Increasing body of evidence ascribes tumorigenesis to the emergence of cancer stem cells (CSCs). The two main theories predicting the origin of CSCs are either transformation of adult stem cells, or de-differentiation of mature cells that is accompanied by the induction of the epithelial to mesenchymal transition (EMT) program [1]. Yet, both are associated with the accumulation of genetic and epigenetic aberrations that underlies cell plasticity [2]. Since accumulating data link mutations in the tumor-suppressor p53 with both CSCs formation and cancer-associated EMT, it is tempting to hypothesize that mutant p53 might facilitate CSCs formation by inducing EMT and cell plasticity. Here, we will evaluate the latter hypothesis by analyzing recent publications, and supporting it by our new data pertaining prostate cancer.
EMT- induced cell plasticity is associated with CSCs features
A recent accepted concept explaining cancer development and relapse is the hierarchical model, which predicts that a small subset of cells within tumors has the ability to both self-renew and to differentiate in various cell types, thus maintaining the ongoing heterogeneous neoplasm [3]. EMT that was first described as critical process in embryogenesis, was shown to be activated in various tumor cells by the EMT- transcription factors, mainly of the SLUG/SNAIL, TWIST and ZEB families. This activation was suggested to yield cells migration and metastasis. Recent studies, however, ascribe EMT, also to enhanced stemness capacity; accordingly, EMT was suggested to be a critical process in mediating CSC phenotype [1,4]. This is mainly due to the cellular changes mediated by the EMT process that is known to induce cancer cells plasticity [5]. Indeed, EMT was found significantly associated with CSCs. For example, in a mouse model for skin cancer, tumor cells that underwent EMT (Epcam− cells) contained higher frequency of tumor propagating cells than that observed in the epithelial cells (Epcam+ cells), implying on higher stemness capacity [6]. Moreover, it was shown that induction of EMT in breast tumor-derived cell lines is associated with a higher capacity to generate mamospheres with frequent CSCs markers, thus supporting the notion that the two are interconnected [7-10]. Several experimental settings have shown that expression of the EMT transcription factors is associated with CSCs. For example, Snail was suggested to be responsible for the transition from asymmetrical to symmetrical division of colorectal CSCs by maintaining high Wnt activity, and thus propagating the CSCs pool [11]. Ectopic expression of TWIST1 in esophageal squamous cell carcinoma caused significant upregulation of OCT4, a main self-renewal mediating factor [12]. Other cancer models for Leukemia [13] and ovarian cancer [14] associated Snail and Twist expression to the CSCs marker CD44. Finally, in pancreatic cancer mouse model, where K-Ras and p53 are mutated (KPC model), Zeb1 was found crucial for EMT, tumorigenic capacity as well as stemness phenotype [15]. Altogether, these evidence strongly link CSCs to EMT, and further suggest that EMT induction may lead to cancer stemness.
Mutant p53 mediates EMT
In a step-wise prostate transformation model we have previously shown that mutant p53 facilitates the expression of EMT related genes, including Twist1 induction and E-cadherin repression. This was accompanied with higher invasion capabilities and morphologically disrupted spheroids [16]. While Wang Z et al. showed that silencing wild-type p53 lead to EMT, migration and metastasis of hepatocellular carcinoma (HCC) cells [17], we further suggest a gain of function activities of mutant p53, that enhances this phenotype to higher extent than p53 inactivation alone [16]. The repression of E-cadherin expression by mutant p53 was also demonstrated in human colon carcinoma cell line model, HCT116. However, in this model, p53 temperature sensitive mutant p53-A143 did not show enhanced invasion [18]. The differences in the observed phenotype can stem from the p53 mutation type or cell types tested. In esophageal cells model, it was suggested that mutant p53R175H cooperates with epidermal growth factor receptor (EGFR) to enhance EMT phenotype upon treatment with EMT inducer, TGFβ. This was demonstrated by lower levels of E-cadherin and higher levels of N-cadherin, Zeb1/2 and Snail, and was associated with inactivation of EGFR-induced senescence, suggesting additional role for mutant p53 and EMT in senescence checkpoint during tumorigenesis [19]. More recently, mutant p53 expressing WAP-T transgenic mice, showed higher expression of an EMT gene signature which was associated with higher tumorigenic grading, associated with enhanced vascularization and metastatic potential [20]. Several studies aimed at understanding the molecular mechanism underlying mutant p53-dependent induction of EMT, suggest that mutant p53 gain of function activities promote EMT by modulating the expression of the key EMT transcription factors. For example, our previous study attributed the induction in Twist1 levels to deregulation of epigenetic mechanisms by mutant p53. Inhibition of BMI-1 protein by mutant p53 led to the reduction in H3K27me3 on Twist1 promoter, and activation of Twist1 expression [16]. In addition, in colorectal cancer cell lines, the induced expression of the EMT transcription factors- Snail and Slug was attributed to the mutant p53-dependent transcriptional upregulation of EFNB2 in response to DNA damage [21]. Other study suggested that in endometrial cancer, mutant p53 represses the expression of miR-130b, a negative regulator of ZEB1, and by that mediates ZEB1 elevation, leading to EMT phenotype [22].
