD95 or APO-1 is a transmembrane protein located on the cell surface), and TNF (tumoral necrosis factor) receptor superfamily member 1A (TNFRSF1A; best known as TNFR1). The signal initiated by these receptors (when excited by their respective ligands) is propagated by caspase 8 and caspase 10. (There is an alternative pathway for the activation of extrinsic apoptosis by caspase 9). These initiator caspases in turn activate the effector caspases that activate endonucleases that degrade the nuclear material, and the proteins of the "core" and of the cytoskeleton, generating the cytomorphological changes already described. These executor caspases are 3, 6 and 7 and this executor pathway is common to extrinsic and intrinsic apoptosis [8].
Figure 4. Signalling Pathways of Regulated Cell Death
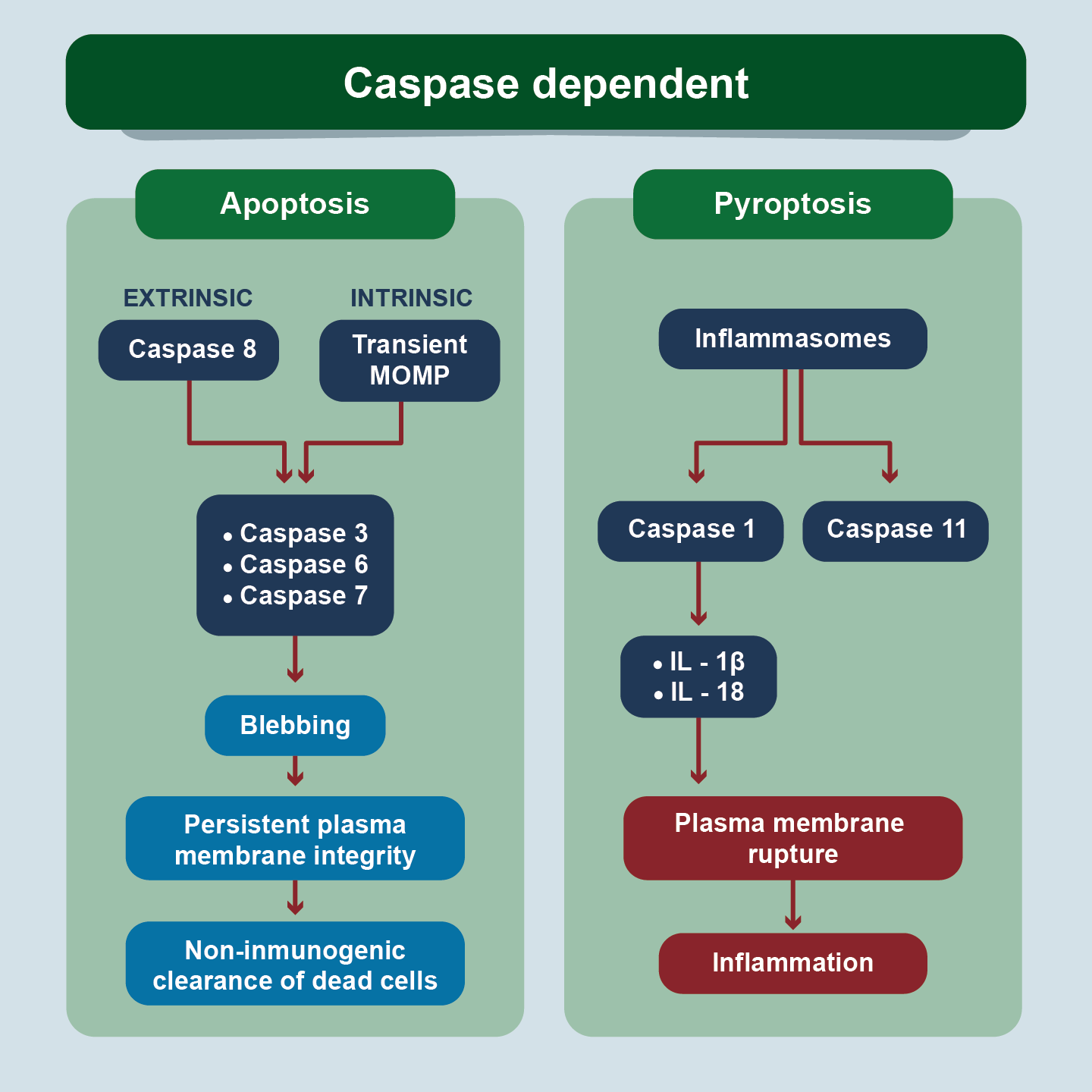
Apoptosis and pyroptosis are caspase dependent events. In apoptosis, the executor caspases produce the cytomorphological findings of cell contraction, chromatin condensation, cytoplasmic blistering, apoptotic body formation, and phagocytosis by adjacent cells. In pyroptosis, a different set of caspases leads to the breakdown of the plasma membrane and inflammation.
Intrinsic apoptosis is a form of RCD initiated by a variety of microenvironmental perturbations including (but not limited to) growth factor withdrawal, DNA damage, endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) overload, replication stress, microtubular alterations or mitotic defects [9]. In response to apoptotic stimuli happens a widespread mitochondrial outer membrane permeabilization (MOMP), which is controlled by pro-apoptotic and anti-apoptotic members of the BCL2, apoptosis regulator protein family (B-cell leukemia/ lymphoma protein 2) [10]. MOMP is mediated by BCL2 associated X, apoptosis regulator (BAX), and/or BCL2 antagonist/killer 1 (BAK1; best known as BAK). MOMP is considered in this event as a point of no return since there is the release of proteins involved in the activation of executor caspases in the cell cytoplasm and acidification of the microenvironment outside the mitochondria. For example, through MOMP cytochrome-C (Cyt-c) involved in the formation of apoptosome (a cytosolic multiproteic platform) is released which maintains substantial catalytic activity. The executor caspases are the same for the extrinsic pathway.
Caspases are a family of cysteine-dependent aspartate-specific proteases that play a critical role in the regulation of cell death, connecting to development, inflammation, and immunity. Caspases is an acronym (cysteine-aspartate proteases). Like many proteases, caspases initially exist as inactive zymogens, namely, procaspases [11]. RCD is therefore categorized into two groups: caspase-dependent (e.g., apoptosis and pyroptosis) and caspase-independent RCD (e.g., necroptosis, ferroptosis, parthanatos, alkaliptosis, and oxeiptosis). In mammalian cells, caspases can be divided into four groups: initiator caspases, effector caspases (CASP3, CASP6, and CASP7), inflammatory caspases (pyroptosis) and the keratinization-relevant caspase. These activated caspases can cleave substrates such as downstream caspases, cellular structural proteins, and immune molecules to cause cell death and inflammation [12]. Anoikis is a particular type of apoptosis, which essentially shares identical pathways as with apoptosis; however, is triggered by inadequate or inappropriate cell-matrix interactions. The architectural state of the cytoskeleton is expected to interfere with the function of integrin, a pro-survival effector. However, the connection between cell architecture alteration and apoptosis remains poorly identified [13].
There are documented nine genes that have essential functions in the intrinsic (APAF1, BCL2, and CASP9), extrinsic (CASP8, FADD, and FAS), both pathways (TP53 and MYC), and in the execution phase (CASP3). The APAF1 gene (Apoptotic peptidase activating factor 1) encodes the Apaf1 protein, which is activated by Cyt-c and regulated by pro-apoptotic molecules (such as Bax) and anti-apoptotic molecules (such as Bcl-X and Bcl-2). As part of the apoptosome, it is responsible for the activation of CASP9, which leads to CASP3-dependent cell death [14]. The FADD gene (FAS-associated via death domain), also known as MORT1, encodes the FADD protein, which acts as an adapter in the extrinsic pathway. It was discovered and associated with interaction with the Fas receptor by Chinnaiyan, et al. [15]. The TP53 tumor suppressor gene (Tumor protein p53) encodes the p53 protein, a transcription factor that has different functions involving cell and genomic stability, senescence, and cell death. The p53 protein targets more than 900 genes, affecting not only apoptosis, but also metabolism, proliferation, and immune response genes [16]. MYC (Myelocytomatosis) a regulator oncogene that codes for a transcription factor. Definition, the proteins they encode, and the function of the other genes have been discussed throughout the text.
