Abstract
Background: Influenza and the common cold are the most frequent infectious diseases in humans. Their diagnosis is mainly symptomatic, and symptom management by over-the-counter drugs is the main treatment. The combination of an analgesic and a nasal decongestant can provide relief from cold and flu symptoms in a single treatment by simplifying therapy and reducing the number of individual doses of the active ingredients. E-Pharma Trento S.p.A. has developed an alternative to conventional pharmaceutical forms, in the form of granules containing a paracetamol/phenylephrine hydrochloride combination, designed to ensure easier and more flexible administration of the drug. This formulation can in fact be dissolved in hot water and administered as an oral solution or swallowed as such, allowing rapid use without water.
A study was thus conducted to characterize the pharmacokinetics of this formulation. The primary objective of this phase 1 study was to assess the pharmacokinetic parameters of the fixed oral combination of 600 mg paracetamol and 10 mg phenylephrine hydrochloride granules administered as an oral solution, evaluated in healthy volunteers after administration of a single dose.
Methods: A phase 1, single dose, pharmacokinetic study was conducted. Subjects were assigned to receive 1 sachet of granules (paracetamol/phenylephrine hydrochloride 600 mg/10 mg), dissolved in hot water under fasting conditions. Main pharmacokinetic parameters were evaluated. Safety and tolerability were also assessed during all ages of the trial.
Results: The developed formulation showed a rapid gastrointestinal absorption reaching the maximum concentration of the two analytes within 30 minutes. The mean maximum plasma concentration observed were 11.06 ± 3.77 µg /ml and 2.15 ± 0.99 ng/ml for paracetamol and phenylephrine, respectively. Overall, the safety and tolerability of the study medication resulted good.
Conclusions: The newly developed formulation of paracetamol/phenylephrine granules administered as an oral solution was able to provide a pharmacokinetic profile substantially consistent to that of other similar oral formulations.
Keywords
acetaminophen, granules, oral administration, pharmaceutical solutions, pharmacokinetics, phenylephrine
Introduction
In humans, the most prevalent infectious syndromes are influenza (flu) and common cold. There is no cure for colds and influenza conditions. Treatments for these illnesses are primarily symptomatic, and their diagnosis is dependent on their symptomatology [1]. Analgesic/antipyretic medications and nasal decongestant medications have a well-established and long-standing clinical utility in treating the symptoms of acute upper respiratory tract infections (URTIs) or the common cold, which are frequently accompanied by pain, fever, and nasal blockage [2-4]. The combination of more active substances can, in fact simplify therapy by reducing the number of individual doses; thus, the compliance of the patient is potentially improved, and medication errors are reduced [5].
Paracetamol is an antipyretic and analgesic medication that is neither a nonsteroidal anti-inflammatory drug (NSAID) nor an opiate. Because it inhibits the cyclooxygenase (COX) pathways, the medication has historically been classified alongside nonsteroidal anti-inflammatory medicines (NSAIDs), even though its precise mode of action is yet unknown [6]. Paracetamol exhibits analgesic and antipyretic effects comparable to those of NSAIDs, but it lacks significant peripheral anti-inflammatory activity. It is believed to inhibit the COX pathway within the central nervous system (CNS), while having minimal effect on peripheral COX enzymes. Moreover, paracetamol does not seem to bind to the active sites of either COX-1 or COX-2. Instead, it reduces the activity of COX through an alternative mechanism [6,7].
The analgesic and antipyretic indications of paracetamol have been demonstrated in many different diseases and conditions such as sore throat, headache, muscle pain, osteoarthritic, back pain, dental pain, pain caused by cancer, after surgery, injuries, post-partum, dysmenorrhea. Paracetamol has been administered as an over-the-counter medication to adults and children for over 60 years for the general treatment of pain and fever [7-9].
Phenylephrine has little to no beta-adrenergic activity and mainly functions as an agonist of the α-1 adrenergic receptor. Because it causes vasoconstriction in both veins and arteries and increases cardiac preload without significantly affecting cardiac myocytes, the drug is therefore the best option for increasing mean arterial pressure. Phenylephrine hydrochloride is also an over-the-counter (OTC) medication commonly used in ophthalmic, nasal and topical formulations,
In the therapeutic doses used for the relief of nasal congestion and sinus pressure, the drug has no substantial stimulant effect on the α-adrenergic receptors of the heart. It stimulates α 1-adrenergic receptors on capacitance blood vessels in the nasal mucosa, leading to vasoconstriction and a decrease in the volume of the nasal mucosa [10-13].
