An 82-year-old male presented to the Oral and Maxillofacial Surgery Department for evaluation and treatment of an expansile lesion of the left mandible. The patient reported occasionally traumatizing the area during mastication causing irritation but otherwise it was asymptomatic. On clinical examination, no ulceration, trauma, destruction of adjacent anatomical structures, or loose teeth were evident. The patient denied any paresthesia or dysesthesia. Radiographic imaging demonstrated a well-circumscribed, expansile, mixed radiopaque/radiolucent mass with well-defined borders. Based on the clinical and radiographic appearance, a working diagnosis of benign fibro-osseous lesion, most consistent with ossifying fibroma, was favored. The lesion was excised and histopathologic evaluation revealed a malignant osteoid producing lesion with areas of neoplastic chondroblastic activity. The cytomorphologic features were characteristic of low-grade central osteosarcoma (LGCO).
osteosarcoma, ossifying fibroma, benign fibro-osseous lesion
Osteosarcomas are osteoid-producing malignant lesions, most commonly found in the metaphysis of long bones in children, with an incidence of 3 cases per million [1]. Approximately 10% of conventional osteosarcomas occur in the head and neck region, with 7% reported in the gnathic bone [2]. Low-grade central osteosarcoma (LGCO) of the jaws represents 1-2% of cases and usually present in patients during the fourth decade [3,4]. Common signs and symptoms of osteosarcoma include cortical expansion, pain, swelling, and paresthesia. The lung is the most common site for metastasis. Histological presentation of LCGO is usually as a well-differentiated tumor, and, due to its rarity, is often misdiagnosed as fibrous dysplasia or other benign fibro-osseous lesions [4].
An 82-year-old male presented upon referral to the Oral and Maxillofacial Surgery Department for evaluation and treatment of a left mandibular expansile lesion. Past medical history was significant for well-controlled type II diabetes and hypertension. The patient reported he had been aware of the lesion for several years but denied experiencing pain or paresthesia. The patient, however, noted the area became symptomatic whenever it was traumatized during mastication.
Clinical examination revealed a non-tender, expansile, exophytic intraoral lesion associated with the left edentulous area of the posterior mandible in the first and second molar region (Figure 1). The surface of the lesion was smooth and exhibited no erythema or ulceration. No facial asymmetry or lymphadenopathy was evident. On palpation, the lesion was firm, non-tender, non-compressible, and fixed to the underlying tissues. Prior to surgery, a panoramic radiograph (Figure 2) and a computed tomography (CT) scan (Figure 3) were obtained. The panoramic radiograph demonstrated a well-defined, well-circumscribed, ovoid, and expansile radiolucent/radiopaque lesion with corticated borders in the posterior left body of the mandible. No destruction of adjacent anatomical structures, root resorption of adjacent teeth or involvement of the inferior alveolar canal were evident. On CT, the lesion measured 2.3 x 2.1 x 2.0 cm and appeared to have an intact cortex with a sclerotic periphery and evidence of internal ossification. The lesion did not appear to be associated with the proximate premolars. The differential diagnosis included benign fibro-osseous lesions (i.e. ossifying fibroma, cemento-osseous dysplasia) and osteosarcoma with a high preference for ossifying fibroma.