There is also a relationship between mitochondria and epigenetic regulation. Mitochondria are cytoplasmic organelles responsible for the generation of cellular energy (ATP) through the process of oxidative phosphorylation (OXPHOS). During OXPHOS, mitochondria generate ROS through the electron transport chain (ETC). Despite being a natural part of the process, an accumulation of ROS due to a shift in equilibrium causes damage to the mitochondrial DNA (mtDNA). When a cell is no longer functioning normally, it initiates regulated cell death, most commonly apoptosis. Mitochondria not only have an essential role in intrinsic apoptosis, but also in providing energy for the complete apoptotic process [17]. Interestingly, much like the nuclear genome (nDNA), mtDNA is also controlled by epigenetic mechanisms. The epigenetic events related to mitochondria are known as mitoepigenetics and are carried out by mtDNA methylation and non-coding RNAs (ncRNAs) [18]. Abnormal mtDNA methylation is associated to pathological conditions, particularly to neurodegeneration (Amyotrophic Lateral Sclerosis) and aging. MicroRNAs (miRNAs) are nc RNAs and have a role in apoptosis, particularly in colon cancer, osteosarcoma, gastric cancer, breast, pancreas, and acute myeloid leukemia (AML). Venetoclax, an inhibitor of the anti-apoptotic protein BCL2, has been described as beneficial in AML in older patients by promoting apoptosis in this entity [19].
PRRs (patterns recognition receptors) are capable of sensing different microbes (PAMPs=pathogen associated molecular patterns) as well as endogenous molecules that are released after cell damage (DAMPs=dangerous associated molecular patterns). This PRR engagement is the prerequisite for the initiation of immune responses to infections and tissue injuries which can be beneficial or detrimental to the host. PRRs include the Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I–like receptors (RLRs), cytosolic DNA sensors and adhesion receptors. Most NLRs are located into the cytosol. They all consist of a central nucleotide-binding oligomerization (NOD) domain, and of C-terminal LRRs (leucine-rich repeat) which possibly mediate ligand binding. At least NLRP1–3 (NLR family, pyrin domain containing) form multiprotein complexes called inflammasomes (Figure 4). Inflammasomes, which were first described by Tschopp and colleagues, consist of one or two NLRs, in most cases of the adapter molecule ASC (apoptosis associated speck-like protein containing a CARD=caspase recruitment domains), and of caspase-1 [20]. Inflammasomes (which are activated by flagellin from pathogens that infect macrophages and by lipopolysaccharides from gram negative bacteria) are responsible for the release of family 1 interleukins (IL-1), particularly IL-1 and IL-18. Because this type of RCD involves IL-1 (historically known as leukocyte pyrogen), Brad Cookson and colleagues called it pyroptosis [21]. Caspase 1,3, 4, 5, and 11 mediate the processing of pro-IL1β, and pro-IL-18 zymogens into IL-1β and IL-18. The activation of inflammasomes requires at least two signals: TLR ligands and IFN (interferon) signals that activate NF-Kβ (nuclear factor kappa-beta= a cytoplasmic factor that translocates to the nucleus), ATP and LPS (lipopolysaccharides).
Several recent breakthroughs indicate that gasdermin D (GSDMD) is the key effector of pyroptosis [22,23]. GSDMD is a protein family that is cleaved by caspases 1 or 11 and its subunits bind to the inner flake of the plasmatic membranes and to the phospholipids of these membranes, producing lipid peroxidation and pore formation that cause lysis of the membranes and cell death. Pyroptosis is morphologically different from apoptosis. There is no DNA fragmentation, but there is nuclear condensation associated with cellular edema and blistering of the plasma membrane, which eventually ruptures. In short, pyroptysis is a GSDMD-mediated form of RCD that plays a role in inflammation and immunity (pyronecrosis is a synonym).
It is a form of caspase-independent RCD induced by disturbances of extracellular or intracellular homeostasis that occurs when the apoptotic machinery is inhibited (Figure 5) [24]. Also known as programmed necrosis, it is activated by stimulation of DRs (Fas and TNFR1), TLR3 and / or TLR4, IFNs, nucleic acid sensors, receptors that respond to retinoic acid, transmembrane proteins, and adhesion receptors. The signals are transmitted by TRIF (TICAM= Toll-like receptor adaptor molecule 1), DAI (Z-DNA binding protein 1= It´s an acid nuclei sensor), STAT3 (signals transducers and activators transcription), PKR (protein kinase receptor). and RIPK1(receptor-interacting serine/threonine kinase 1, a regulator of FASLG-induced T-cell necroptosis, a Fas ligand). RIPK3 is an intermediate molecule common to several signaling pathways, which activates the effector molecule pMLKL (mixed linage pkinase domain-like pseudokinase). The mechanism by which pMLKL produces necroptosis is not completely clear. Apparently, it has to do with the heat shock protein contest [25,26]. Morphologically it shares findings with necrosis such as: membrane permeabilization, cellular edema and loss of cellular integrity. It is activated in viral infections, neurodegenerative diseases, tissue injury and response to chemotherapy. The balance between membrane injury and repair capacity defines the rate of necroptosis.
Figure 5. Signalling Pathways of Regulated Cell Death
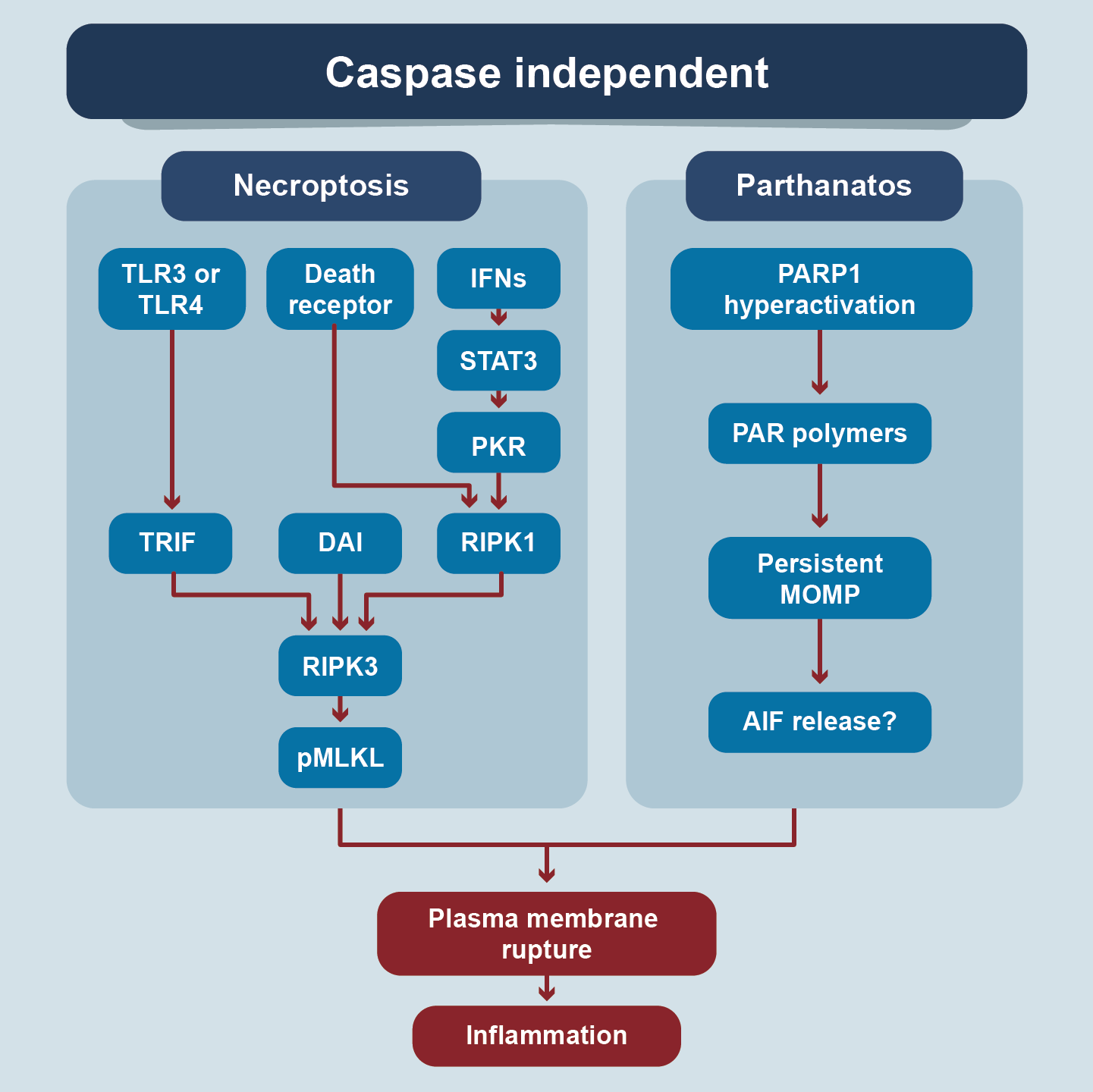
These types of RCD are caspases independent. All have different triggers and mediator molecules but converge in the production of plasma membrane rupture and inflammation.
