Abstract
Immune checkpoints are protein molecules that act as key modulators of the immune response of T cells. They are receptors located on the surfaces of these cells that interact with ligands, allowing them to exchange signals with other cells.
This exchange of signals is known as checkpoints, but it occurs due to the interaction of proteins (ligands and receptors). This molecular synapse between cells can generate an activation or an inhibition of an event. T cells (adaptive immunity) are cells that roam the body in search of foreign cells that have antigens that can be recognized by these receptors. If during this exchange of signals, the T cell recognizes a normal or healthy cell of the individual, it will proceed to "check" other cells. A neoplastic cell should be recognized as not "healthy" or abnormal, but since they are mutated cells of the individual, they are not always recognized as foreign and can generate ligands of the tumor cell that activate the checkpoints so that it recognizes or interprets them as "normal" and does not generate an attack by the T cells on the tumor cell. Therefore, the ligand(s) of the tumor cells generate, as an evasion mechanism, an inactivating signal to the T cell checkpoints, to avoid being recognized as pathological and to avoid an immune cell attack to stop their growth. ICIs (immune checkpoint inhibitors) are monoclonal antibodies that block either the T cell receptor or the ligand that initiates the signal by interacting with the receptor. In this way the signal will be blocked, and the T cell will be able to differentiate the neoplastic cell from a normal cell and implement a cytolytic and immune attack. This therapeutic modality is known as immunotherapy. Cancer experts believe that these treatments can enhance the efficacy of drug treatments and reduce or eliminate the often-debilitating adverse drug effects that come with traditional chemotherapy. The work aims to define the physiological functions of checkpoints in cell reproduction, maintaining homeostasis and immune tolerance, their activation by ligands produced by tumors to evade immune surveillance, and their inhibition as antineoplastic therapy (ICIs). It also proceeds to discuss secondary effects that appear when immune tolerance is broken and very briefly other non-neoplastic indications that have been emerging for these ICIs.
Keywords
T cells, checkpoints, Immune checkpoint inhibitors (ICIs), immunotherapy, cancer
Introduction
In cellular and molecular biology, checkpoints are chemical processes or substances that can temporarily block or delay a biochemical event. Checkpoint mutations are associated with downregulation of some cancers. Checkpoints are protein molecular receptors located on the surface of T cells (and other immune and tumor cells), which interact with ligands (allowing the exchange of information with other cells). They thus modulate the immune response of T cells. These surface receptors are part of the immune system, and their fundamental role is to prevent and/or avoid a disproportionate reaction that damages or destroys healthy cells. These check points distinguish between healthy cells and foreign, diseased, or senescent cells that need to be removed. Given the number of antigens to which the immune system is exposed, if there were no checkpoints to regulate these responses, there would be no immune tolerance and many autoimmune diseases would occur. The activation of the checkpoints by means of ligands produces a switch-off signal, so that the T cell does not initiate a cytolytic or immune attack against those cells that generate said ligands.
It should be remembered that T lymphocytes (cellular immunity) together with B lymphocytes (humoral immunity through antibodies) are the primary cells of the adaptive immune response (also called acquired immunity) which is the second line of defense against infectious, inflammatory, tumorous, or immune insults. The first line of defense is the innate immune response. The key cells of this innate immune response are macrophages, monocytes, and neutrophils [1-4]. T cells develop in the thymus, and different subsets of T cells have different roles in adaptive immunity. For example, cytotoxic T cells (natural killer) directly attack and eliminate infected or "foreign" cells, while helper T cells increase the response and help the functions of other cells. Regulatory T cells (also called suppressor T cells) suppress the immune response. As T cells become activated and expand, the expression of checkpoint receptors is upregulated and they act on activated T cells, on reg T cells, and on exhausted T cells. In activated T cells they control and contract this cell population. In T reg cells, CTLA-4 and PD-1 promote their suppressive function and increase exhausted T cells, promoting tolerance and decreasing autoimmunity. Manipulation of these checkpoints could be a therapeutic target in autoimmune diseases (checkpoint agonists inducing T cell depletion).
Examples of checkpoints that inhibit T cells and generate a turn-off signal are CTLA-4 (cytotoxic T-lymphocyte antigen-4 or CD152), PD-1 (programmed cell death protein 1 or CD279), LAG-3 (lymphocyte-activation gene 3), TIM-3 (T cell immunoglobulin and mucin domain 3), VISTA (V-domain immunoglobulin suppressor of T cell activation). These checkpoints are what maintain immune tolerance and reduce autoimmunity. CTLA-4 blocks the activation of autoreactive T cells in the immune response early in lymph nodes, during antigen presentation in the priming phase, where dendritic cells present antigens and activate T cells, and at the junction between B7 and the T cell. Instead, PD-1 (receptor)/PD-L1 (ligand) axis acts in the effector phase of cancer immunity, later in peripheral tissues (see later). There are immune checkpoints that generate an ignition signal favoring the expansion, survival of T cells, IL-2 production, decrease immunological tolerance and favor the appearance of autoimmunity. Such are: D-40, OX-40 (CD134), 4-1 BB (CD137), GITR (glucocorticoid-induced tumors necrosis factor [TNF] receptor), and ICOS (inducible T cell co-stimulator (CD278).