Mutant p53 expression is correlated with CSCs features
It was recently claimed that tissues containing an increased frequency of highly dividing stem cells permit the increased incidence of mutations, and thus are prone to develop tumors [23,24]. As p53 serves as an important guardian of the genome, it is expected that it will play a major role in ensuring the quality and stability of stem cells genome [25]. Thus, it is not surprising that mutations in p53 may in turn lead to the formation of aberrant stem cells, referred as CSCs. Indeed, mutant p53 knock-in mice models have shown that bone marrow-derived hematopoietic and mesenchymal stem cells (MSCs) populations exhibit an augmented frequency of CSCs that are capable of inducing tumors in vivo [26,27]. This notion is further substantiated by our previous observation that mutant p53 facilitates the in vitro reprograming of mouse embryonic fibroblast (MEFs) towards the generation of induced pluripotent stem cells (iPSC). This facilitated process lead to the generation of malignant iPSC that in that respect can be regarded as CSCs [28]. These data emphasize the role of mutant p53 in mediating harmful de-differentiation, and by that accelerating cancer stem cells malignancy. Accordingly, recent evidence suggest that in various cancer types the expression of mutant p53 is correlated with CSCs features. For example, p53 protein derived of a deletion mutation that disrupts p53 DNA binding domain, was found accumulated in multipotent stem cells of brain sub-ventricular zone and in transit-amplifying progenitors, which are suggested to be the cellular origin of glioma that accounts for the development of glioblastoma [29]. In other cancer models mutant p53 was associated with enhanced CSCs features. For example, mutant p53R248W/P72R in human osteosarcoma cells promoted CSCs properties such as formation of larger sarco-spheres and enhanced expression of stemness genes (e.g. CD133, ABCG2, Nanog) [30]. Accordingly, in glial and breast-derived cancer cells, mutant p53 expression was found correlated with enhanced spheres formation and induction of the stemness markers CD133 and CD44 [31]. Additionally, we and others showed that colorectal cancer cells expressing mutant p53 are associated with larger sub-population of cells expressing CSCs markers and drug resistance [32, 33]. Interestingly, recently, we observed that mutant p53 expressing MSCs-derived highly aggressive tumor cells, express an embryonic gene signature that may testify that such CSCs underwent re-programing [27]. Altogether, these data further support the role for mutant p53 gain of function in facilitating the oncogenic characteristics of CSCs.