The fixed paracetamol/phenylephrine combinations, administered principally as capsules or oral solutions, are approved and have been used for decades throughout Europe to relieve the symptoms of the common cold and flu when associated with stuffy nose (nasal congestion) and sinuses (sinusitis), with a well-established and accepted risk/benefit ratio. The formulation of the product Paracetamol/phenylephrine hydrochloride 600 mg/10 mg has been developed in the form of granules to be dissolved in hot water and administered as an oral solution or swallowed as such, allowing rapid use without water.
This formulation allows, in fact, the patient to choose between a traditional intake with a glass of water or an administration directly into the mouth without the use of liquids. A Phase 1 study was therefore undertaken to evaluate its pharmacokinetic profile.
The main objective of the study was to assess the pharmacokinetic profile in healthy subjects of this combination of 600 mg paracetamol and 10 mg phenylephrine HCl formulated as granules and administered as a single oral dose in solution form under fasting conditions. The secondary objective was to investigate the safety and tolerability of the product based on safety clinical and laboratory examinations and registration of adverse events and/or adverse drug reactions.
Materials and methods
Subjects
A total of 36 healthy male and female volunteers, aged 18 to 55 years and with a body mass index (BMI) ranging from 18.5 to 30 kg/m², were deemed eligible for participation based on medical history, physical examination, and laboratory tests. Main exclusion criteria included history of drug abuse or use of illegal drugs; alcohol abuse; regular consumption of beverages or food containing methylxanthines; pregnancy; known hypersensitivity or intolerance to NSAIDs; presence or a history of clinically significant cardiovascular, renal, hepatic, pulmonary, metabolic, endocrine, hematological, gastrointestinal neurological, psychiatric or other diseases; any chronic disease which might interfere with resorption, distribution, metabolism or excretion of the drug; history of difficulty in swallowing; positive serologic findings for HIV antibodies, HBsAg, and/or HCV antibodies, vaccination within 14 days prior to screening visit. All volunteers provided written informed consent.
Ethics
The trial was performed in accordance with the Declaration of Helsinki and its last amended [14] and met the ethical requirements set in the Regulation (EU) No. 536/20141 [15]. The trial was also performed in accordance with ICH Topic E8. Note for Guidance on General Considerations for Clinical Trials [16] and ICH Topic E6. Guideline For Good Clinical Practice E6(R2) Step 5 [17]. Before the start of the study, the protocol and other appropriate documents (CRF, information for subject and informed consent) were submitted to the national Ethics Committee For Clinical Trials (ECCT) in accordance with local and European legal requirements.
Study design
This was a phase 1 single-dose, pharmacokinetic trial conducted in healthy volunteers. Each subject received an oral single dose of granules for oral solution (1 sachet =600 mg of paracetamol and 10 mg of phenylephrine hydrochloride) solved in very hot water under fasting conditions. Volunteers abstained from consuming any food or beverages other than water, starting at 9:00 PM the evening before administration and continuing until approximately 4 hours after administration, coinciding with lunchtime the following day. Water was provided ad libitum until 1 hour before and from 1 h after the drug administration on day 1. Concomitant medication was generally not allowed for the duration of the trial, except the allowed contraceptive methods as defined in exclusion criterion.
Criteria for evaluation
The primary endpoint of the trial was the assessment of main pharmacokinetic parameters considering AUC(0-t) and Cmax of paracetamol and phenylephrine, while the evaluation of tmax was the secondary endpoint. AUC(0-∞), AUCres, MRT, and t½ of analytes were also calculated as additional endpoints.
Determination of plasma concentrations
Blood samples for analysis of paracetamol and phenylephrine hydrochloride in plasma (total number of 18 blood samples, 12.4 ml each) were drawn at the following times: 0:00 (pre-dose), 0:10, 0:20, 0:30, 0:40, 0:50, 1:00, 1:10, 1:20, 1:30, 1:45, 2:00, 3:00, 4:00, 6:00, 8:00, 10:00, 12:00. All blood samples were collected into tubes using an anticoagulation agent. After the end of the trial the plasma samples were transported frozen to the bioanalytical center for the bioanalytical procedures.