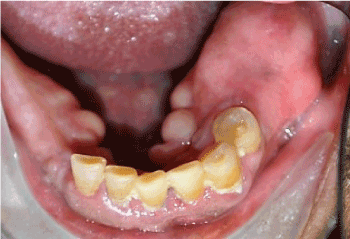
Figure 1. Expansile, exophytic lesion associated with edentulous area of the left posterior mandible, in the region of the first and second molars
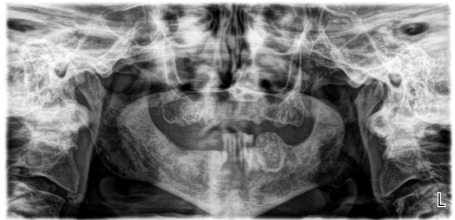
Figure 2. Panoramic radiograph demonstrating a well-defined, well-circumscribed, expansile, exophytic lesion within the body of the left posterior mandible without destruction of adjacent anatomical structures
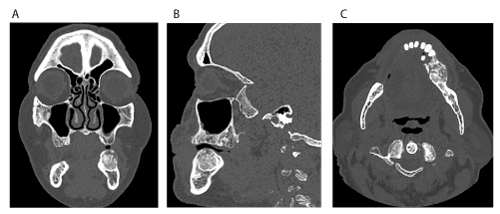
Figure 3. Computed tomography (CT) imaging. (A) Coronal view. (B) Sagittal view. (C) Axial view. Note the sclerotic periphery, evidence of internal ossification and benign appearance
An excisional biopsy was performed in the operating room under general anesthesia. A full thickness mucoperiosteal flap was raised to expose the lesion. The lesion was adherent to the soft tissue in several areas and a soft tissue-like exophytic growth was noted in its anterior superior aspect (Figure 4). The lesion was excised using a reciprocating saw with 5 mm margins observed past the visually notable change from white sclerotic bone into normal bony architecture. The remaining mandibular teeth were removed as part of the patient’s overall prosthetic plan.
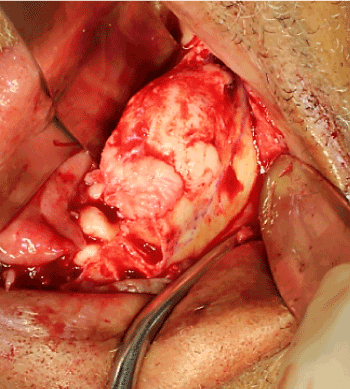
Figure 4. Intraoperative appearance of the lesion once exposed through a full thickness mucoperiosteal flap. A soft tissue-like growth is evident in the anterior superior aspect of the lesion
The gross specimen consisted of a 4.1 x 2.2 x 2.0 cm mass of dense bone (Figure 5). Serial sectioning revealed a homogenous tan-white cut surface with no hemorrhage or necrosis grossly identified. No soft tissue extension was appreciated.
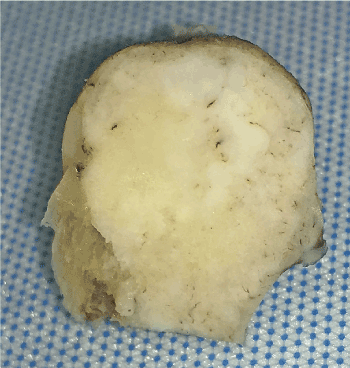
Figure 5. Gross specimen exhibiting a tan-white variegated smooth cut surface and lack of hemorrhage and necrosis
Histopathologic evaluation of the heamotoxylin and eosin (H&E) stained biopsy revealed an intramedullary fibro-osseous lesion (Figure 6A) exhibiting hypercellularity, mitotic activity, and mild to moderate cytologic atypia in the stromal cells. Trabeculae of abnormal woven bone was surrounded by atypical cells with osteoid formation. Lobules of neoplastic cartilage with large, pleomorphic, and hyperchromatic nuclei were evident (Figures 6B & 6C) along with mineralization of the malignant cartilage. Spindle-shaped atypical osteoblasts were seen producing neoplastic osteoid which tended to merge with the neoplastic chondroid matrix (Figure 6C). Histologic sections additionally revealed areas where the lesion invaded native bone, while in other areas it focally permeated through the cortex with focal periosteal growth. The histopathologic findings supported a diagnosis of low-grade central osteosarcoma of the mandible. The amount of the chondroblastic component present in the lesion was not sufficient to further sub-classify it as a chondroblastic osteosarcoma. Areas of transition from benign intramedullary fibro-osseous tissue to osteoid-producing spindle-shaped atypical cells representing osteosarcoma were appreciated (Figures 7A, 7B, & 7C). The diagnosis was discussed with the patient, however, he declined further clinical investigation to exclude metastatic disease.