Mutant p53 plays a role in facilitating CSCs features of prostate tumor cells in association with inducing the EMT process
When focusing on better understanding mutant p53 gain of function activities in promoting cancer development, we recently suggested that mutant p53 is associated with higher expression of CSCs markers in colorectal cancer [33]. Moreover, in a prostate transformation model, epithelial prostate cells were immortalized (EP156T) and introduced with either mutant p53R175H or inactivation of p53 by the dominant negative p53 peptide GSE56, to study p53 role in prostate carcinogenesis. Results obtained indicated that mutant p53 induced EMT in a gain of function manner [16]. As induction of the EMT program was suggested to be associated with CSCs, we hypothesized that mutant p53 augmented the oncogenic CSCs features through EMT. Analysis of the expression of CD44high/CD24low CSCs markers [34] in EP156T prostate indicated that in mutant p53R175H expressing cells the CD44high/CD24 low sub-population is larger compared with wild-type p53 and GSE56 expressing cells. This observation was further corroborated in additional prostate cancer cell line system, DU145, which endogenously expresses mutant p53P223L,V274F [16], confirming the conclusion that prostate cancer cell-lines, expressing mutant p53, harbor larger CD44high/CD24 low sub-population (Figure 1). These observations suggest that mutant p53 gain of function is associated with an increased tumorigenic CSCs sub-population. In accordance with our observation that EP156T and DU145 cells expressing mutant p53 are associated with higher EMT features [16], we found that while the CSCs sub-population was enriched for EMT transcription factors Twist and SLUG, the E-Cadherin expression was decreased (Figure 2), thus inferring that the cellular sub-population that gained CSCs features was most likely dictated by EMT.
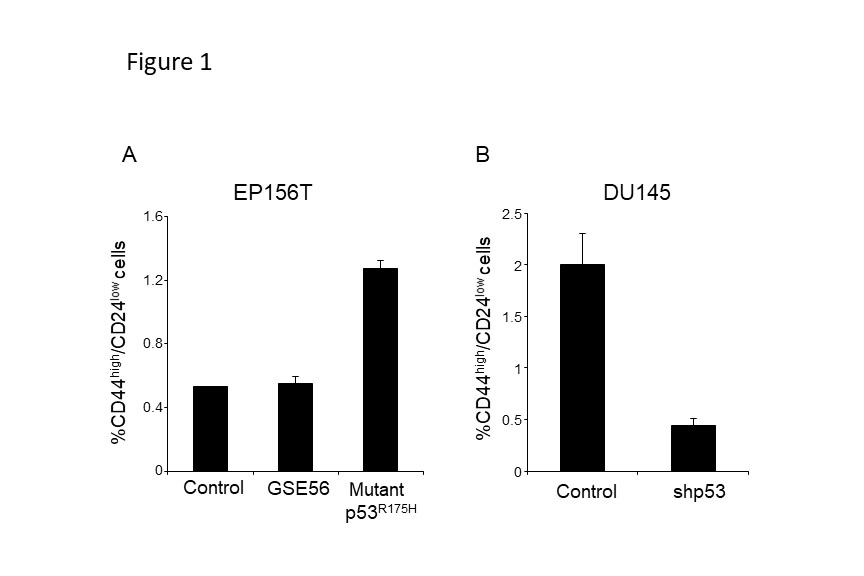
Figure 1: Mutant p53 expressing cells contain larger CD44high/CD24low sub-population. The EP156T (A) and the DU145 (B) cells were established and maintained as described previously [16]. Briefly, epithelial prostate cells were immortalized (EP156T) and introduced with either mutant p53R175H or inactivation of p53 by the dominant negative p53 peptide GSE56, or control vector. The DU145 cell line, endogenously expressing mutant p53P223L,V274F, were stably introduced with shRNA against p53 to knock-down mutant p53P223L,V274F expression. The established cells were immunostained with anti-CD44-APC conjugated (eBioscience) and anti-CD24-PE conjugated (BD Biosience) antibodies followed by evaluation of the size of CD44high/CD24low sub-population by FACS analysis. FACS procedure was performed as described previously [29]. Graph presenting an average of three experiments.