Autophagy is an evolutionarily conserved degradation pathway and has been implicated in human disease and aging [27]. The process of autophagy involves the sequential formation of three unique membrane structures, namely the phagophore, autophagosome, and autolysosome (Figure 6). Morphologically, it is characterized by the appearance of large intracellular vesicles, blistering of plasma membranes, and depletion of cytoplasmic organelles in the absence of chromatin condensation. Over 40 autophagy-related genes/proteins (ATGs) play key roles in autophagic membrane dynamics and processes. As a dynamic recycling system, the bulk and nonselective autophagy process is generally considered as a pro-survival mechanism in response to multiple types of cellular stresses.Nevertheless, autophagy can selectively degrade pro-survival proteins related to other types of RCD, thereby tipping the balance from life to death [28]. In 2013, Beth Levine described autophagy as a subtype of autophagy-dependent cell death induced by nutrient deprivation or by Tat-Beclin 1, an autophagy-inducing peptide fusing amino acids from BECN1(autophagy protein) and HIV Tat protein (a diglycine linker) [29]. Autophagy-dependent cell death probably plays a pathogenic role in neurotoxicity and hypoxia-ischemia-induced neuronal death,indicating that this type of RCD can possibly be targeted for neuroprotection [30]. Dysregulation of autophagy has been associated with embryological lethality, developmental disorders, cancer, and cardiovascular disorders.
Figure 6. Autophagy
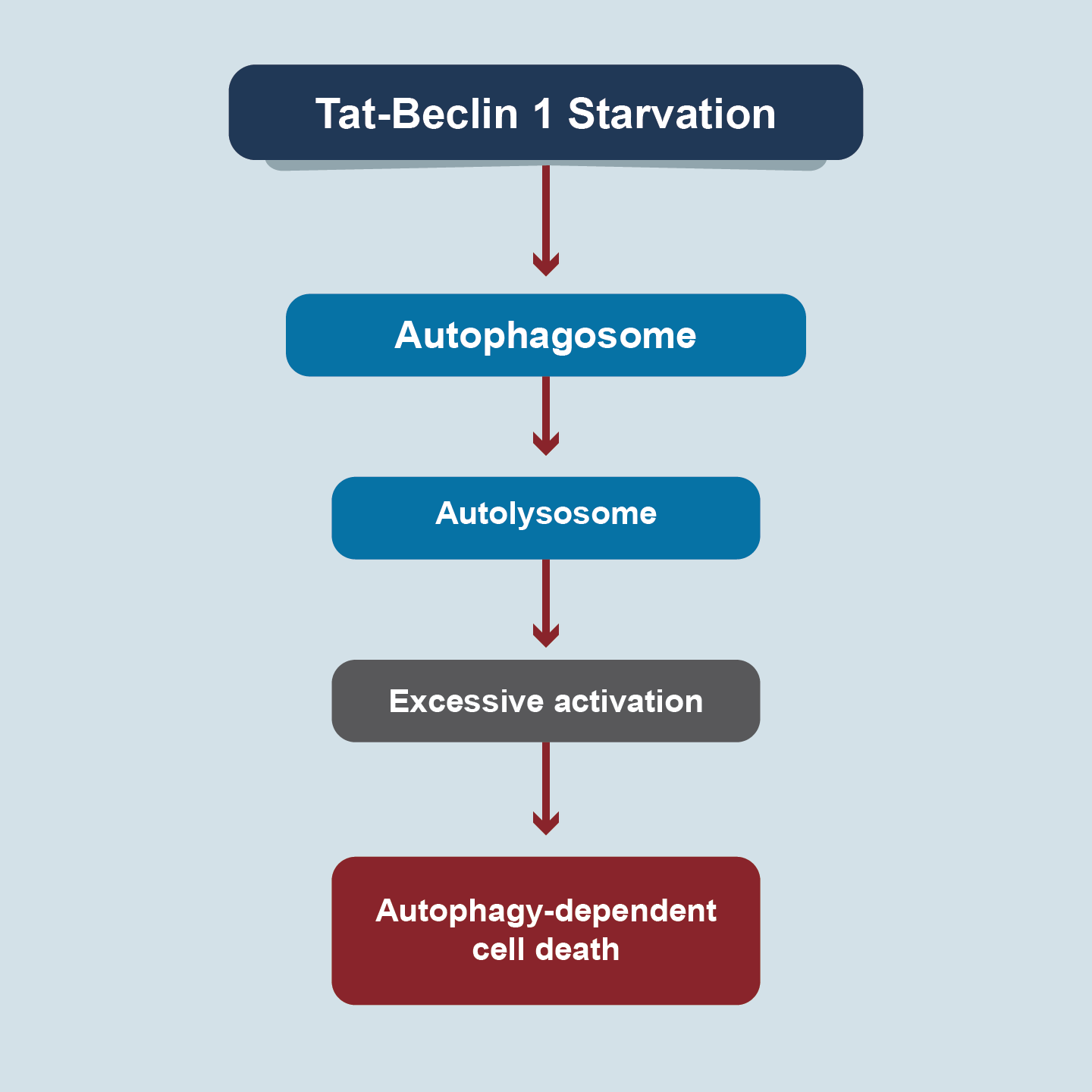
Autophagy-dependent cell death is driven by the molecular machinery of autophagy.
The term was coined by Valina and Ted Dawson in 2009 [31]. Parthanatos is a poly [ADP-Ribose] polymerase 1 (PARP1)-dependent RCD that is activated by oxidative stress-induced DNA damage and chromatinolysis [5]. Neither apoptotic body nor small DNA fragments are formed (as in apoptosis). Neither does cellular edema occur but there is a rupture of plasma membranes. PARP1 is a chromatin-associated nuclear protein that plays a critical role in the repair of DNA single-strand or double-strand breaks. PARP1 can recognize DNA breaks and use nicotinamide adenine dinucleotide (NAD+) and ATP to trigger poly (ADP-ribose)-sylation and initiate DNA repair. Apoptosis-inducing factor mitochondria-associated 1 (AIFM1, also known as AIF) is required for parthanatos execution [32]. AIF is activated because the hyperactivity of PARP mediates the synthesis of PAR, a molecule that moves from the nucleus to the cytoplasm, binds to mitochondrial proteins releasing AIF. AIF produces chromatinolysis (AIF translocates to the nucleus, generating DNA fracture). MIF (macrophage migration inhibitory factor) is a precipitant of parthanatos by catalyzing DNA cleavage. ROS, hypoxia, hypoglycemia, and inflammatory keys produce DNA alkylation. Parthanatos is associated with various pathological conditions such as cardiovascular, renal, metabolic disorders, transient cerebral ischemia. and neurodegenerative disorders.
The term “lysosomal cell death” was coined in 2000 [33]. Lysosome-dependent cell death (LCD) is a type of RCD mediated by hydrolytic enzymes that are released into the cytosol after lysosomal membrane permeabilization[34]. Lysosomes are acidic cellular organelles that can degrade nucleic acids, proteins, lipids, carbohydrates, entire organelles (e.g., mitochondria), and invading pathogens. The process necessarily passes through the permeabilization of the lysosomal membrane. Lysosomal hydrolases, for example, cathepsins (cysteine peptidases) play a major role in this type of RCD. Blocking his expression could be a therapeutic goal in the future. Abundant iron deposits in lysosomes permeabilize their membranes, increasing the deposition of this toxic metal in the cytosol, which is associated with oxidative injury and this contributes to lysosomal storage disorders, diseases associated with age (cardiovascular and neurodegenerative disorder), and response to intracellular pathogens [35]. Based on the cellular context, LCD can adopt necrotic, apoptotic, autophagic, or ferroptotic-like features, adding complexity to this cell death pathway [36].
Entotic cell death is a form of cell cannibalism in which one cell engulfs and kills another cell. The term entosis was coined in 2007 by Joan Brugge [37]. Entosis and entotic cell death occur mostly in epithelial tumor cells in the contexts of aberrant proliferation, glucose starvation, matrix deadhesion,or mitotic stress [38,39]. The underlying mechanisms are not well-understood. Cell adhesion and cytoskeletal rearrangement pathways play a central role in the control of entosis induction [40]. Generally, the cells that engulf entotic cells (to be internalized) are of the same cell lineage (homotypic) or of a different lineage (heterotypic) but they are viable non-phagocytic cells. More than phagocytosis, what occurs is a cellular invasion. The elimination process is independent of the Bcl 2 protein and caspases. Engulfed cells undergo phagocytosis and lysosomal degradation. Unlike autophagia, entosis represents a process of self-control. This form of RCD is observed in several human neoplasms, generally associated with the tumor stage, operating as an onco-suppressive process. Until now, there are no agents that can induce or inhibit it [41].
NETosis, was first observed in neutrophils by Arturo Zychlinsky in 2004 [42]. Netotic cell death is a form of RCD driven by NET release. NETs are extracellular net-like DNA-protein structures released by cells in response to infection or injury.NETs can also be generated by other leukocyte populations (e.g., mast cells, eosinophils, and basophils), epithelial cells, and cancer cells in response to various stresses [43]. Elevated NETosis not only acts against the spread of infection by trapping pathogenic microorganisms (e.g., bacteria and viruses), but also promotes DAMPs release, thus possibly contributing to the pathogenesis of autoimmune disorders (e.g., systemic lupus erythematosus, rheumatoid arthritis, asthma, vasculitis, and psoriasis), ischemia-reperfusion injury, and tumor development [44]. Netosis is a dynamic process that involves multiple signals and steps that lead to the translocation of a series of by-products from the cytoplasm to the nucleus, which generates citrullination of histones, chromatin de-condensation, destruction of the nuclear envelope and liberation of chromatin fibers that form the net [45]. But this leads to cell death.