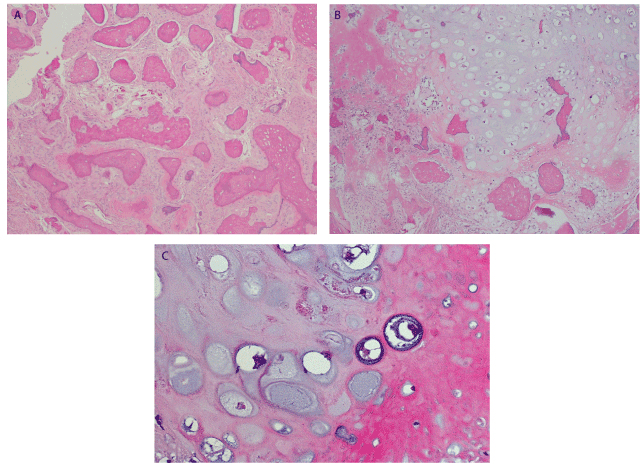
Figure 6. (A) H&E stain x100 showing areas reminiscent of an intramedullary benign fibro-osseous lesion. (B) Tumor producing areas of chondroblastic matrix growing in lobules with significant hypercellularity and cytologic atypia. Neoplastic bone is seen merging with neoplastic hyaline cartilage. H&E stain x100. (C) Spindle-shaped atypical osteoblasts are seen producing neoplastic osteoid which appears as the eosinophilic amorphous glassy material. The chondroblastic matrix exhibits chondrocytes with hyperchromatic and pleomorphic nuclei. The coarse, lace-like neoplastic osteoid is seen merging into neoplastic chondroblastic matrix. H&E stain x400
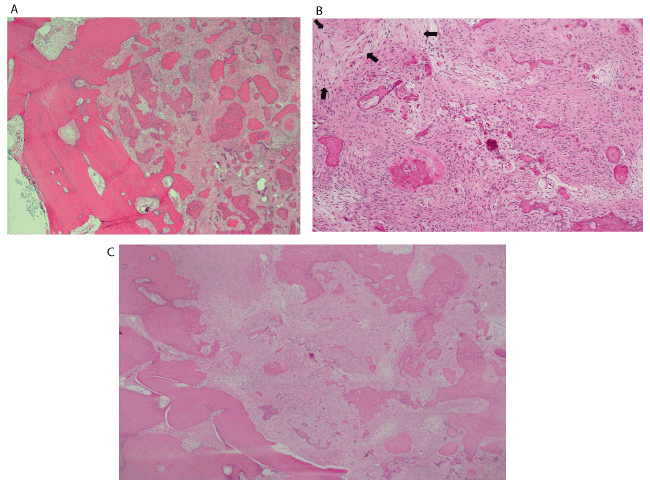
Figure 7. A, B, and C. H&E stains at 4x, 10x, and x40 showing a transition zone from reminiscent intramedullary benign fibro-osseous lesion to osteoid-producing spindle-shaped atypical cells, representing osteosarcoma
Osteosarcomas are the most common primary malignant tumor of bone when hematopoietic malignancies are not considered.
Osteosarcomas of the jaws represent about 7% of all skeletal osteosarcomas and occur at an average age of 33 to 39 years; however, the range of presentation is from 16 to 69 years of age [1]. These tumors present more often in males with an equivalent predilection in the mandible and maxilla [1]. Clinical findings include but are not limited to expansion of the bone, cortical bone destruction, tooth mobility, and anesthesia/paresthesia.
Radiographically, osteosarcomas of the jaws are classically described as having a “sun-ray”, “sunburst” or “moth-eaten” appearance but can also present as mottled radiopaque or mixed radiopaque/radiolucent lesions with ill-defined borders. Invasion and destruction of adjacent anatomical structures is a common finding. Extracortical bone, an induced periosteal reaction to the tumor, is frequently found at right angles to the cortex where the periosteum appears elevated (known as Codman triangle); however, cortical bone destruction is characteristic. Adjacent teeth may exhibit spiking root resorption and widening of the periodontal ligament space when involved by the tumor.