Two signals are required for an immune cell to become activated. Signal 1 is controlled by T lymphocytes, which recognize a specific antigen (ligand) in their receptor by forming a complex with the major histocompatibility complex (MHC), and signal 2 occurs because a receptor also recognizes its corresponding ligand but in antigen presenting cells (APC) releasing molecules known as B7, which is where the signal is encoded [5]. To give an example, if an inhibitory checkpoint such as CTLA-4 adheres to B7 molecules like glue, signal 2 is hijacked, blocking communication with the T cell, which will not initiate a cytolytic or immune attack.
Immune Tolerance
T cells constitute a very important and potent effector compartment of the immune system. There is central and peripheral tolerance. Immune tolerance refers to that T-cell responses are strictly regulated to avoid inappropriate immune responses, such as autoimmune reactions. Central tolerance in the thymus acts as the first control during T-cell development to eliminate autoreactive T-cell clones. The nuclear factor AIRE expressed in medullary thymic epithelial cells facilitates ectopic expression of tissue-restricted antigens in the thymus and thereby plays an important role in the negative selection of autoreactive T cells in the thymus [6,7]. Therefore, there is a role for central tolerance in eliminating autoreactive T cells and thus preventing autoimmune reactions. However, in part due to lack of self-tissue antigen expression in the thymus, altered expression of self-antigens, or low affinity expression of self-antigens, some autoreactive T cells still manage to escape negative selection, leave the thymus, and enter the peripheral immune repertoire. Hence, peripheral regulation of T-cell responses is crucial to prevent inappropriate responses to self-antigens [8]. Therefore, T cell checkpoints define a balance between tolerance and autoimmunity. In fact, the immune regulatory functions of the "classical" checkpoints (CTLA-4, PD-1, TIM-3, TIGITI and LAG-3) were discovered in the setting of autoimmune disease modeling and their blockade or deficiency resulted in the induction, activation. exacerbation of these diseases. Subsequently, they were described as influencing the results in the setting of tumors or chronic viral infection. Hence the importance of co-inhibitory molecules (ICIs) in the regulation of peripheral tolerance and autoimmunity, and their role in anti-tumor immunity.
Regulation and Checkpoints of Cell Cycle
Regulation
The cell cycle is an ordered series of events that occur in the cell leading to the duplication of its DNA, and division of the cytoplasm and corresponding organelles to produce two daughter cells. These events are the sequential expression of different genes [9]. The regulation of the cell cycle is vital for the division to operate in an orderly manner and to avoid errors. This regulation is a process that controls the rate of cell growth and division. Without this necessary control, the cells would grow in a disorderly and abnormal way. Cell cycle checkpoints ensure that progression from one phase to the next occurs until conditions are favorable. Otherwise, checkpoints will stop progression. Prematurely entering a subsequent phase can be catastrophic and lead to cell death. The fundamental idea is that each daughter cell is a precise and complete copy of the genome. Furthermore, chromosomes must duplicate once, and only once, before mitosis. In other words, cycle checkpoints represent a solution to ordering and monitoring DNA synthesis and cell division [10]. During certain phases of the cycle, there are proteins that are activated or synthesized. For example, enzymes involved in DNA synthesis are induced in G1 and S phase, while tubulin is induced in G2 phase. A human cell takes an average of 24 hours to divide. Regulatory molecules promote progress or impede it according to monitoring conditions. Control mechanisms are both internal and external.
An internal control mechanism is formed by the MPF (maturation promoting factor) that is found in the cytoplasm of mitotic cells. This diffusible protein induces mitosis and is made up of 2 sub-units. The largest is P45 and the smallest is P32. P45 has kinase activity, that is, it transfers phosphate groups from ATP to serine and threonine residues on protein substrates. The three most relevant targets for mitosis of this factor are microtubules, whose phosphorylation is essential for the transition from the stability interface to the instability interface of the metaphase. The second objective is the phosphorylation of Histone H-1 that leads to chromosomal condensation and the third is the phosphorylation of nuclear laminas that leads to disaggregation of the nuclear envelope. P32 (cyclin) is an activator of MPF kinase, so as the interface proceeds from S and G2, cyclin increases to a level sufficient to bind MPF. The term cyclin was coined because the concentration of this regulatory protein rises and falls (cycles) in a predictable pattern with each cell cycle. When it becomes active it causes the cell to enter M phase. The degradation of cyclin completes the mitotic cycle. The external control mechanisms are usually orchestrated by hormones. For example, TGF (transforming growth factor) is a protein (chelone) that is produced in epithelial tissues. In some epithelium-derived tumors they can inhibit the cell cycle. EPO (erythropoietin) produced in the liver and kidneys stimulates the production of erythrocytes in the bone marrow, which have a half-life of 120 days. If their number decreases or hypoxic conditions occur, EPO is produced. PDGF (platelet-derived growth factor) induces smooth muscle cells, fibroblasts, and endothelial cells to heal. Other factors that come into play are fibroblast-derived growth factor, nerve-derived growth factor, and epidermal growth factor. These three factors produce tyrosine kinase activity [11].