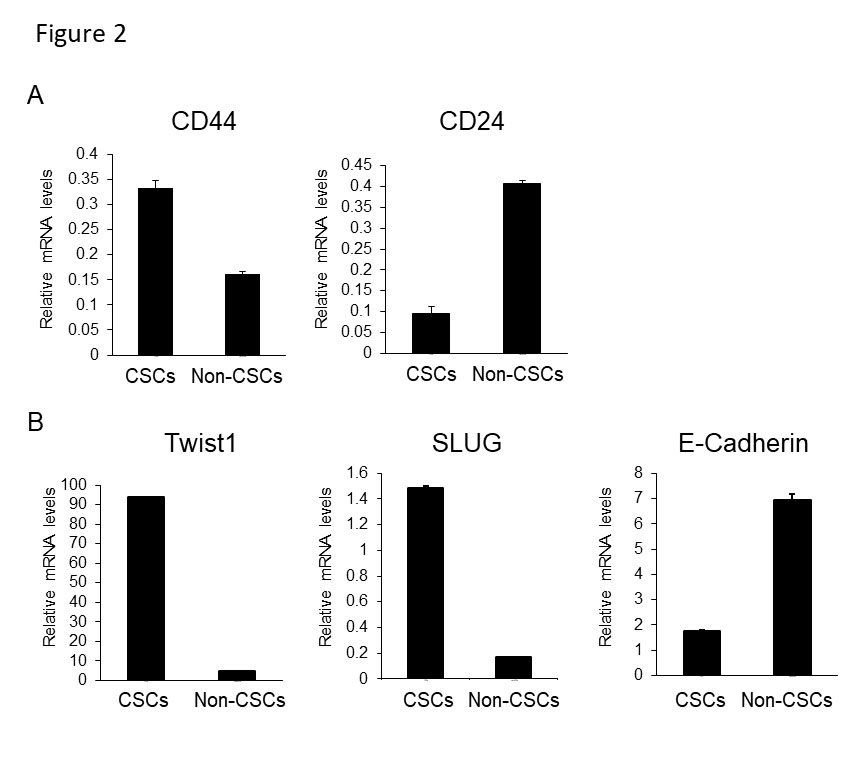
Figure 2: CD44high/CD24low sub-population is associated with EMT features in mutant p53P223L,V274F expressing DU145 cells . The DU145 cells were immunostained with anti-CD44 and anti-CD24 antibodies (as performed in Figure 1), and the CD44high/CD24low sub-population was sorted out of the whole DU145 cell population by FACS. (A) To ensure sorting efficiency, CD44 and CD24 mRNA expression levels were measured in the CD44high/CD24low (CSCs) sub-population, in comparison to the CD44low/CD24high (non-CSCs) sub-population, by quantitative real time PCR (qRT-PCR). (B) mRNA levels of EMT transcription factors, Twist and SLUG, and the expression of the epithelial marker, E-Cadherin, were measured by qRT-PCR in the CSCs sub-population, and in the non-CSCs sub-population as a control. FACS sorting procedure and qRT-PCR was conducted as previously described [29]. Graph presenting an average of three experiments.
As these observations are mutant p53 dependent, it is tempting to speculate that mutant p53 mediates CSCs features of cancer cells, via induction of EMT program. This hypothesis is supported by a recently published paper suggesting that mutant p53 facilitates CSCs features of glioblastoma and breast cancer cells by activating YAP/TAZ signaling [31] that was already proposed as regulator of EMT process [35]. Once established, this important hinge between EMT and CSCs formation in a mutant p53 dependent manner, may be regarded as a milestone in the understanding of the molecular events underlying the augmented oncogenic aggressiveness of CSCs.
Acknowledgements
Research in the laboratory of Varda Rotter is supported by a Center of Excellence grant from Flight Attendant Medical Research Institute (FAMRI) and Israel Science Foundation ISF-MOKED. V.R. is the incumbent of the Norman and Helen Asher Professorial Chair for Cancer Research at the Weizmann Institute.