Alkaliptosis is a new type of RCD driven by alkalinization of cells described in 2018 [46]. The action of an opioid analgesic known as JTC801 can lead to cell death of cells of neoplastic lineage of the pancreas, kidney, prostate and brain and intracellular alkalinization depends on the inhibition of the IKKβ (inhibitor kinase of NF-Kβ)-NF-Kβ axis, underregulating carbonic anhydrase which participates in fine-tuning the pH. The clinical significance of this type of RCD remains to be defined.
Oxeiptosis was introduced in 2018 by Andreas Pichlmair [47]. It is a type of caspase-independent RCD induced by oxygen radicals (ozone and H2O2) that activate an enzymatic pathway, namely: KEAP1-PGAMS-AIFM1, a pathway that is usually protective against oxidative cell injury, but that if it becomes hyperactive due to oxygen radicals induce RCD. Its role in humans is unknown.
In 2012, the term ferroptosis was formally used by Brent Stockwell to describe a lipotoxicity iron-dependent form of non-apoptotic RCD [48]. It is characterized by small, dysmorphic mitochondria with diminished crista and a condensed and ruptured outer membrane, but without any of the characteristics of cells that are dying from necrosis and / or apoptosis. It occurs by a process of lipid peroxidation catalyzed by iron that targets the polyunsaturated fatty acids (PUFAs) of cell membranes [49]. GPX4 is a member of the GPXs (glutathione peroxidases) family that is a cytoprotective of bio-membranes against lipid peroxidation. If there is cysteine depletion, there is depletion on GSH (reduced glutathione) and consequently on GPX4, favoring lipid peroxidation. In ferroptosis the iron that accumulates (intra or extracellularly) producing ROS, cysteine depletion and lipid peroxidation. Ferroptosis can be associated with autophagy particularly in pancreatic tumors [50]. Excessive ferroptosis is likely to occur in certain human diseases, especially neurodegenerative and iron overload disorders, calling for its therapeutic suppression. For example, iron chelating agents and lipophilic antioxidants. In contrast, the induction of ferroptosis constitutes a potential strategy in cancer therapy particularly in pancreatic tumors[51].
It is also worth mentioning that multiple terms have been described trying to identify new forms of RCD or subtypes within a form of death. For example, methousis refers to RCD with massive accumulation of single-membrane fluid-filled vacuoles derived from macropinosomes (structures formed by micropinocytosis or ‘cell drinking’ is a type ofendocytosisthat involves the nonspecific uptake of extracellular material, such as soluble molecules, nutrients, and antigens). Autophagy has several subtypes such as mitophagy, which is a selective process that segregates and degrades mitochondria; reticulophagy when inappropriate or non-functional portions of the ER are removed; pexophagy when peroxisomes are removed; nucleophagy the nucleus; ribophagy refers to degradation of damaged ribosomes and lysophagy of lysosomes [52]. Clockophagy is a new autophagy pathway that removes ARNT (aryl-hydrocarbon nuclear receptor translocator-like protein), which is a fundamental transcription factor in the circadian clock of mammals. The circadian rhythm is an endogenous mechanism that controls various cellular processes such as iron metabolism, oxidative stress, and cell death [53]. It is obvious that several mechanisms can act together in the same pathology or process and, also, the opposite, that several pathologies share the same mechanism [54-57]. Immunogenic cell death (ICD) is a functionally peculiar form of RCD that is sufficient to activate an adaptive immune response specific for endogenous (cellular) or exogenous (viral) antigens expressed by dying cells. These agents are able to stimulate the timely release of a series of DAMPs, whose recognition by PRRs expressed by innate and adaptive components of the immune system warns the organism of a situation of danger, resulting in the elicitation of an immune response generally associated with the establishment of immunological memory [58]. ICD as a type of RCD that is sufficient to activate an adaptive immune response in immunocompetent hosts.
Cellular senescence refers to pathophysiological processes by which cells lose their proliferative capacity while remaining viable and metabolically active. Chronic cellular senescence has been associated with aging, loss of life years, tissue deterioration, atherosclerosis, and osteoarthritis [59].
Mitotic catastrophe is a regulated onco-suppressive mechanism that prevents proliferation and / or survival of cells that are not capable of completing mitosis or have problems in the mitotic machinery to complete the mitotic circle or a failure in mitotic check points. The morphological change is a single nuclear change, with multinucleation, macro-nucleation or micro-nucleation [60].
Terminal differentiation is a state between life and cell death difficult to define [61].
Immunity
The “inflammatory process” is incredibly complex, diverse and involves the participation of many cell and molecular products, interacting in complex combinations, activation of immune responses and PRRs. Briefly, the innate immune system is comprised of various anatomical barriers to infection, including physical barriers (bronchial epithelium and cilia), chemical barriers (alkalinity of bronchial mucus), and biological barriers (oropharyngeal flora) [62]. Added to these barriers are soluble factors and phagocytic cells that form the first line of defense against pathogens. Soluble factors include the complement system, the reactants acute phase proteins and chemical messenger proteins called cytokines [63]. The complement system (a biochemical network of more than 30 proteins in plasma or cell surfaces) is a key component of innate immunity. The system develops responses that kill invading pathogens by direct lysis (cell disruption) or promoting phagocytosis. These proteins also produce inflammatory responses, which are an important part of innate immunity. The acute phase reactant proteins are a class of plasma proteins that are important in inflammation. Cytokines secreted by immune cells in the early stages of inflammation stimulate the synthesis of acute phase reactant proteins by the liver [64]. The cytokines have an important role in regulating the immune response; some cytokines directly interfere with pathogens. Interferons (IFNs) have antiviral activity [65]. Soluble factors are important to recruit phagocytic cells to local areas of infection, such as monocytes, macrophages, and neutrophils (myeloid cells) that engulf and digest invading microorganisms through a process called phagocytosis. In addition, neutrophils also form extracellular traps, which are chromatin networks containing antibacterial proteins that can trap and kill extracellular bacteria. These cells express the PRRs identifying PAMPs (diverse biochemical signatures) that are unique to pathogenic microorganisms but preserved through several families of pathogens. Immediate immune response is non-specific and does not have “immunological memory”, which means that the same response (same time and same intensity) must be orchestrated each time the system re-encounters the antigen.
Adaptive immunity (also called acquired immunity), a second line of defense against pathogens, takes several days, even weeks to develop to the fullest. However, adaptive immunity is much more complex because it involves an antigen specificity and “immunological memory”. Exposure to a specific antigen stimulates production of immune cells that target the pathogen for destruction [63]. Immunological “memory” means that the immune response to a second exposure of the same pathogen is faster and stronger because the antigens are “remembered”. The primary mediators of this response are B and T lymphocytes. B lymphocytes produce antibodies which are specialized proteins that recognize and bind to foreign proteins or pathogens, in order to neutralize or facilitate destruction by macrophages. The response mediated by antibodies is called humoral immunity. In contrast, cellular immunity is mediated by T lymphocytes, which develop in the thymus. Different subsets of T cells have different roles in immunity adaptive. For example, cytotoxic T (natural killer) cells directly attack and kill infected cells, while helper T cells increase the response and help the function of other cells [62]. Regulatory T cells (also called suppressor T) suppress the immune response [62]. In addition to its vital role in innate immunity, the complement system modulates the adaptive immune response and is an example of interaction between the two immune systems [64]. Obviously, both response systems work together to protect the body from infection and disease.
Knowledge of the interaction of SARS CoV-2 with the human innate immune system is limited and much of that knowledge is by extrapolation of the mechanisms used by other homologous coronaviruses. Virus ligands connect to host cell PRRs in the cytosol via RLRs or at extra-cytoplasmic or endosomal receptors via TLRs, activating a cascade of signals that trigger cytokine secretion (e.g., IFN I / III, TNF-α, IL-1, IL-6, IL-8) which (physiological conditions) induce antiviral programs in target cells and enhance the adaptive immune response [66]. But coronaviruses have mechanisms that evade innate immunity, which evade or antagonize the recognition, signaling or secretion of molecules. For example, the nonstructural proteins of the virus produce changes in the viral RNA that "resemble" the mRNA of the host, thus avoiding the recognition by the RLRs, avoiding their degradation and favoring the transcription of the viral RNA [67]. Coronaviruses encode an endoribonuclease (NSP15) that blocks recognition by MDA-5 (molecules that sense and recognize ss-RNAs) [68]. Also, the propagation of signals through the cytoplasm can be blocked, although this mechanism is not clearly understood in SARS-CoV-2. There is a dysregulated response in the function of INFs that not only prevents recognition but could be involved in the pathogenesis of the disease, since hyperinflammation and cytokine storm (CS) could not be counteracted [69]. It is essential to understand how the balance between antiviral immune programs and inflammatory immune programs is lost, in order to find effective markers of disease activity and design therapeutic strategies.