Plasma concentrations were analyzed using liquid chromatography by tandem mass spectrometry (LC-MS/MS). For paracetamol, the calibration range was 150.06 - 20008.00 ng/ml with an inter-assay precision of 1.70 - 7.37% CV; for phenylephrine, the calibration range was 10.00 - 2000.00 pg/ml with an inter-assay precision of 2.17 - 3.09% CV. All plasma samples were stored for at least 3 months.
Pharmacokinetic analysis
Pharmacokinetic parameters were derived from plasma concentration–time data using a noncompartmental approach and summarized by descriptive statistics, namely arithmetic and geometric means, standard deviation (SD), coefficient of variation (CV), median and ranges (lower and upper). These parameters included AUC(0-t), AUC(0-∞), Cmax, tmax, AUCres, MRT and t½. The descriptive statistic was used for the evaluation of secondary and additional endpoints.
Safety
Safety clinical and laboratory examinations, assessed at the beginning and at the end of the trial, and registration of adverse events and/or adverse drug reactions were recorded during the trial. Descriptive statistics were used to summarize all safety data: clinical (physical) examination, standard laboratory examinations (clinical blood chemistry, hematology, urinalysis), monitoring of vital signs (pulse rate, blood pressure), ECG monitoring, and registration of adverse events and/or adverse drug reactions during the trial. The pharmacokinetic and statistical evaluation were carried out by means of the validated statistical software package SAS for Windows, version 9.4.
Results
Subject disposition and characteristics
A total of 36 subjects were randomized and 35 subjects completed the trial according to the protocol. One subject terminated the trial prematurely and was not replaced. All available samples (35 study completers and one drop-out) were analyzed. The baseline demographic characteristics of the study population are summarized in Table 1.The mean ± SD age was 36.5 ± 9.7 years, body weight was 69.0 ± 10.7 kg, and body mass index was 24.6 ± 2.7 kg/m2.
(n=35) |
Mean ± SD |
Min – Max |
Age (years) |
36.5 ± 9.7 |
20.0 - 52.0 |
Height (cm) |
167.0 ± 9.9 |
147.0 - 188.0 |
Weight (kg) |
69.0 ± 10.7 |
45.0 - 90.0 |
BMI (kg/m2) |
24.6 ± 2.7 |
19.2 - 29.8 |
male : female |
20:15 |
BMI: Body Mass Index |
Table 1. Baseline demographic data of subjects
Pharmacokinetic parameters
The plasma concentration time profiles and all pharmacokinetic parameters of paracetamol and phenylephrine are summarized in Figure 1 and in Table 2. The developed formulation showed a rapid gastrointestinal absorption reaching the maximum concentration of the two analytes within 30 min. For paracetamol the mean maximum plasma concentration (Cmax) observed was 11.06 ± 3.77 µg/ml with a mean AUC(0-∞) of 27.91 ± 6.09 µg.h/ml; while for phenylephrine the mean Cmax was 2.15 ± 0.99 ng/ml with a mean AUC(0-∞) of 1.67 ± 0.49 ng.h/ml.