Histologically, for a diagnosis of osteosarcoma, malignant mesenchymal cells must produce osteoid. These cells may also produce chondroid and fibrous connective tissue in varying extents. The spindle-shaped malignant cells exhibit pleomorphism, a high nucleus to cytoplasmic ratio, and hyperchromatic nuclei.
LGCO of the jaw represents less than 2% of reported cases and is less aggressive than conventional osteosarcoma [6]. The treatment of choice for LGCO is surgical excision with wide margins of 2 cm, either with or without adjuvant therapy [5]. The 5-year survival rate after excision is 60-70% [1]. The most important prognostic factor is achieving complete removal of the lesion. The most common site for metastasis is the lung. Transformation into a high grade osteosarcoma after recurrence is approximately 15% [6].
Osteosarcomas of the jaw can be easily misdiagnosed as benign fibro-osseous lesions such as fibrous dysplasia, cemento-osseous dysplasia, and ossifying fibromas, especially when osteoid is not easily identified in histologic sections. Roughly 4% of reported cases of LGCO have benign clinical and radiographic characteristics [4]. However, LGCO can be clinically differentiated from benign lesions by its ability to cause neurosensorial alterations, a characteristic not seen in benign fibro-osseous lesions. Immunohistochemical expression of murine double-minute type 2 (MDM2) and cyclin-dependent kinase 4 (CDK4) can aid in the diagnosis of challenging cases of low-grade osteosarcomas since fibro-osseous lesions do not express these markers [7].
Fibrous dysplasia is a developmental, sporadic condition in which normal bone is replaced by cellular fibrous connective tissue with irregular bony trabeculae due to a postzygotic, activating mutation in the guanine nucleotide-binding proteins/α-subunit (GNAS) gene [1]. It may present as monostotic or polyostotic, with or without café-au-lait spots, and/or enDOMRine disturbances. Monostotic fibrous dysplasia is often diagnosed during the second and third decades of life with predilection for the maxilla [1]. Males and females are equally affected. In contrast, polyostotic fibrous dysplasia is commonly diagnosed by 10 years of age and females are affected more often than males. Clinical presentation is that of a painless swelling that has been present for months to years. Radiographically, fibrous dysplasia is described as a ground-glass opacification with poorly defined borders blending into adjacent normal bone. This radiographic presentation is not seen in ossifying fibroma, cemento-osseous dysplasia, or osteosarcoma. Adjacent teeth may exhibit narrowing of the periodontal ligament space and an ill-defined lamina dura [1].
Histologically, fibrous dysplasia consists of irregular trabeculae of woven (immature) bone in a background of a cellular fibrous stroma [1]. The abnormal bony trabeculae is thin, curvilinear, and disconnected. Osteoblastic rimming is not generally seen, which is a common characteristic of ossifying fibroma. Peritrabecular clefting (retraction of the stroma from the bony trabeculae) is evident, a feature not seen in ossifying fibromas or osteosarcoma. Calcified spherules commonly present in ossifying fibroma are rarely seen in fibrous dysplasia. Additionally, fibrous dysplasia lacks the cellular atypia and infiltrative pattern characteristic of LGCO. If required for breathing or other concerns, conservative treatment with osseous recontouring can be performed, though not required since fibrous dysplasia tends to stabilize upon growth completion. Although there have been some reports of malignant transformation of fibrous dysplasia (particularly longstanding polyostotic fibrous dysplasia) into osteosarcoma, the reported incidence is 0.4% to 6.7% [4].