Myeloid cells orchestrate the immune response to infections. CDCs (conventional dendritic cells) such as moCDs (monocyte-derived CDs), pDCs (plasmacytoid CDs), and macrophages are part of this cell population. This initial response to the SARS-CoV-2 virus appears to be dysregulated leading to the characteristic Covid-19 syndromes such as ARDS (acute respiratory distress syndrome), CS, and lymphopenia [70]. The analysis of PBMCs (peripheral blood mononuclear cells) shows activated CD4+ and CD14+ HLA-DR and these immune signatures correlate with the progression of the disease and with pathogenesis not only in the lung, but also in the terminal ileum and kidneys. However, the relationship between disease severity and myeloid cells requires further study.
ILCs (innate lymphoid cells) do not express receptors for antigens such as B or T cells and can be divided into two large groups. NKs (cytotoxic natural killer) and ILCs-1,2,3) (non-cytotoxic helper). In Covid-19 the number of NK is reduced. There appears to be an altered maturation or migration and the reduced values correlate with elevated levels of IL-6 as well as with high values of TNF-α. Treatment with monoclonal antibodies that target IL-6 or TNF-a could restore NK function in Covid-19 [71].
CD4 T helps B cells make antibodies and orchestrate the response of other immune cells. CD8 T kill virus-infected cells to reduce viral load. The dysregulated T response also generates immunopathology in Covid-19 and lymphopenia (particularly decreased CD8 T levels) correlate with the severity of the disease. The mechanism is not known. SARS-CoV-2 does not infect T cells as it does make the coronavirus in MERS-CoV (7) [72]. The humoral response (B cells) is critical for clearing cytopathic viruses and for the immune memory response necessary to protect against exogenous reinfection. Response B to SARS-CoV-2 is robust and fast. IgA, IgG, IgM, and neutralizing IgG rise in titers between 7-14 days after the onset of symptoms and persist in the weeks following viral clearance (Figure 7) [73]. The antibodies are directed fundamentally against the protein S and N of the virus and in protein S, specifically against RBD (receptor binding domain), a domain that is highly immunogenic and the antibodies are vigorously neutralizing by blocking the interaction of the RBD domain with the receptor (ACE-2) of the virus. Neutralization is virus-specific and depends on epitopes within the RBD [74].
Figure 7. Antibody-Mediated Immunity in SARS-CoV-2
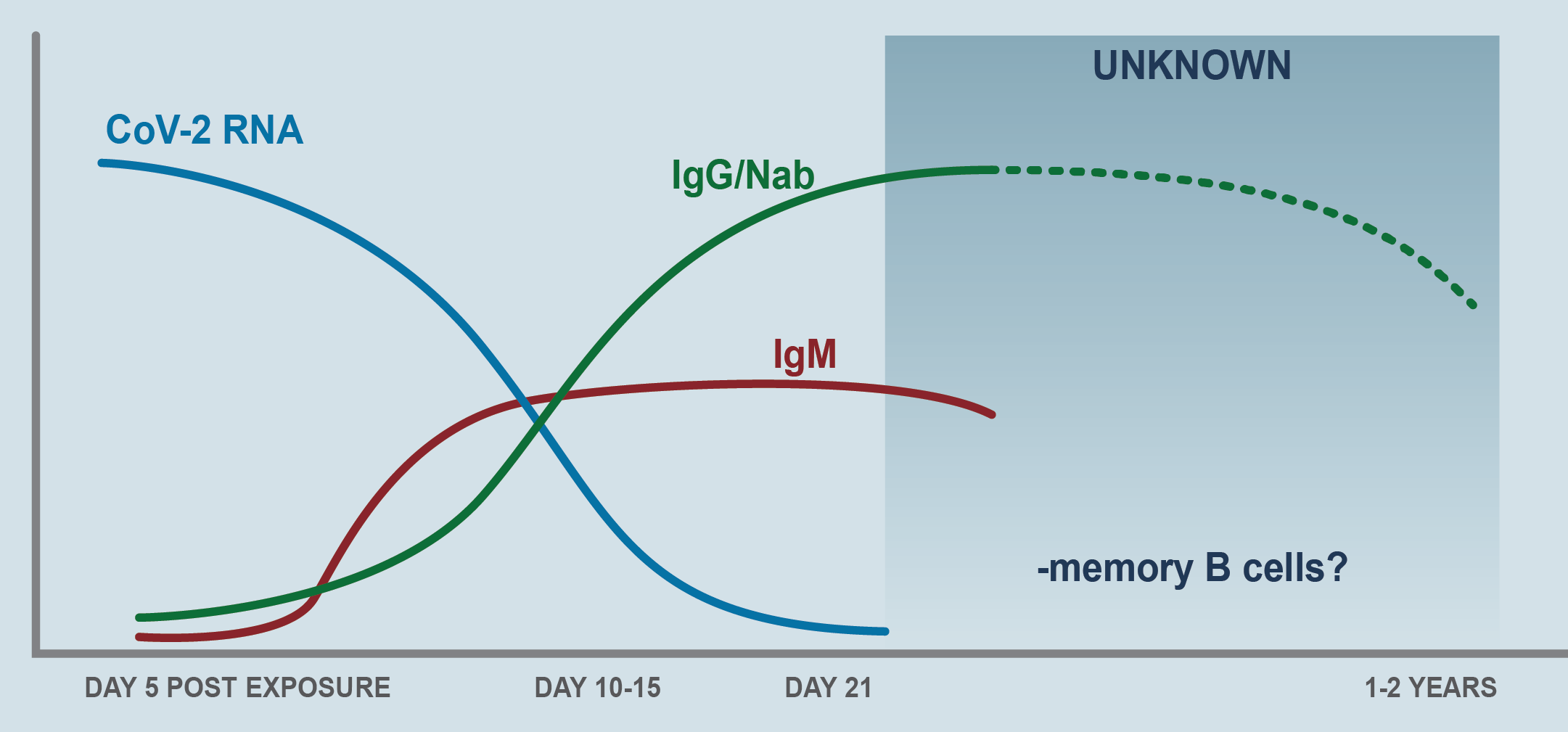
Virus-specific IgM and IgG are detectable in serum between 7 and 14 days after the onset of symptoms. Viral RNA is inversely correlated with neutralizing antibody titers. Higher titers have been observed in critically ill patients, but it is unknown whether antibody responses somehow contribute to pulmonary pathology. The SARS-CoV-1 humoral response is relatively short lived, and memory B cells may disappear altogether, suggesting that immunity with SARS-CoV-2 may wane 1–2 years after primary infection (Adapted from figure 4 of Vabret N, et al).
The duration of the neutralizing coverage of antibodies in Covid-19 is not known. Although it is true that some studies have drawn attention, in patients with mild disease, to the rapid reduction of antibody levels [75], the study carried out in Iceland showed that the antibodies do not decline in at least the first 4 months that the trial lasted [76]. The SARS-CoV-1 humoral response is relatively short lived, and memory B cells may disappear altogether, suggesting that immunity with SARS-CoV-2 may wane 1–2 years after primary infection. It is not clear whether antibodies to SARS-CoV-2 could contribute to lung disease via antibodies-dependent enhancement (ADE). This phenomenon is observed when non-neutralizing virus-specific IgG facilitate entry of virus particles into Fc-receptor (FcR) expressing cells, particularly macrophages and monocytes, leading to inflammatory activation of these cells [77].
Mitochondrial dysfunction and RCD
The human lung is composed of approximately 40 cell types, regionally and spatially located through the organ, and contains several levels of mitochondria [78]. The mitochondrial genome is inherited through the maternal germ line and has been inherited from aerobic prokaryotes bacteria more than two billion years ago and retains many of the morphological and biochemical characteristics of their bacterial ancestors. They are an icon of convoluted double-crested structures that are present in all cells of the body and have their own genome, transcriptome, and proteome [79]. According to the endosymbiotic theory, the mitochondria are descendants of old bacteria that entered symbiotic relation with the cells of the host [80]. Conceptualized as “cell batteries” (and consistent with this analogy subject to change), mitochondria are complex cellular organelles assembled from proteins encoded by two distinct genomes: nuclear chromosomal DNA and the mitochondrial genome, i.e., mitochondrial DNA (mtDNA). Despite the small size of the mtDNA (16.5 kb in humans), it encodes 13 potential oxidative phosphorylation subunits (OXPHOS) which interact with more than 70 units encoded by nuclear DNA: their concerted action is necessary to produce ATP, which is required for all active cellular processes [81]. Historically, the major role of mitochondria has been to catalyze the oxidation of metabolites for ATP production, via OXPHOS. OXPHOS involves transferring high energy electrons derived from NADH and FADH2 to the mitochondrial complex I and II, then to complex III and finally to complex IV, where they react with oxygen to form water. During this process, the protons are pumped through the inner mitochondrial membrane resulting in a proton-motive force which is used by the FOF1 ATP synthase [82]. Mitochondria continually oxidize fatty acids and consume end products of glucose, glutamine, and amino acids for the ATP production. Lung mitochondria preferably use glucose-derived substrates such as pyruvate for oxidative energy production. It also has its own electron transport chain (ETC), complex IV cytochrome c-oxidase (COX subunit IV-2) sensitive to oxygen and doubly active (binding oxygen) compared to COX from other tissues [83].