|
E-Pharma formulation |
(granules administered as oral solution) |
PK Parameter |
N |
Arithmetic mean |
SD |
CV |
median |
Paracetamol |
|
AUC (0-t) (µg*h/ml) |
35 |
26.91 |
5.83 |
21.7 |
25.44 |
AUC (0-∞) (µg*h/ml) |
35 |
27.91 |
6.09 |
21.8 |
26.33 |
Cmax (µg/ml) |
35 |
11.06 |
3.77 |
34.1 |
9.97 |
tmax (h) |
35 |
0.43 |
0.23 |
53.4 |
0.33 |
AUCres (%) |
35 |
3.55 |
1.4 |
39.4 |
3.36 |
MRT (h) |
35 |
3.26 |
0.5 |
15.4 |
3.19 |
t ½ (h) |
35 |
2.43 |
0.83 |
34.3 |
2.25 |
Phenylephrine |
|
AUC (0-t) (ng*h/ml) |
35 |
1.62 |
0.49 |
30 |
1.65 |
AUC (0-∞) (ng*h/ml) |
35 |
1.67 |
0.49 |
29.6 |
1.72 |
Cmax (ng/ml) |
35 |
2.15 |
0.99 |
46.2 |
1.98 |
tmax (h) |
35 |
0.37 |
0.16 |
45 |
0.33 |
AUCres (%) |
35 |
3.02 |
1.71 |
56.8 |
2.7 |
MRT (h) |
35 |
1.59 |
0.57 |
35.7 |
1.68 |
t ½ (h) |
35 |
2.4 |
1.53 |
63.7 |
1.95 |
AUC(0-t): Area under the plasma concentration-time curve calculated by the trapezoidal rule from zero to last observed concentration at time t; AUC(0-∞): Area under the plasma concentration-time curve from zero extrapolate to infinity (AUC(0-∞) = AUC(0-t) + Clast/λz where Clast is the last concentration above the limit of quantification and λz is the terminal elimination constant); AUCres: Residual area in percent; Cmax: Observed maximum plasma concentration; CV: Coefficient of variation; MRT: Mean residence time; SD: Standard deviation; tmax: Observed time point of maximal concentration; t1/2: Plasma concentration half-life calculated according to t1/2= ln(2)/ λz. |
| |
|
|
|
|
|
Table 2. Pharmacokinetic parameters of paracetamol and phenylephrine after an oral single dose of 600 mg of paracetamol and 10 mg of phenylephrine HCl
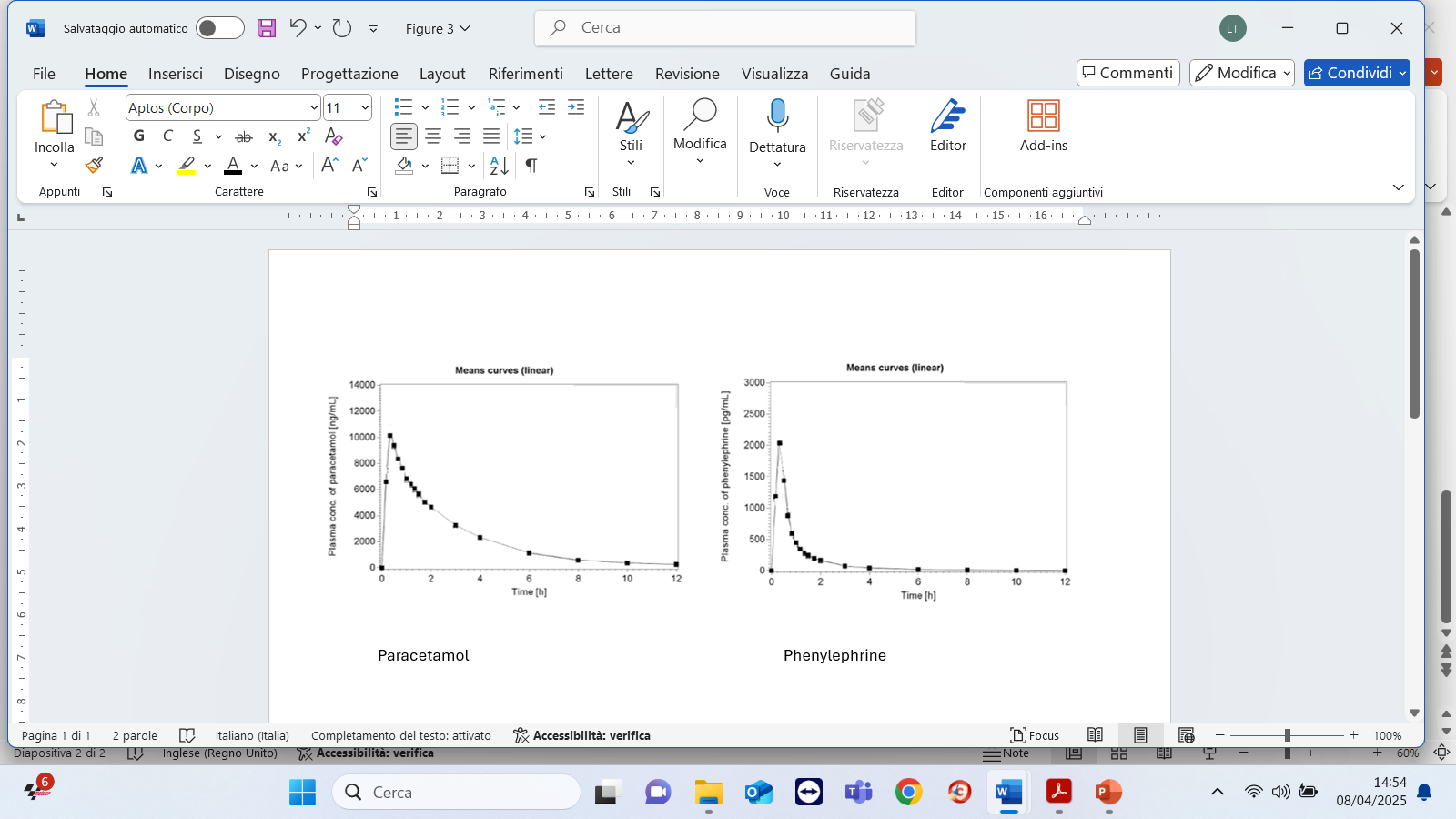
Figure 1. Mean paracetamol and phenylephrine plasma concentration-time profile
Safety
Thiry-five healthy volunteers were exposed to an oral single dose of paracetamol/phenylephrine hydrochloride 600/10 mg corresponding to 1 sachet of granules for oral solution. During the trial, a total of two non-serious adverse events (AEs) were registered in two subjects (Table 3): one increased in white blood cell count and one syncope. Both AEs were considered not or unlikely related to product administration. No serious adverse events (SAEs) were registered. All laboratory and clinical screening revealed no indications for adverse events or poor tolerability.