Cemento-osseous dysplasia is a reactive benign fibro-osseous lesion which presents in the tooth-bearing areas of the jaws. Focal and florid cemento-osseous dysplasia are most commonly seen in the posterior mandible while periapical cemento-osseous dysplasia is most common in the anterior mandible. All three types have a predilection for middle-aged African American females. Most cases are asymptomatic and associated teeth are vital. Bony expansion and/or pain are not generally reported. Radiographically, cemento-osseous dysplasia begins as a well-defined radiolucent lesion which then develops into a radiopaque/radiolucent lesion and, as it continues to mature, becomes a radiopaque lesion with a radiolucent rim. This radiographic evolution can also be seen in ossifying fibroma and, less commonly, in osteosarcoma. Histologically, cemento-osseous dysplasia exhibits a mixture of woven bone, lamellar bone, and cementum-like particles in a background of cellular fibrous connective tissue with scattered hemorrhage [1]. The cementum-like particles of cemento-osseous dysplasia do not present with brush borders as those in ossifying fibroma. Furthermore, osteoblastic rimming is not as prominent as in ossifying fibroma and the bony trabeculae in cemento-osseous dysplasia do not typically exhibit prominent peritrabecular clefting as seen in fibrous dysplasia. Asymptomatic cases of cemento-osseous-dysplasia do not require treatment; however, follow up during routine dental examination is recommended. Sarcomatous transformation of cemento-osseous dysplasia has been reported though extremely rare and the mechanism remains unknown [1,9,10-12].
Ossifying fibroma is a benign slow growing expansile neoplasm of bone in which pain and paresthesia are rarely reported. The lesion is typically encapsulated; however, as it approaches 2-3 cm, it begins to lose its encapsulation [5]. Ossifying fibromas are most often found in women in the third and fourth decades of life with predilection for the tooth-bearing areas of the mandible, especially the premolar and molar regions [1]. The radiographic appearance is that of an ovoid or spherical, well circumscribed, well-demarcated radiopaque lesion with buccal or lingual cortical expansion. Adjacent teeth may demonstrate root divergence or root resorption. Bowing of the inferior border of the mandible may be appreciated in large mandibular lesions. Early lesions may appear completely radiolucent; however, as they mature, they develop a more radiolucent/radiopaque appearance and, eventually, may present as completely radiopaque. Based on radiographic presentation, ossifying fibromas can be misdiagnosed as focal cemento-osseous dysplasia.
Histologically, the ossifying fibroma is mainly composed of a fibrous stroma and bone elements (i.e. woven or lamellar bony trabeculae, cementum-like spherules) exhibiting osteoblastic rimming and varying degrees of maturation. The cementum-like spherules exhibit brush borders that blend into adjacent connective tissue, a characteristic not seen in fibrous dysplasia, cemento-osseous dysplasia, or osteosarcoma [1]. Encapsulated lesions are typically treated by enucleation and curettage. As the lesion begins to lose its encapsulation, surgical resection with a 5 mm margin is recommended [5]. Once completely removed, 12% of ossifying fibroma cases have been reported to recur [1]. Ossifying fibromas are considered to have no malignant potential with only one previous case of malignant transformation into osteosarcoma reported in the literature [8].
The lesion described in this case report presented as a longstanding, asymptomatic, well demarcated, well-circumscribed, expansile, exophytic lesion without significant cortical destruction and no evident extracortical bone formation. Destruction of adjacent anatomical structures was not appreciated. The clinical characteristics and radiographic presentation were those of a benign fibro-osseous lesion. Surgically, the extent of the lesion was grossly visualized with an apparent abrupt change in bone architecture between lesional and normal bone. A surrounding capsule could not be grossly identified. Due to its growth, radiographic appearance, and intraoperative appearance, cemento-osseous dysplasia was ruled out as a possible diagnosis. Since the lesion lacked a ground-glass appearance on imaging and was well-circumscribed in an 82-year-old patient without prior history of gnathic expansion or bone recontouring, fibrous dysplasia was not further considered. An ossifying fibroma was favored based on clinical and radiographic presentation; however, a low-grade osteosarcoma was also included in the differential diagnosis prior to microscopic evaluation.