Additional critical functions of mitochondria such as regulation, proliferation, differentiation, cell death, redox and calcium homeostasis have been revealed during the last two or three decades. Recently, research has pointed to mitochondria as controlling the immune responses and determinants of immune cell phenotypes and their functions, including CD4+ T cell differentiation and CD8+ memory T cell formation [84]. Mitochondrial aerobic glycolysis is required for effective activation of T cells through the generation of mtROS, which are necessary for optimal activity of nuclear factor of activated T cell (NF-AT) and proximal T-cell receptor-mediated signaling [85].
While elevated levels of ROS produce tissue damage and promote aging, low ROS levels enhance defense mechanisms by inducing adaptive immune response (mitochondrial hormesis or mithormesis) [86]. Other fundamental concepts of the activity and function of mitochondria are mitochondrial biogenesis and mitophagy. Both refer to the phenomena of fusion and fission of the mitochondria. In fusion two mitochondria bind to generate other mitochondria while in fission is divided to generate two. This phenomenon allows the cells to replace metabolically dysfunctional mitochondria with fresh, undamaged mitochondria and allows the cells a healthy pool of mitochondria, since they do not form of “novo”, but the biogenesis results in growth and division of pre-existing mitochondria [87]. Mitophagy is a selective encapsulation by double membrane autophagosomes that is delivered to lysosome for degradation and removes damaged mitochondria that generate excessive amounts of ROS that can damage the mitochondria or other cellular components themselves [88,89]. Metabolically active cells (e.g., type II pneumocytes) also known as alveolar epithelial cells (AECs) have developed robust mitochondrial quality control programs.
Within the mitochondrial proteome are key proteins such as mitochondrial antiviral signaling protein (MAVS), which is the first protein located in the mitochondria involved in innate immune response and inflammatory response [90]. MAVS interacts with many molecules that play a role in apoptosis, mitochondrial dynamics, autophagy and proteosomal degradation. NOD-like receptor (NLRX1) is a member of the NLR family of PRRs that has a unique N-terminal domain (which explains the letter X1 in the acronym) and is the first, and only so far, of the PRRs located in mitochondria and therefore, it establishes a connection between mitochondrial functions and innate immunity [91]. Alveolar macrophages are essential for lung defense of the host through its ability to survive and regulate the innate and adaptive immune response. They are believed to be maintained in a relatively “quiescent state” with active suppression of inflammatory response to harmless antigens to prevent collateral damage to lung tissue [92,93].
The presence of two genomes in human cells - the nuclear genome and the tiny mtDNA - is a curiosity of evolution. Altered mtDNA diseases (more than 400 mutations or deletions in the 16,569-bas-pair mitochondrial chromosomes that contain only 37 genes), are heterogeneous disorders with well-known genetic causes [94]. The possibility of mitochondrial replacement in certain individualized cases is a viable therapeutic option. In fact, on December 15, 2016, the Human Fertilization and Embryology Authority in the United Kingdom approved the use - in certain specific cases - of an in vitro fertilization (IVF) technique involving donation of mitochondria [95]. The discovery of pathogenic defects of mtDNA occurred in the 1980s, but since then much research has revealed a common diseases number, in which mitochondrial dysfunction as a pathogenic and/or perpetuating mechanism of the process underlies. Mitochondria regulate four mechanisms of cell death: extrinsic apoptosis, intrinsic apoptosis, necrosis/necroptosis and pyroptosis [96,97].
CS is a severe inflammatory consequence in Covid-19, associated with deleterious systemic events such as oxidative stress, dysregulation in iron homeostasis (hyperferritinemia), hypercoagulability and thrombus formation. Biomarkers of inflammation and thrombosis are predictors of mortality. For example, lymphopenia, increased D-dimers, and IL-6 (endothelial vascular damage). Some researchers propose that infection with SARS-CoV-2 causes mitochondrial dysfunction of both intracellular and extracellular mitochondria [98] (Figure 8).
Figure 8. Mitochondria dysfunction in pathogenesis of COVID-19
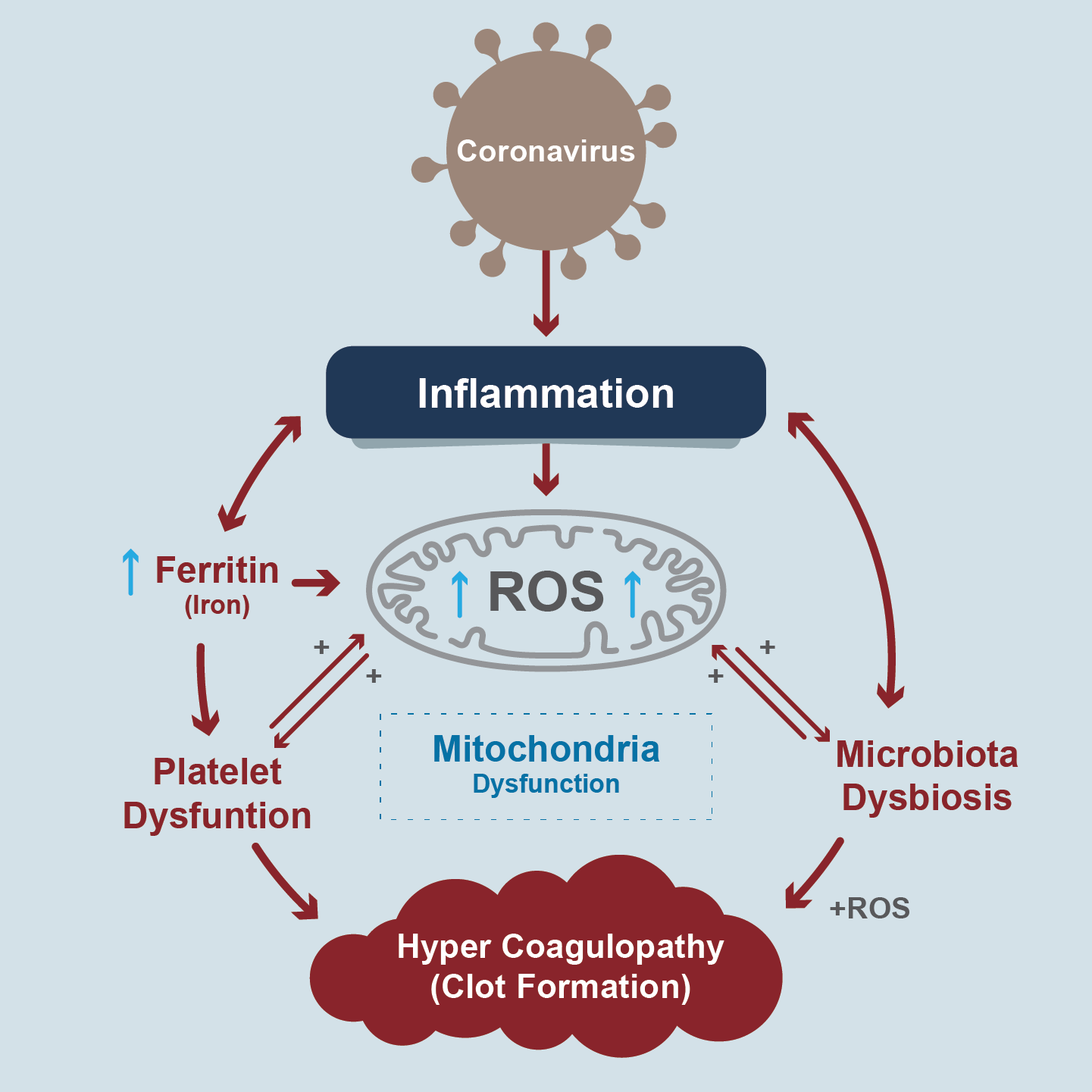
A hypothetical scheme describing events initiated by the COVID-19 pro-inflammatory surge of cytokines and ferritin levels leading to oxidative stress and cellular damage (Adapted from figure 1 of Saleh J, et al)
CS through TNF-α, IL-6 and IL-10 increase the production of mitochondrial ROS and, in turn, mitochondrial ROS increases the production of cytokines establishing a dangerous vicious circle. These cytokines prevent oxidative phosphorylation in mitochondria, and therefore the production of ATP, and favor apoptosis, but also, since the mitochondria are damaged, they release mtDNA that behaves like a DAMP and activates more inflammation, increasing the production of more ROS and perpetuating the vicious circle. These 3 cytokines are hallmarks of severe disease in Covid-19. They, and damaged mitochondria, could be markers of severe disease of clinical interest and therapeutic targets in critical disease [99].