Checkpoint of cell cycle
These checkpoints stop the progress of the cell cycle if the DNA of any chromosome is damaged or if certain critical processes of cell division have not been completed. The proteins that make up the cell cycle checkpoint machinery do not play an active role in normal cell cycle events. They are called on the scene only when abnormalities occur. The monitoring of the normality of the cycle is maintained through a system of sensors that recognize damage to DNA or cell division. If the sensors detect such damage, they activate a response to stop the cycle, while the damage is repaired, or the defect is corrected. The fundamental idea is not to enter the next phase with damaged sensitive information. If the cell damage is beyond the capacity to repair, the checkpoint machinery will activate a signal for an indefinite or permanent state of progress suspension or a signal to generate apoptosis [12].
At the G1 checkpoint, cells check whether the internal and external conditions for cell division are adequate. Size, nutrient availability, DNA integrity are evaluated. If the cell does not receive the "go ahead" after monitoring, at the G1 checkpoint, it can leave the cell cycle and enter a resting phase called G0. Some will remain permanently in G0 or resume splitting when conditions improve or are resolved. The main restriction to enter the cell cycle is damaged DNA. Following DNA replication in S phase, the cell enters a growth phase known as G2. Multiple checkpoints can be induced at the phase transition from G2 to M in response to damaged or incompletely replicated DNA. The cell will suspend reproduction to allow repairs. In this phase, G2 checkpoints also monitor the level of proteins and growth factors that are essential for the next phase of the cell cycle. If the damage is irreparable, the cell undergoes programmed cell death so that the damaged DNA does not pass on to the daughter cells. M-phase checkpoint (spindle checkpoint) is a spindle checkpoint that checks metaphase where all the chromosomes should have lined up at the mitotic plate and is under bipolar tension. All sister chromatids are examined, and chromosomes should/have aligned at the mitotic plate and be under bipolar tension [13]. Here, the cell examines whether all the sister chromatids will correctly attach to the spindle microtubules. Because the separation of the sister chromatids during anaphase is an irreversible step, the cycle will not proceed until all the chromosomes will firmly attach to at least two spindle fibers from opposite poles of the cell. If the chromosomes are not attached to the spindle apparatus, M- checkpoint will stop further cell cycle (Figure 1).
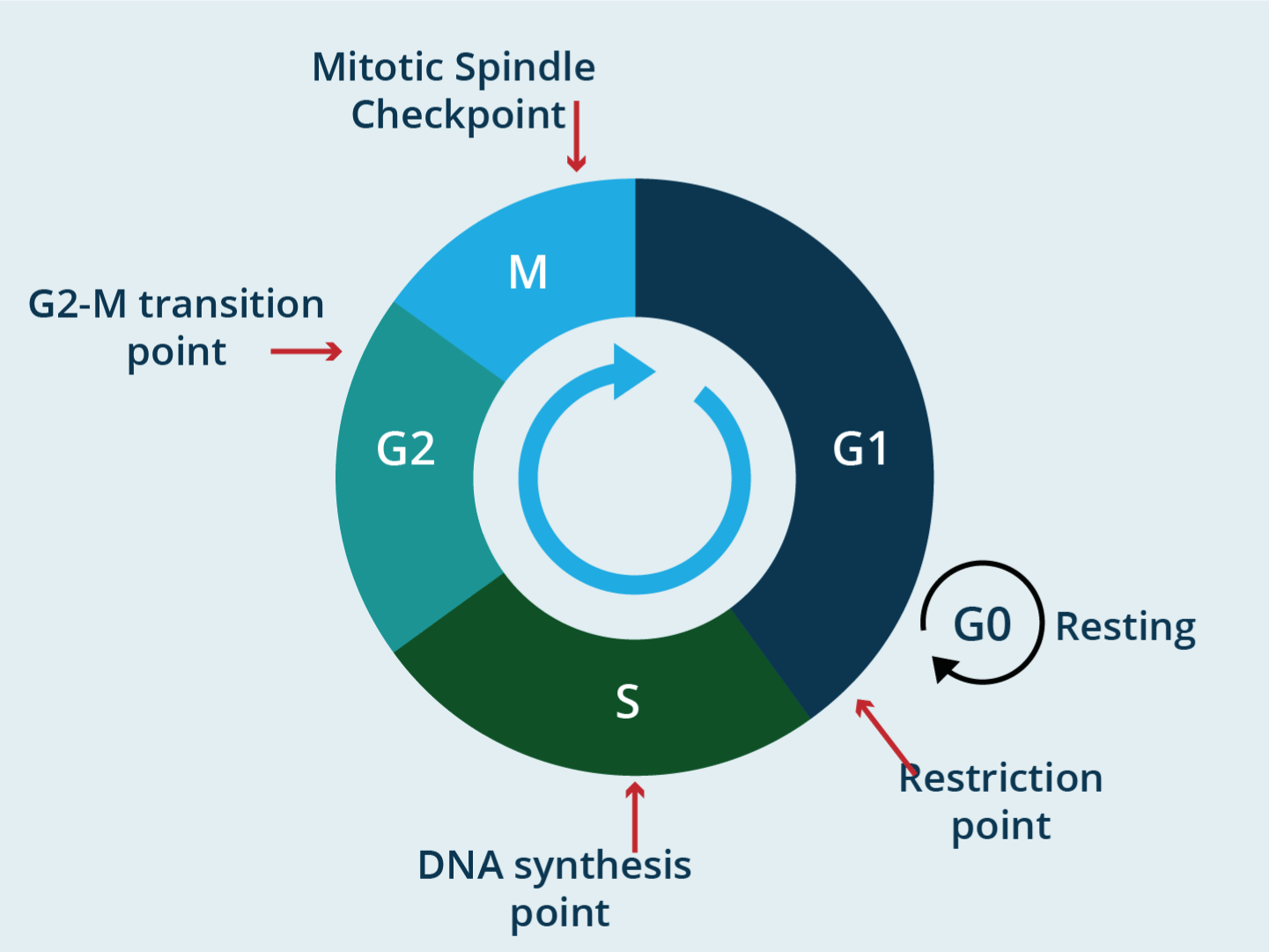
Figure 1. Title: the cell cycle checkpoints
Legend: A checkpoint is one of the several points in eukaryotic cell cycle at which the progression of cell cycle to the next stage can be halted until conditions are favorable