System Organ Class/PT |
E-Pharma formulation |
(granules administered as oral solution) |
Total number of subjects with TEAEs |
2 (5.6%) |
Investigations |
1 (2.8%) |
White blood cell count increased |
1 (2.8%) |
Nervous system disorders |
1 (2.8%) |
Syncope |
1 (2.8%) |
PT: preferred term TEAEs: treatment emergent adverse events; The coding is according MedDRA dictionary (Version: 25.0) |
Table 3. Number of treatment emergent adverse events by System Organ Class, Preferred Term and treatment
Discussion
The present study was designed to assess the pharmacokinetic profile a formulation of paracetamol/phenylephrine HCl formulated as granules and administered as an oral solution (1 sachet = 600 mg/10 mg) in male and female healthy volunteers. The two active ingredients are well known both in monotherapy and in combination therapy. They have been used for many years as OTC drugs and their individual pharmacokinetic profiles have been well characterized in humans. Paracetamol is rapidly and completely absorbed after oral administration, with peak plasma concentrations usually reached between 10 to 60 minutes [18,19], nevertheless, the rate absorption of paracetamol is known to depend on gastric emptying [20] and formulation [21,22].
It was reported that conventional 500 mg tablet of paracetamol administered at healthy volunteers produced a mean plasma Cmax of approximately 8-9 µg/ml after about 1 hour, with an AUC(0-∞) ranged between 24.6 and 28.5 μg.h/ml [21,22]. Paracetamol is distributed throughout most body tissues, with the exception of adipose tissue. Its plasma protein binding is low, and approximately 10% to 20% of the drug binds to red blood cells [6,18,19]. Its elimination half-life varies from about 1 to 3 hours [19].
After being mostly metabolized in the liver, the glucuronide and sulfate conjugates of paracetamol are mainly excreted in the urine. Less than 5% is excreted as unchanged paracetamol [6,18,19]. Phenylephrine is readily and completely absorbed after oral administration with peak plasma concentrations usually reached between 30 minutes and 2 h. It has relatively low bioavailability of 38% as a result of first-pass conjugation within the gut wall [18,23].