Hyperferritinemia is a marker of severe systemic disease and adverse outcomes in Covid-19. It comes from ferritin released from exhausted cells that are dying. This increased iron reacts with oxygen produces ROS which damages the mitochondrion [100]. Under physiological conditions, the mitochondria use iron for 3 essential activities: synthesis of the heme group, biogenesis of iron-sulfur conglomerates and mitochondrial ferritin deposition, necessary to facilitate the "redox" functions in mitochondria. Normally, hepcidin (a key iron regulating hormone) sequesters it in enterocytes and macrophages, keeping it at the intracellular level and preventing the efflux and inflammation that this implies. But when there is a systemic iron overload, more cellular oxidative stress is generated, more cytokine release, reduction of cellular oxygen consumption, lipid peroxidation and a shift of the aerobic (pyruvate) to anaerobic (lactate) metabolism by means of lactic dehydrogenase (LDH) , an enzyme that is upregulated in Covid-19 [101]. Hyperferritinemia produces RCD due to ferroptosis. Iron oxides interact with proteins of the serum coagulation cascade. This, and the endothelial dysfunction produced by the virus (endotheliosis) leads to the formation of thrombi and generates dysbiosis of bronchopulmonary and intestinal microbiota [102]. Iron chelators, ferroptosis inhibitors, hepcidin and erythropoietin modulators could be future therapeutic targets.
Platelets are cells without a nucleus with the primary function of homeostasis and wound healing. Having no nucleus, platelet health depends on the health of your mitochondria (5-8 mitochondria per platelet). By having dysfunctional mitochondria, platelets undergo apoptosis, and this induces clots 50 times faster than normal platelets, increasing the formation of thrombi in Covid-19. In fact, thrombocytopenia is a marker of severity and poor prognosis. Cardiolipin is a mitochondrial phospholipid that participates in maintaining the integrity of the membranes of these organelles, particularly in the inner sheet. In patients with coagulopathy and thrombocytopenia in Covid-19, IgA anticardiolipin antibodies are detected in the serum, which could be used as a marker of mitochondrial alteration [103].
1) Among the fundamental processes of cell life are not only survival, proliferation, and differentiation, but also RCD, which plays a major role in development, tissue homeostasis, inflammation, immunity, and multiple pathophysiological conditions. RCD may not only play a role in maintaining homeostasis but also in "unjustified" cell loss.
2) Despite the biodiversity of pathways that lead to RCD and their distinctive immunological changes and consequences, the interrelationships between different forms of RCD remain unknown, and can be activated in a unique, mixed, serial, parallel or "concert" manner. to generate a scheduled result. Therefore, blocking one pathway could lead to activation of another.
3) Possibly for this reason, 20 years of drug study have not generated the expected results and pharmacological research should be redirected towards the simultaneous inhibition of multiple signals or the "upstream" blocking of the initiation of the various CDR programs, thus as the critical point of "no return" for each subtype to try to stop, reverse or speed up the process as appropriate.
4) There are few significant human diseases in which RCD may not be relevant or its control of potential therapeutic value. Hopefully, the enormous effort being put into analysing the mechanisms and regulation of RCD will lead to the identification of new targets for drug action in many of these disease states. Covid-19 is a good and recent example.
This work was only carried out by the author. Author AA contributed on the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
None.
None.
- Wyllie A H (1998) A tidy death. The promise of apoptosis research. Odyssey 4: 47-51.
- Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239-257. [Crossref]
- Singh R, Letai A, Sarosiek K (2019) Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Re Mol Cell Biol 20: 175-193. [Crossref]
- Linkermann A, Stockwell BR, Krautwald S, Anders HJ (2014) Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 14: 759-767. [Crossref]
- Vanden Berghe T, Linkermann H, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15: 135-147. [Crossref]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, et al. (2012) Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19: 107-120. [Crossref]
- Lee EW, Seo J, Jeong M, Lee S, Song J (2012) The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep 45: 496-508. [Crossref]
- Yang G, Elbadawi M, Efferth T (2020) Multiple cell death modalities and their key features (Review) World Acad Sci J 2: 39-48.
- Czabotar PE, Lessene G, Strasser A, Adam JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49-63. [Crossref]
- Galluzzi L, Keep O, Kroemer G. (2016) Mitochondrial regulation of cell death: a phylogenetically conserved control. Microb Cell 3: 101-108. [Crossref]
- Galluzzi L, Lopez-Soto A, Kumar S, Kroemer G (2016) Caspases connect cell-death signaling to organismal homeostasis. Immunity 44: 221-231. [Crossref]
- Tang D, Kang R, Vanden Berghe T, Vandenabeele P, Kroemer G (2019) The molecular machinery of regulated cell. Cell Research 29: 347-364.
- Paoli P, Giannoni E, Chiarugi P (2013) Anoikis molecular pathways and its role in cancer progression. Biochem Biophys Acta 1833: 3481-3498. [Crossref]
- Cecconi, F, Gruss P (2001) Human Genome and Diseases: Apaf1 in developmental apoptosis and cancer: How many ways to die? CMLS Cell Mol Life Sci 58: 1688-1697.
- Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ, et al. (2011) The holo-apoptosome: activation of procaspase-9 and interactions with caspase-3. Structure 19: 1084-1096. [Crossref]
- Nguyen TAT, Grimm SA, Bushel PR, Li J, Li Y, et al. (2018) Revealing a human p53 universe. Nucleic Acids Res. 46; 8153-8167. [Crossref]
- Frezza C, Cipolat S, Martins de Brit O, Micaroni M, Beznoussenko GV, et al. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177-189.
- Iacobazzi V, Castegna A, Infantino V, Andria G (2013) Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab 110: 25-34. [Crossref]
- Cavalcanti GC, Schaan AP, Fonseca-Cabral, Santana-da-Silva MN, Pinto P, et al. (2019) A cell´s fate: an overview of the molecular biology and genetics of apoptosis. Int J Mol Sci 20: 4133. [Crossref]
- Opitz B, van Laak V, Eitel J, Suttorp N (2010) Innate immune recognition in infectious and non-infectious diseases of the Lung. Am J Respir Crit Care Med 181: 1294-1309. [Crossref]
- Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907-1916. [Crossref]
- Chen X, He W, Hu L, Li J, Fang Y, et al. (2016) Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26: 1007-1020.
- Ding J, Wang K, Liu W, She Y, Sun Q, et al. (2016) Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111-116. [Crossref]
- Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517: 311-320. [Crossref]
- Schock SN. Chandra NV, Sun Y, Irie T, Kitagawa Y, et al. (2017) Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ 24: 615-625. [Crossref]
- Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A (2018) Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J Immunol 200: 2748-2756. [Crossref]
- Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176: 11-42. [Crossref]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349-364. [Crossref]
- Liu Y, Shoji-Kawata S, Sumpter Jr RM, Wei Y, Ginet V, et al. (2013) Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA 110: 20364-20371. [Crossref]
- Kriel Jurgen, Loos Ben (2019) The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ 26: 640-652. [Crossref]
- David KK, Andrabi SA, Dawson TM, Dawson VL (2009) Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 14: 1116-1128. [Crossref]
- Mashimo M, Kato J, Moss J (2013) ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci USA 110: 18964-18969. [Crossref]
- Franko J, Pomfy M, Prosbova T (2000) Apoptosis and cell death (mechanisms, pharmacology and promise for the future). Acta Medica (Hradec. Kralove) 43: 63-68. [Crossref]
- Aits S, Jaattela M (2013) Lysosomal cell death at a glance. J Cell Sci 126: 1905-1912. [Crossref]
- Terman A, Kurz T (2013) Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 18: 888-898. [Crossref]
- Gao H, Bai Y, Jia Y, Zhao Y, Kang R, et al. (2018) Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun 503: 1550-1556. [Crossref]
- Overholtzer M, Mailleux AA, Mouneimne G, King RW, Cibas ES, et al. (2007) A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131: 966-979. [Crossref]
- Hamann JC, Surcel A, Chen R, Albech JG, Robinson DN (2017) Entosis Is Induced by Glucose Starvation. Cell Rep 20: 201-210. [Crossref]
- Durgan J, Tseng YY, Hamann AC, Domat MC, Collison L, et al. (2017) Mitosis can drive cell cannibalism through entosis. Elife 6: e27134. [Crossref]
- Durgan J, Florey O (2018) Cancer cell cannibalism: multiple triggers emerge for entosis. Biochim Biophys Acta Mol Cell Res 1865: 831-841. [Crossref]
- Hartman ML (2020) Non-apoptotic cell death signaling pathways in Melanoma. Int J Mol Sci 21: 1-29. [Crossref]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhl Y (2004) Neutrophil extracellular traps kill bacteria. Science 303: 1532-1535. [Crossref]
- Kazzaz NM, Sule G, Knight JS (2016) Intercellular interactions as regulators of NETosis. Front Immunol 7: 453. [Crossref]
- Branzk N, Papayannopoulos V (2013) Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol 35: 513-530. [Crossref]
- Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, et al. (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18: 1386-1393. [Crossref]
- Song X, Zh S, Zeh HJ, Kang R, Tang S (2018) JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 154: 1480-1493. [Crossref]
- Holze, C, Michaudel C, Mackwiah C, Haas DA, Benda C, et al. (2018) Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol 19: 130-140. [Crossref]
- Friedmann Angeli JP, Schneider M, Conrad M (2014) Inactivation of the ferroptosis regulator GPx4 triggers acute renal failure in mice. Nat Cell Biol 16: 1180-1191. [Crossref]
- Kang R, Tang D (2017) Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep 5: 153-159.