It should be obvious therefore, that any pharmacological manipulation to block or stimulate checkpoints in the treatment of diseases can potentially have secondary effects on any organ or tissue of the human economy.
Immunotherapy
Immunotherapy is a therapeutic modality of cancer treatment that uses the ability of an individual’s immune system to fight cancer and includes various strategies.
Inhibitors of checkpoints (ICIs)
It should be clear that the activation of checkpoints will generate a shutdown signal in normal cell division in response to DNA damage or some critical process of cell division (for genome protection), and in the immune system it will generate the same signal in T cells so that they do not initiate cytolytic or immune attacks against their own cells. Neoplastic cells can use this tool. Obviously, it is a mechanism to evade immune control using the checkpoint activation machinery in its favor. The key is the activation of the checkpoints. Therefore, inhibition of checkpoints is the key to this immunotherapy tool, since by inhibiting checkpoints the switch-off signal of T lymphocytes is blocked, which will recognize neoplastic cells as "foreign" or abnormal and will initiate an attack, to destroy tumor cells. Unlike conventional chemotherapy (inhibiting the cell cycle), in immunotherapy the drug that blocks the checkpoints does not attack the tumor cell directly, but instead uses the patient's acquired immunity machinery (T cells) to attack the tumor and stop its growth, that is, it takes advantage of the autoimmune functions of the host (Figure 2).
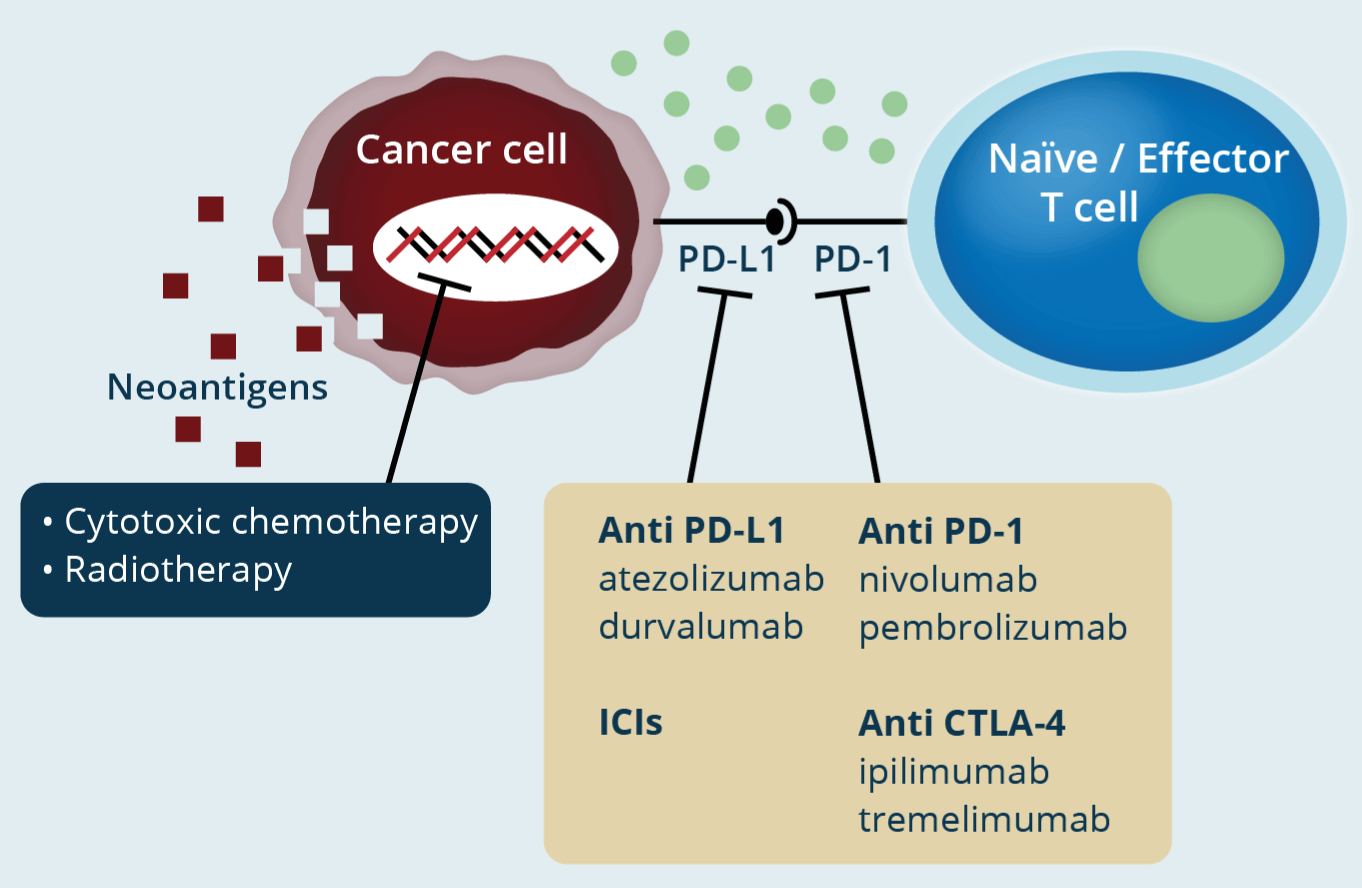
Figure 2. Title: Immune checkpoint inhibitors in cancer treatment
Legend: In conventional chemotherapy/radiotherapy, the genetic material of the cell is attacked, producing cytotoxicity to kill the neoplastic cell. With ICIs, the patient's T lymphocytes will be activated to generate an immune attack (immunotherapy) (modified from Onoi K, et al.)
Neoplastic cells are produced daily and almost all of them are eliminated through the host's immune response. It is pertinent currently to review the concept of the cancer-immunity cycle that involves 7 phases. 1) The release of cancer antigens by the death of neoplastic cells. 2) Presentation of said antigens to T cells by professional antigen presenting cells (e.g., APC). 3) Activation of cytotoxic T cells (priming phase). 4) Migration of T cells. 5) Infiltration of T cells in the micro tumoral environment. 6) recognition of neoplastic cells. 7) attack and elimination through cytokines that induce apoptosis of cancer cells (effector phase) [14]. Neoplastic cells with low immunogenicity (almost no cancer antigens present) can evade this autoimmune response and survive for long periods of time (equilibrium phase) (immune cold). Other mechanisms that suppress the host's immune response and the expected cycle are the accumulation of mutations in neoplastic cells, the induction of T regs cells and MDS cells (myeloid derived suppressor cells) and the expression of part of the tumor of molecules that stimulate or activate immune checkpoints (ligands), all of which will result in uncontrolled tumor growth (escape phase). In addition, certain types of tumors are detected only after they have proliferated and developed a system that prevents their elimination through an autoimmune response.