The areas reminiscent of an intramedullary benign fibro-osseous lesion within the tumor suggested that it may have arisen from a pre-existing benign fibro-osseous lesion. Furthermore, the radiographic presentation of a well-demarcated, expansile, mixed radiopaque/radiolucent lesion with no localized destruction of adjacent structures further suggested that this lesion possibly arose within a pre-existing benign fibro-osseous lesion. A longstanding, undiagnosed ossifying fibroma was favored since some areas in the periphery of the lesion showed spicules of woven bone with focal osteoblastic rimming and small, rounded to curved, mineralized spherules with a peripheral brush border in a background of bland fibrous stroma (Figure 6A).
We suspect this is the second reported case of malignant transformation of an ossifying fibroma to a low-grade central osteosarcoma. A thorough search using PubMed search engine only returned one other case report in which the patient had multiple recurrences of an ossifying fibroma prior to malignant transformation [8]. Only six cases were found in the literature in which osteosarcoma arose in patients with cemento-osseous dysplasia [8-12]. In three cases, the radiographic presentation was that of an ill-defined destructive mandibular lesion and the patient presented with pain and loss of function. In the remaining three cases, the patient was asymptomatic but the radiographs revealed significant bone destruction. In only two of these six cases was osteosarcoma believed to have arisen from within cemento-osseous dysplasia. In the other four cases, osteosarcoma was concurrent with but did not arise from within the area affected by cemento-osseous dysplasia, leading the authors to consider a collision or synchronous tumor rather than malignant transformation. Numerous articles were found in which fibrous dysplasia demonstrated malignant transformation into osteosarcoma, and interestingly most of these cases involved the polyostotic type [13].
The exact molecular mechanism by which benign fibro-osseous lesions undergo malignant transformation into osteosarcoma has not been determined, however, various pathways have been proposed and are outlined in Table 1 [27]. One of the more common mechanisms is the over-activation of signaling pathways to include insulin-life growth factors I and II (IGF-I, IGF-II), connective tissue growth factor (CTGF), and transforming growth factor (TGF) [14]. As with other malignancies, suppression of genetic mutation within the various tumor suppressor genes and pathways has been associated with osteosarcoma, chiefly the retinoblastoma tumor suppressor gene (Rb) and the p53 pathway [15]. Furthermore, oncogenes associated with osteosarcoma, c-myc, c-fos, and c-jun have been strongly associated with the development of osteosarcoma [16]. In 1990 Wu JX, et al. established a strong fos protein expression in 30 cases of human osteosarcoma, specifically c-fos and c-jun. C-fos is a transcription factor that, when dimerized with c-jun, falls in the activator protein-1 (AP-1) family and influences the transformation and differentiation of both osteoblasts and osteoclasts [17,18]. Hirotaka, et al. looked at viral induction of osteosarcomas in animal models (mice and chicken embryos) primarily through the fos gene (specifically v-Fos and c-Fos) [19]. Hirotaka and others demonstrated that overexpression of the fos gene is linked with the development of osteosarcomas [20-22].
Table 1. Various pathways of malignant transformation
|
Cemento-osseous dysplasia |
Fibrous dysplasia |
Ossifying fibroma |
Gnathic osteosarcoma |
Genetics/Molecular |
Unknown |
Sporadic condition results from postzygotic, activating mutations in the Guanine nucleotide-binding protein/α-subunit (GNAS) gene, which encodes the alpha subunit of a stimulatory G protein. Such mutations have not been detected in ossifying fibroma or cemento-osseous dysplasia. 1
Constitutive activation of G-protein signaling impairs osteoblastic differentiation in skeletal progenitor cells, stimulates melanin production in melanocytes, and causes hyperplasia and hyperfunction of various enDOMRine cell types. In addition, mutated osteoblasts overexpress interleukin (IL)-6, which stimulates osteoclastic activity and may contribute to bone lesion expansion [1].