For an oral 10 mg phenylephrine HCl dose administered in adults alone or with other actives including paracetamol, the mean total exposure (AUC 0-∞) reported across a series of published studies [24-28] ranged from 0.82 to 2.49 ng.h/ml, while the mean maximum exposure (Cmax) ranged from 0.87 to 3.69 ng/mL. Both parameters were found to be reduced in pediatric patients [29].
It should be noted that, as with other drugs undergoing extensive first-pass metabolism, phenylephrine has shown wide inter- and intra-individual variability in bioavailability in all studies. Furthermore, according to some authors [25,27,28,30], the presence of paracetamol, especially at high doses, may further alter the metabolism of phenylephrine, with a significant impact on its pharmacokinetics. This explains the wide range of plasma concentrations of phenylephrine hydrochloride 10 mg found in the different pharmacokinetics studies. The mean elimination half-life of phenylephrine is between 2.1 to 3.4 hours. Both unchanged phenylephrine and its metabolites are excreted almost entirely in urine [23].
As previously reported, the proposed formulation in granules administered as an oral solution (600 mg paracetamol/10 mg phenylephrine hydrochloride) showed a rapid gastrointestinal absorption reaching the maximum concentration of the two analytes in a short time. The observed parameters in terms of rate and extent of paracetamol absorption (i.e., Cmax. 11.06 ± 3.77 µg, tmax: 0.43 h AUC(0-∞) 27.91 ± 6.09 µg h/ml) were, as expected, slightly different compared to conventional 500 mg tablet being, in our case, a 600 mg oral solution. The studied formulation made it possible to achieve a higher peak concentration in a shorter time.
Regarding phenylephrine the absorption kinetics (i.e., Cmax 2.15 ± 0.99 ng/ml, tmax 0.37h, AUC(0-∞) 1.67 ± 0.49 ng h/ml) was consistent with the above-mentioned published literature data. In addition, safety results confirmed that the product was well tolerated. Only two not serious treatment emergent adverse events in two patients (5.6%) were observed and no clinically important laboratory changes or trends was evidenced. The tolerability results obtained are also in line with literature data.
It should be emphasized, in fact, that in all the clinical studies considered [24-29] involving different oral formulations of phenylephrine administered alone or in combination with other active ingredients including paracetamol, despite the wide variability of the pharmacokinetic parameters obtained, no serious or severe adverse events were reported.
All the authors reported that the tested products were well tolerated and showed a good safety profile, comparable to placebo. In particular, at least in healthy volunteers, including pediatric patients, cardiovascular assessments of pulse and blood pressure and ECG did not show clinically relevant alterations in any of the studies. However, as combination therapy with paracetamol and phenylephrine has been shown to potentially raise blood pressure more than phenylephrine alone, it should be used with caution in individuals with cardiovascular conditions.
Conclusion
The developed formulation of paracetamol/phenylephrine hydrochloride 600 mg/10 mg granules administered as an oral solution can provide a pharmacokinetic profile substantially consistent to that of other similar formulations.
Authors’ contributions
Matteo Mezzena, Leila Tosi and Marco Anelli contributed equally to the preparation of the manuscript. All authors read and approved the final version of the manuscript.
Conflicts of interest
The authors certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
References
- Eccles R (2005) Understanding the symptoms of the common cold and influenza. Lancet Infect Dis 5: 718-725. [Crossref]
- Allan GM, Arroll B (2014) Prevention and treatment of the common cold: Making sense of the evidence. CMAJ 186: 190-199. [Crossref]
- De Sutter AI, Eriksson L, van Driel ML (2022) Oral antihistamine‐decongestant‐analgesic combinations for the common cold. Cochrane Database Syst Rev 1: CD004976. [Crossref]
- Eccles R, Martensson K, Chen SC (2010) Effects of intranasal xylometazoline, alone or in combination with ipratropium, in patients with common cold. Curr Med Res Opin 26: 889-899. [Crossref]
- Wei Q, Zhou J, Li H, Wang L, Wu Y, et al. (2023) Medication adherence with fixed-dose versus free-equivalent combination therapies: Systematic review and meta-analysis. Front Pharmacol 14: 1156081. [Crossref]
- Gerriets V, Anderson J, Patel P, Nappe TM (2025) Acetaminophen. 2024 Jan 11. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025 Jan.