Specific checkpoints and their inhibitors
ICIs are monoclonal antibodies that bind to the checkpoint receptor or to the ligand. There are specific inhibitors of the PD-1/PD-L1 or PD-L2 axis. The activation of this axis in cancer turns off the signals of the T cells and the neoplastic cell is not considered foreign, pathological, or dysfunctional. Monoclonal antibodies by blocking the receptor (PD-1) or the ligand (PD-L1 or PD-L2), or both, prevent the turn-off signal of T cell recognition activity and this orchestrates cytolytic and immune attack (Figure 3). The same happens with monoclonal antibodies that block the CTLA-4 checkpoint. In 2018, the Nobel Prize in Medicine was awarded to two immunologists who generated the concept of immunotherapy based on ICIs: James Allison and Tasuku Honjo [15]. Some tumors express high levels of PD-1 or CTLA-4 allowing them to escape immune attack. Elevated expression of PD-1 and/or its ligands in various types of tumors has been associated with a poor prognosis, but it also has predictive value for response to monoclonal antibodies [16]. Also, PD-L1 and PD-L2 can be expressed on non-neoplastic cell surfaces such as macrophages and dendritic cells. Just to exemplify, PD-1 inhibitors are Nivolumab (Nivolumab is a genetically engineered, fully human immunoglobulin (Ig) G4 monoclonal antibody directed against the negative immunoregulatory human cell surface receptor programmed death-1 (PD-1) with immune checkpoint inhibitory and antineoplastic activities), Pembrolizumab (Pembrolizumab is a potent, highly selective, fully humanized immunoglobulin (Ig) G4-kappa monoclonal antibody against PD-1 with potential immune checkpoint inhibitory and antineoplastic activities), and PD-L1 inhibitors Atezolizumab, Avelumab and Durbalumab, and CTLA-4 inhibitor Ipilimumab (Ipilimumab is an immunomodulatory monoclonal antibody directed against the cell surface antigen CTLA-4 and also a type of immune checkpoint inhibitor. MW :148 kD). Combined anti-PD-1 and anti-PDL-1 or anti-PD-L1 and anti-LAG3 therapy has been implemented in some tumors. In advanced prostate cancer that expresses ARV7 (muted androgen receptor), which makes it resistant to androgen therapy, blockade of both checkpoints (CTLA-4 and PD-L1) has been used. The basic action will occur in the CD8+ T cells that are in the tumor microenvironment (TME). The most notable activity of CD4+ T cells in TME is to expand and differentiate CD8+ T cells into T cytotoxic lymphocytes. With the participation of DCs, these T lymphocytes recognize and lyse tumor cells via granzins/perforins and FasL/TRAIL molecules [17-20].