|
Mutations in the tumor suppressor gene HRPT2 (which encodes the parafibromin protein) have been identified in patients with hyperparathyroidism-jaw tumor syndrome. This rare syndrome is characterized by parathyroid adenoma or carcinoma, ossifying fibromas of the jaws, renal cysts, and Wilms tumors. In addition, investigators have found HRPT2 gene mutations in a few sporadic cases of ossifying fibroma of the jaws. However, the potential pathogenetic role of HRPT2 mutations in ossifying fibroma remains poorly understood [1].
|
Studies have demonstrated a complex genetic profile among osteosarcomas, with alterations frequently detected in p53, RB1, and chromosome 21q [1].
Loss of retinoblastoma (RB1) tumor suppressor gene common occurrence along with p53 abnormalities [2]. |
Guanine nucleotide-binding protein/α-subunit (GNAS) testing |
Guanine nucleotide-binding protein/α-subunit (GNAS) absent in ossifying fibromas, cemento-ossifying fibromas, and cemento-ossifying dysplasias [2].
|
GNAS testing exhibits low sensitivity [1].
|
Guanine nucleotide-binding protein/α-subunit (GNAS) absent in ossifying fibromas, cemento-ossifying fibromas, and cemento-ossifying dysplasias [2].
|
Guanine nucleotide-binding protein/α-subunit (GNAS) absent in ossifying fibromas, cemento-ossifying fibromas, and cemento-ossifying dysplasias [2].
|
Immunohistochemistry |
N/A |
Strong reactivity for osteocalcin [2].
|
Weak reactivity for osteocalcin [2]. |
Immunohistochemical expression of MDM2 and CDK4 also may help to distinguish low-grade osteosarcoma from benign fibro-osseous lesions and other benign bone tumors [1].
Osteoid is immunoreactive for osteocalcin and osteonectin in a majority of cases [2]. |
Associated syndromes |
N/A |
McCune-Albright Syndrome, Jaffe-Lichtenstein Syndrome [1]. |
N/A |
Paget disease of bone, Li-Fraumeni Syndrome, Hereditary Retinoblastoma, Rothmund-Thompson syndrome, Osteoblastoma, Hereditary multiple exostosis, Ollier Disease, Werner Syndrome, and Congenital or hypoplasitc or absent thumbs [1,2]. |
Risk Factors |
|
History of radiation therapy [1]. |
|
Radiation exposure, alkylating agents [1]. |
Despite this preponderance of research, the molecular mechanisms by which osteosarcoma arises from benign conditions have yet to be elucidated. Regarding fibrous dysplasia, it was once thought that radiation was a crucial factor in its malignant transformation, during the 1960s when this was a mainstay of therapy. However, when radiation therapy was discontinued as treatment, cases were still reported [23,24]. A few authors have suggested that surgical treatment causing irritation is a potential mechanism, though obviously this only applies to cases that were surgically treated and these cases had a small sample size of five patients [25,26]. Consequently, more work needs to be completed to better identify mechanisms of malignant transformation, though the rarity makes that a difficult proposition.
There are no financial disclosures from any of the above authors. The views expressed herein are those of the authors and do not reflect the official policy of the Department of the Army, Department of Defense ,or the U.S. Government.
- Neville BW, Damm DD, Allen CM, Chi AC (2016) Oral and Maxillofacial Pathology. Missouri: Elsevier. 592-604, 614-617.
- Wenig B (2016) Atlas of Head and Neck Pathology. Pennsylvania: Elsevier. 286-289.
- Tabatabaei SH (2015) Diagnostic challenges of low-grade central osteosarcoma of the jaw: A literature review. J Dent Shiraz Univ Med Sci 16(2): 62-67. [Crossref]
- Diniz AF (2007) Low-grade central osteosarcoma of the mandible: A case study report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 103(2): 246-252.
- Marx RE, Stern D (2012) Oral and maxillofacial pathology: a rationale for diagnosis and treatment. Illinois: Quintessence. 851-859.