- Ayoub SS (2021) Paracetamol (acetaminophen): A familiar drug with an unexplained mechanism of action. Temperature 8: 351-371. [Crossref]
- Eccles R (2006) Efficacy and safety of over‐the‐counter analgesics in the treatment of common cold and flu. J Clin Pharm Ther 31: 309-319. [Crossref]
- Jóźwiak-Bebenista M, Nowak JZ (2014) Paracetamol: Mechanism of action, applications and safety concern. Acta Pol Pharm 71: 11-23. [Crossref]
- PubChem (2025) Phenylephrine.
- Richards E, Lopez MJ, Maani CV. Phenylephrine. 2023 Oct 30. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025 Jan.
- Gelotte CK, Zimmerman BA (2015) Pharmacokinetics, safety, and cardiovascular tolerability of phenylephrine HCl 10, 20, and 30 mg after a single oral administration in healthy volunteers. Clin Drug Investig 35: 547-558. [Crossref]
- Johnson DA, Hricik JG (1993) The pharmacology of α‐adrenergic decongestants. Pharmacotherapy 1993 13: 110S-115S. [Crossref]
- World Medical Association (2013) World medical association declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 310: 2191-2194.
- Regulation (Eu) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC.
- ICH Topic E 8. General considerations for clinical trials. Step 5. Note for guidance on general considerations for clinical trials (CPMP/ICH/291/95). March 1998.
- Guideline for good clinical practice E6 (R2), Step 5 (EMA/CHMP/ICH/135/1995). Adopted by CHMP on 15 December 2016. Effective since: 14 June 2017.
- PubChem (2025) Acetaminophen.
- Martindale online (2025) Paracetamol.
- Prescott LF (1980) Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmacol 10: 291S-298S. [Crossref]
- Ibanez Y, Rodríguez JM, Luján M, Grattan TJ, Martin AJ, et al. (2006) A pharmacokinetic study investigating the rate of absorption of a 500 mg dose of a rapidly absorbed paracetamol tablet and a standard paracetamol tablet. Curr Med Res Opin 22: 1893-1897. [Crossref]
- Portolés A, Puerro M, Terleira A, Rodrı́guez A, Caturla MC, et al. (2003) A new high-absorption-rate paracetamol 500-mg formulation: A comparative bioavailability study in healthy volunteers. Curr Ther Res 64: 401-411. [Crossref]
- Hengstmann JH, Goronzy J (1982) Pharmacokinetics of3H-phenylephrine in man. Eur J Clin Pharmacol 21: 335-341. [Crossref]
- Gelotte CK, Zimmerman BA (2015) Pharmacokinetics, safety, and cardiovascular tolerability of phenylephrine HCl 10, 20, and 30 mg after a single oral administration in healthy volunteers. Clin Drug Investig 35: 547-558. [Crossref]
- Gelotte CK (2018) An open-label, randomized, four-treatment crossover study evaluating the effects of salt form, acetaminophen, and food on the pharmacokinetics of phenylephrine. Regul Toxicol Pharmacol 95: 333-338. [Crossref]
- Janin A, Monnet J (2014) Bioavailability of paracetamol, phenylephrine hydrochloride and guaifenesin in a fixed-combination syrup versus an oral reference product. J Intl Med Res 42: 347-359. [Crossref]
- Atkinson HC, Stanescu I, Anderson BJ (2014) Increased phenylephrine plasma levels with administration of acetaminophen. N Engl J Med 370: 1171-1172. [Crossref]
- Atkinson HC, Stanescu I, Salem II, Potts AL, Anderson BJ (2015) Increased bioavailability of phenylephrine by co-administration of acetaminophen: results of four open-label, crossover pharmacokinetic trials in healthy volunteers. Eur J Clin Pharmacol 71: 151-158. [Crossref]
- Gelotte CK, Parasrampuria DA, Zimmerman BA (2023) Single-dose pharmacokinetics and metabolism of the oral decongestant phenylephrine HCl in children and adolescents. Pulm Ther 9: 139-150. [Crossref]
- Atkinson HC, Potts AL, Anderson BJ (2015) Potential cardiovascular adverse events when phenylephrine is combined with paracetamol: Simulation and narrative review. Eur J Clin Pharmacol 71: 931-938. [Crossref]