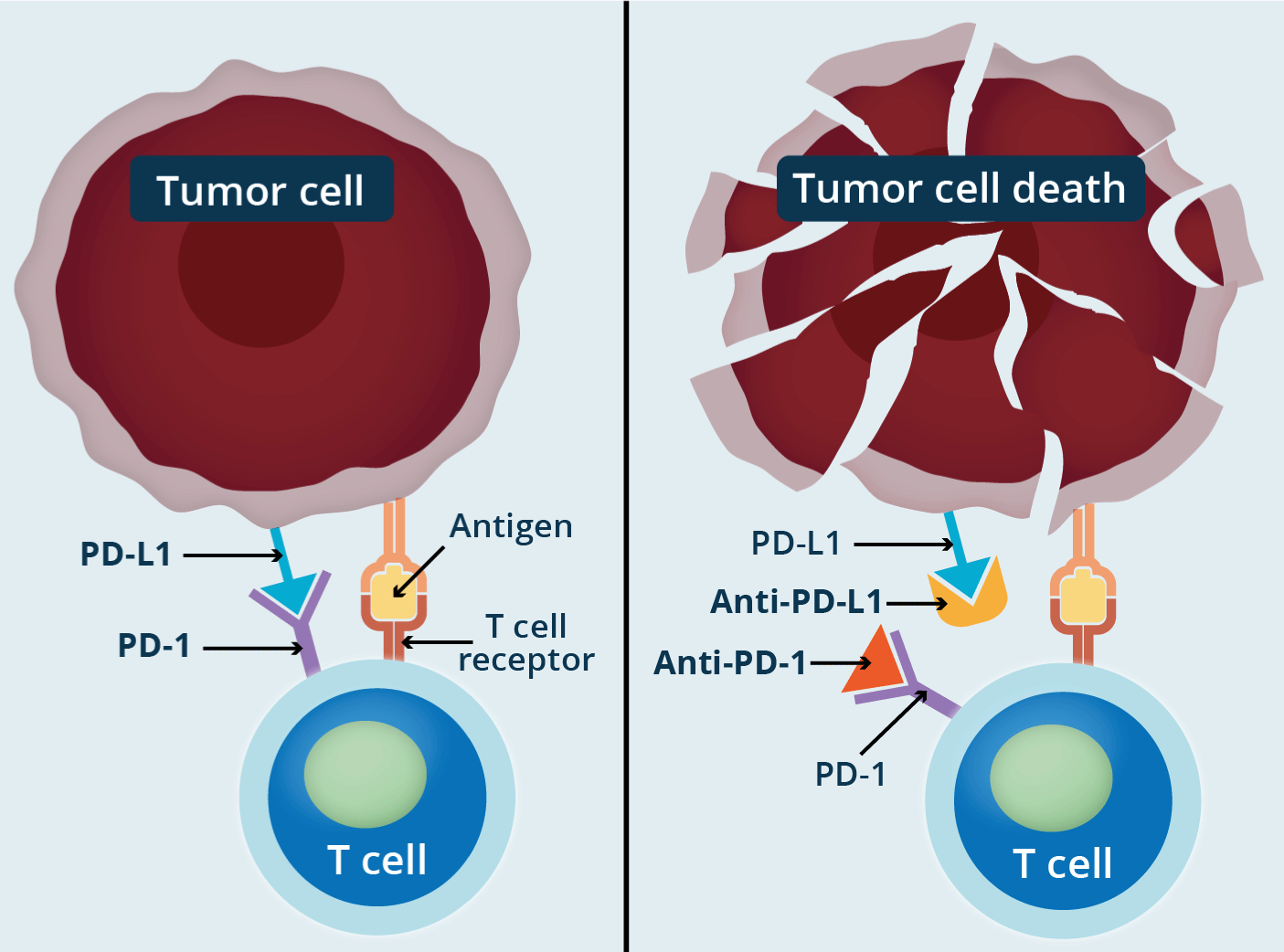
Figure 3. Title: How do immune checkpoint inhibitors work against cancer?