References
- Shibue T, Weinberg RA (2017) EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14: 611-629. [Crossref]
- Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ (2005) Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer 5: 899-904. [Crossref]
- Kreso A, Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14: 275-291. [Crossref]
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA (2018) EMT in cancer. Nat Rev Cancer 18: 128-134. [Crossref]
- Goding CR, Pei D, Lu X (2014) Cancer: pathological nuclear reprogramming? Nat Rev Cancer 14: 568-573. [Crossref]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, et al. (2018) Identification of the tumour transition states occurring during EMT. Nature 556: 463-468. [Crossref]
- Lin Y, Wang Y, Shi Q, Yu Q, Liu C, et al. (2017) Stabilization of the transcription factors slug and twist by the deubiquitinase dub3 is a key requirement for tumor metastasis. Oncotarget 8: 75127-75140. [Crossref]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133: 704-715. [Crossref]
- Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, et al. (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3: e2888. [Crossref]
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, et al. (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145: 926-940. [Crossref]
- Hwang WL, Jiang JK, Yang SH, Huang TS, Lan HY, et al. (2014) MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat Cell Biol 16: 268-280. [Crossref]
- Izadpanah MH, Abbaszadegan MR, Fahim Y, Forghanifard MM (2017) Ectopic expression of TWIST1 upregulates the stemness marker OCT4 in the esophageal squamous cell carcinoma cell line KYSE30. Cell Mol Biol Lett22: 33. [Crossref]
- Kidan N, Khamaisie H, Ruimi N, Roitman S, Eshel E, et al. (2017) Ectopic Expression of Snail and Twist in Ph+ Leukemia Cells Upregulates CD44 Expression and Alters Their Differentiation Potential. J Cancer 8: 3952-3968. [Crossref]
- Lupia M, Angiolini F, Bertalot G, Freddi S, Sachsenmeier KF, et al. (2018) CD73 Regulates Stemness and Epithelial-Mesenchymal Transition in Ovarian Cancer-Initiating Cells. Stem Cell Reports 10: 1412-1425. [Crossref]
- Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, et al. (2017) The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 19: 518-529. [Crossref]
- Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, et al. (2011) Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ 18: 271-281. [Crossref]
- Wang Z, Jiang Y, Guan D, Li J, Yin H, et al. (2013) Critical roles of p53 in epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma cells. PLoS One 8: e72846. [Crossref]
- Roger L, Jullien L, Gire V, Roux P (2010) Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J Cell Sci 123: 1295-1305. [Crossref]
- Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, et al. (2010) Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 70: 4174-4184. [Crossref]
- Lenfert E, Maenz C, Heinlein C, Jannasch K, Schumacher U, et al. (2015) Mutant p53 promotes epithelial-mesenchymal plasticity and enhances metastasis in mammary carcinomas of WAP-T mice. Int J Cancer 136: E521-E533. [Crossref]
- Alam SK, Yadav VK, Bajaj S, Datta A, Dutta SK, et al. (2016) DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ 23: 707-722. [Crossref]
- Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, et al. (2013) Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 32: 3286-3295. [Crossref]
- Tomasetti C, Li L, Vogelstein B (2017) Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355: 1330-1334.
- Tomasetti C, Vogelstein B (2015) Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347: 78-81. [Crossref]
- Molchadsky A, Rotter V (2017) p53 and its mutants on the slippery road from stemness to carcinogenesis. Carcinogenesis 38: 347-358. [Crossref]
- Hanel W, Marchenko N, Xu S, Yu SX, Weng W, et al. (2013) Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ 20: 898-909. [Crossref]
- Koifman G (2018) Identification of a mutant p53-dependent embryonic gene signature associated with augmented tumorigenesis of stem cells. [Under review]
- Sarig R, Rivlin N, Brosh R, Bornstein C, Kamer I, et al. (2010) Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med 207: 2127-2140. [Crossref]
- Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, et al. (2009) Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 15: 514-526. [Crossref]
- Di Fiore R, Marcatti M, Drago-Ferrante R, D'Anneo A, Giuliano M, et al. (2014) Mutant p53 gain of function can be at the root of dedifferentiation of human osteosarcoma MG63 cells into 3AB-OS cancer stem cells. Bone 60: 198-212. [Crossref]
- Escoll M, Gargini R, Cuadrado A, Anton IM, Wandosell F (2017) Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 36: 3515-3527. [Crossref]
- Hosain SB, Khiste SK, Uddin MB, Vorubindi V, Ingram C, et al. (2016) Inhibition of glucosylceramide synthase eliminates the oncogenic function of p53 R273H mutant in the epithelial-mesenchymal transition and induced pluripotency of colon cancer cells. Oncotarget 7: 60575-60592. [Crossref]
- Solomon H, Dinowitz N, Pateras IS, Cooks T, Shetzer Y, et al. (2018) Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 37: 1669-1684. [Crossref]
- Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL (2008) CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer 98: 756-765. [Crossref]
- Park JH, Shin JE, Park HW (2018) The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol Cells 41: 83-92. [Crossref]