- Sinha R (2010) Low-grade osteosarcoma of the mandible. J Maxillofac Oral Surg 9(2): 186-190. [Crossref]
- Dujardin F (2011) MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Molecular Pathology 24: 624-637. [Crossref]
- Lee YB, Kim NK, Kim JY, Kim HJ (2015) Low-grade osteosarcoma arising from cemento-ossifying fibroma: A case report. J Korean Assoc Oral Maxillofac Surg 41: 48-51. [Crossref]
- Triantafillidou K (2012) Ossifying fibroma of the jaws: A clinical study of 14 cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol 114: 193-199.
- Lopes MA (2010) Clinico-pathologic conference: Case 1. Head and Neck pathology 4: 329-333. [Crossref]
- Olusanya AA (2012) Concurrent cemento-osseous dysplasia and osteogenic sarcoma: Report of two cases. Case Reports in Medicine. 1-4. [Crossref]
- Schneider L (1999) Malignant spindle cell tumor arising in the mandible of a patient with florid osseous dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 88: 69-73.
- Lopes MA, Kim HS, Mariano FV, Corrêa MB, Rabelo NT, et al. (2010) Clinico-pathologic conference: case 1. High-grade osteosarcoma (OS) and florid cemento-osseous dysplasia (FCOD). Head Neck Pathol 4(4): 329-333.
- Rikhof B, De Jong S, Suurmeijer AJ, Meijer C, Van Der Graaf WT (2009) The insulin-like growth factor system and sarcomas. J Pathol 217: 469. [Crossref]
- Scholz RB, Kabisch H, Weber B, Roser K, Delling G, et al. (1992) Studies of the RB1 gene and the p53 gene in human osteosarcomas. Pediatr Hematol Oncol 9: 125. [Crossref]
- Wu JX, Carpenter PM, Gresens C, Keh R, Niman H, et al. (1990) The proto-oncogene c-fos is over-expressed in the majority of human osteosarcomas. Oncogene 5: 989-1000.
- David JP, Mehic D, Bakiri L (2005) Essential role of RSK2 in c-Fos-dependent osteosarcoma development. J Clin Invest 115(3): 664-672. [Crossref]
- Eferl R, Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859-868. [Crossref]
- Hirotaka Watanabe (1997) Chondrocytes as a specific target of ectopic Fos expression in early development. Proceedings of the National Academy of Sciences 94(8): 3994-3999. [Crossref]
- Grigoriadis AE, Schellander K, Wang ZQ, Wagner EF (1993) Osteoblasts are target cells for transformation in c-fos transgenic mice. J Cell Biol 122: 685-701. [Crossref]
- Pearman AT, Chou WY, Bergman KD, Pulumati MR, Partridge NC (1996) Parathyroid hormone induces c-fos promoter activity in osteoblastic cells through phosphorylated cAMP response element (CRE)-binding protein binding to the major CRE. J Biol Chem 271: 25715-25721.
- Papachristou DJ, Batistatou A, Sykiotis GP, Varakis I, Papavassiliou AG (2003) Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone 32: 364-71. [Crossref]
- Tanner HC, Dahlin DC, Childs DS (1961) Sarcoma complicating fibrous dysplasia: Probable role of radiation therapy. Oral Surg Oral Med Oral Pathol 14: 837-846. [Crossref]
- Altay M, Bayrakci K, Yildiz Y (2004) The development of osteosarcoma following radiotherapy for fibrous dysplasia. Acta Orthop Traumatol Turc 38: 353-356.
- Picci P, Sieberova G, Alberghini M (2011) Late sarcoma development after curettage and bone grafting of benign bone tumors. Eur J Radiol 77: 19-25.
- Ramasamy R, Fazekasova H, Lam EW (2007) Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: Impact on in vivo tumor growth. Leukemia 21: 304-310.
- Braodhead ML, Clark JCM, Meyers DE (2011) The molecular pathogenesis of osteocarcoma: A review. Sarcoma. [Crossref]