Checkpoint proteins, such as PD-L1 on tumor cells and PD-1 on T cells, help keep immune responses in check. The binding of PD-L1 to PD-1 keeps T cells from killing tumor cells in the body (left panel). Blocking the binding of PD-L1 to PD-1 with an immune checkpoint inhibitor (anti-PD-L1 or anti-PD-1) allows the T cells to kill tumor cells (right panel) (National Cancer Institute).
T-cell exhaustion in tumor immunity
Described more than two decades ago, T cell depletion primarily affects T cells CD8+. Depletion alters cellular metabolism, chemokine activity, receptor expression, and cytokine signaling. The cells become dysfunctional but a characteristic of the depletion of these lymphocytes is the high expression of checkpoints, so by blocking these checkpoints the function and response of CD8+ is restored. Exhaustion has opposite pathways in cancer and autoimmunity. Its presence is associated with a poor immune response to cancer, but with a better prognosis in autoimmune disease (Figure 4) [10]. Dysfunctionality occurs due to chronic exposure to an antigen, in turn induced by the lack of help to CD8+ by CD4+ T cells, and by exposure to immunosuppressive cytokines. This also occurs in chronic viral infection [21]. These CD8+ T lymphocytes are unable to lyse tumor cells and have a decreased ability to produce potent effector cytokines (TNF-α, IFN-Ɣ, IL-2) along with increased expression of checkpoints such as CTLA-4, PD-1, TIM-3, TIGITI, LAG-3. Very recently, there has been a growing interest in transcriptional factors that initiate, amplify, and maintain the depletion state. GATA-3 is a transcription factor of zinc rings that manages the dysfunctional state of these cells. CTLA-4 in cancer is expressed not only in neoplastic cells but also in CD4+, CD8+ and Treg T cells. Its role is clear in that it acts as a brake on the T response in tumor immunity [23]. Similar happens with PD-1. Originally identified as a T-cell receptor associated with programmed cell death (hence the acronym PD), its anti-tumor immune response was later recognized. Its ligands (PD-L1 and PD-L2) expressed in various tumors and myeloid cells dampen the antitumor immune response. In fact, tumor-derived PD-L1 can directly inhibit CD8+ T cells and promote tumor escape from immune recognition [24].
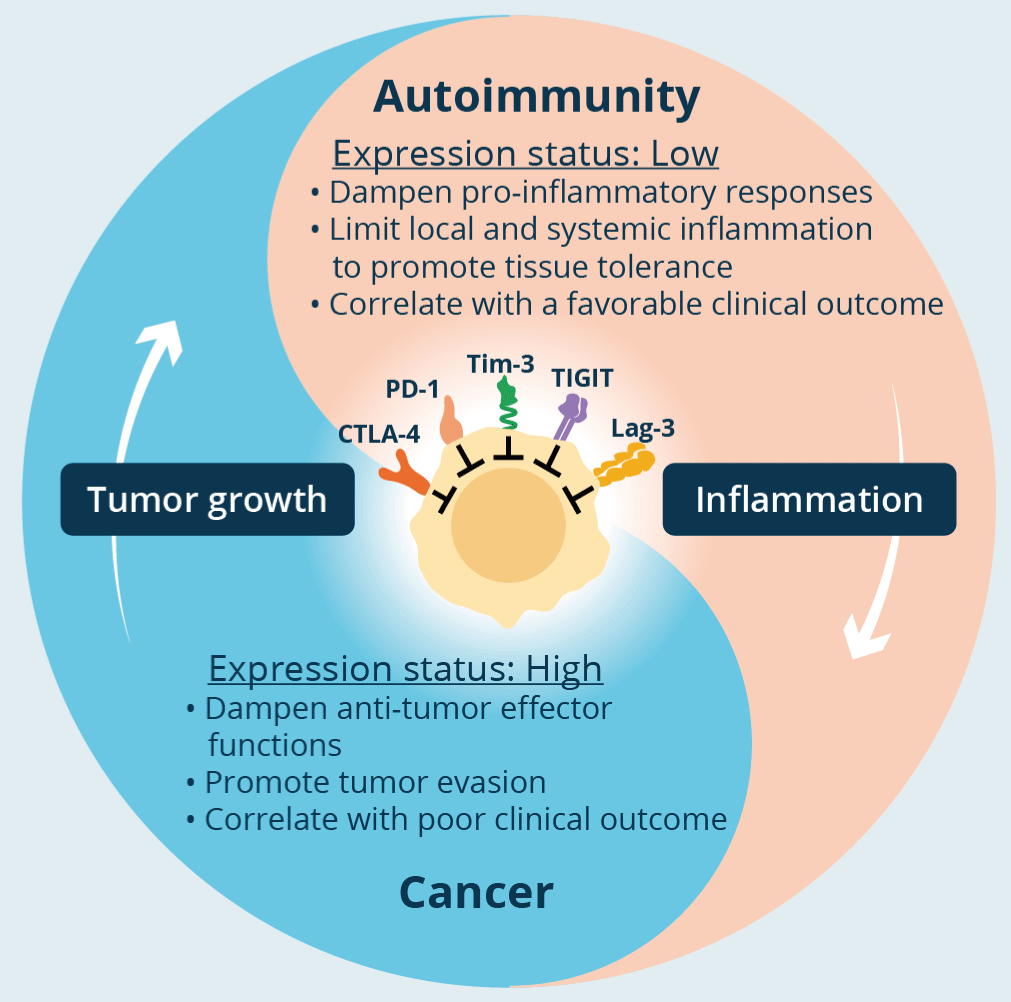
Figure 4. Title: T-cell exhaustion in cancer and autoimmunity
Legend: In the tumor, co-inhibitory receptors on T cells dampen T-cell effector functions thereby enhancing tumor progression and correlating with worse clinical outcome. In autoimmunity, these receptors play a role in reducing local and systemic tissue inflammation, maintaining tissue tolerance, and their increased expression is associated with a good clinical outcome (from Schnell).