- Stockwell BR, Friedmann-Angeli B, Bayir H, Toyokuni S, Woerpel KA, et al. (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273-285. [Crossref]
- Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, et al. (2020) Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol 27: 420-435. [Crossref]
- Yang M, Chen P, Liu J, Zhu S, Kroemer G, et al. (2019) Clockopaghy is a novel selective autophagy process favoring ferroptosis. Sci Adv 5: eaaw2238. [Crossref]
- D´Arcy MS (2020) Cell death: a review of the major forms of apoptosis, necrosis, and autophagy. Cell Biol International 43: 582-592. [Crossref]
- Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN (2019) Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev 99: 1765-1817.
- Woznicki JA, Flood P, Bustamante-Garrido M, Stamov P, Moloney D, et al. (2020) BCL-G regulates secretion of inflammatory chemokines but is dispensable for induction of apoptosis by INF-Ɣ and TNF-α in intestinal epithelial cells. Cell Death Dis 11: 68. [Crossref]
- Chen Y, Hua Y, Li X, Arsalan IM, Zhang W, et al. (2020) Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol 11: 42. [Crossref]
- Williams JA, Ding WX (2020) Role of autophagy in alcohol and drug-induced livery injury. Food Chem Toxicol 136: 111075. [Crossref]
- Galluzzi L, Buqué A, Keep O, Zitvogel L, Kroemer G. (2017) Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17: 97-111. [Crossref]
- Sharpless NE, Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15: 397-408. [Crossref]
- Dominguez-Brauer C, Thu KL, Mason JL, Blaser H, Bray MR, et al. (2015) Targeting mitosis in cancer: emerging strategies. Mol Cell 60: 524-536. [Crossref]
- Kaestner L, Minetti G. (2017) The potential of erythrocytes as cellular aging models. Cell Death Differ 24: 1475-1477. [Crossref]
- Parkin J, Cohen B (2001) An overview of the immune system. Lancet 357: 1777-1789. [Crossref]
- Delves PJ, Roitt IM (2000) The immune system. First of two parts. N Engl J Med 343: 37-49. [Crossref]
- Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20: 34-50. [Crossref]
- Mak TW, Saunders ME (2004) Innate immunity. The immune response: basic and clinical principles. Amsterdam: Elsevier Academy Press. pp: 79-92.
- Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, et al. (2019) IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest 130: 3625-3639. [Crossref]
- Zhao X, Sehgal M, Hou Z, Cheng J, Shu S, et al. (2018) Identification of Residues Controlling Restriction versus Enhancing Activities of IFITM Proteins on Entry of Human Coronaviruses. J Virol 92: e01535-17. [Crossref]
- Hackbart M, Deng X, Baker SC (2020) Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Nat Acad Sci USA 117: 8094-8103.
- Vabret N, Britton GJ, Gruber C, Hegde S, Lim J, et al. (2020) Immunology of COVID-19: current state of the science. Immunity 52: 910-941. [Crossref]
- Mehta P, McAuley DF, Brown M, Sanchez MR, Tattersall RS, et al. (2020) HLH Across Specialty Collaboration, UKCOVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395: 1033-1034. [Crossref]
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, et al. (2018) Innate Lymphoid Cells: 10 Years On. Cell 174: 1054-1066. [Crossref]
- Zheng M, Gao Y, Wang G, Song G, Liu S, et al. (2020) Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 17: 533-535. [Crossref]
- Hsueh PR, Huang LM, Chen PJ, Kao CL, Yang PC (2004) Chronological evolution of IgM, IgA, IgG and neutralizing antibodies after infection with SARS-associated coronavirus. Clin Microbiol Infect 10: 1062-1066. [Crossref]
- To KKW, Tsang OTY, Leung WS, Tam AR, Wu TC, et al. (2020) Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis 20: 565-574.
- Ibarrondo FJ, Fulcher JA, Goodman-Meza D, Elliott J, Hoffman C, et al. (2020) Rapid decay of Anti-SARS-CoV-2 antibodies in persons with mild disease. N Engl J Med 383: 1085-1087. [Crossref]
- Gudbjartsson DF, Norddahl GL, Melsted P, Gunnarsdottier K, Holm H, et al. (2020) Humoral immune response to SARS-CoV-2 in Iceland. N Engl J Med 383: 1724-1734. [Crossref]
- Taylor A, Foo SS, Bruzzone R, Dinh LV, King NJC, et al. (2015) Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev 268: 340-364. [Crossref]
- Natarajan V, Parinandi NL (2014) Mitochondrial function in lung health and disease. J Clin Invest 126: 809-820. [Crossref]
- Cloonan SM, Choi AM (2016) Mitochondria in lung disease. J Clin Invest 126: 809-820. [Crossref]
- Gray MW, Burger G, Lang BF (1999) Mitochondrial evolution. Science 283: 1476-1481.
- Chinnery PF, Zeviani M (2016) Mitochondrial matchmaking. N Engl J Med 375: 1894-1896. [Crossref]
- Scheffler IE (2007) Mitochondria (2nd Edn) Hoboken: Wiley-Liss.
- Huttemann M, Lee I, Gao X, Pecina P, Pecinova A, et al. (2012) Cytochrome c-oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J 26: 3916-3930. [Crossref]
- Bialas AJ, Sitarek P, Milkowska-Dymanowska J, Protrowski WJ, Gorski P (2016) The role of mitochondrial and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cel Longev 2016: 7808576. [Crossref]
- Kang MJ, Shadel GS (2016) A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis (Seoul) 79: 207-213. [Crossref]
- Ristow M, Schmeisser K (2014) Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 12: 288- 341. [Crossref]
- Jornayvaz FR, Shulman GI (2010) Regulation of mitochondrial biogenesis. Essays Biochem 47: 69-84. [Crossref]
- Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872- 884. [Crossref]
- Bhatia-Kissova I, Camougrand N (2013) Mitophagy is not induced by mitochondrial damage but plays a role in the regulation of cellular autophagic activity. Autophagy 9: 1897-1899. [Crossref]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-KB and IRF 3. Cell 122: 669-682. [Crossref]
- Arnoult D, Soares F, Tattoli I, Girardin SE (2011) Mitochondria in innate immunity. EMBO Rep 12: 901-910. [Crossref]
- Vlahos R, Bozinovski S. (2014) Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol 5: 435. [Crossref]
- Byrne AJ, Mathie SA, Gregory LG, Lloyd CM (2015) Pulmonary macrophages: key players in the innate defence of the airways. Thorax 70: 1189-1196. [Crossref]
- Falk MJ, Decherney A, Kahn JP (2016) Mitochondrial replacement techniques - implications for the clinical community. N Engl J Med 374: 1103-1106. [Crossref]
- Phimister EG (2017) Mitochondrial donation clearing the final regulatory hundle in the United Kingdom. N Engl J Med 376: 171-173. [Crossref]
- Shenoy S (2020) Coronavirus (Covid-19) sepsis: revising mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflammation Res 7: 1-9. [Crossref]
- Dard L, Blanchard W, Hubert CL, Lacombe D, Rossignol R (2020) Mitochondrial function and rare diseases. Mol Aspects Med 71: 100842. [Crossref]
- Phua J, Weng L, Ling L, Egi M, Lim CM, et al. (2020) Intensive Care management of coronavirus disease 2019 (COVID-19): challenges and recommendations. Lancet Respir Med 8: 506-517. [Crossref]
- Jo EK, Kim JK, Shin DM, Sasakawa C (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13: 148-159. [Crossref]
- Huang C, Wang Y, Li X, Ren L, Zhao J, et al. (2020) Clinical´s features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497-506.
- Young A, Oldford C, Mailloux RJ (2020) Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol 28: 1-7. [Crossref]
- Edear M, Saleh J, Pegssornaux C (2020) Iron: innocent bystander or vicious culprit in Covid-19 pathogenesis? Int J Infect Dis 97: 303-305. [Crossref]
- Saleh J, Peyssonraux C, Singh KK, Edear M (2020) Mitochondria and microbiota dysfunction in Covid-19 pathogenesis. Mitochondrion 54: 1-7.