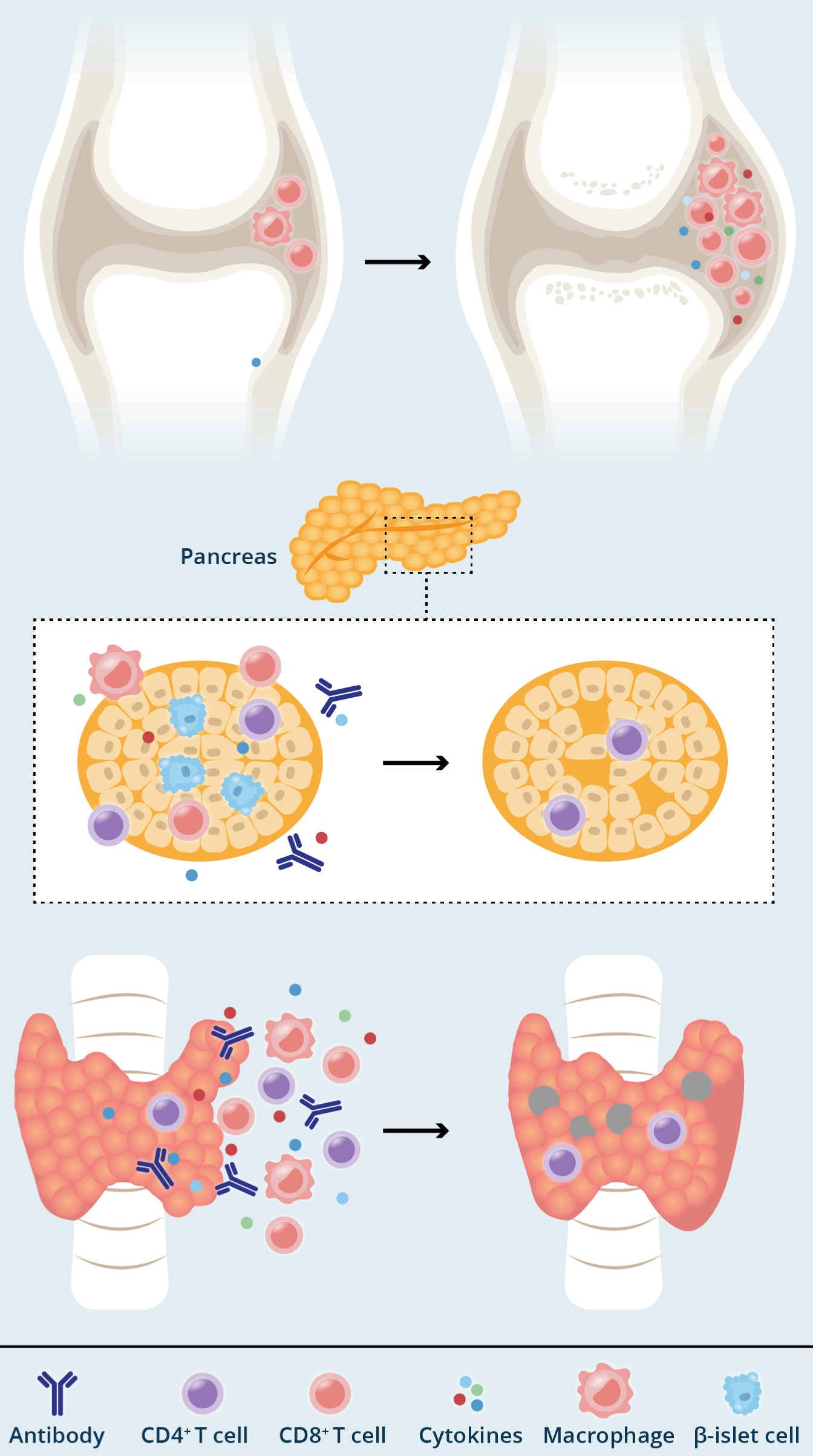
Figure 5. Title: Mechanisms of chronic immune-checkpoint inhibitor-mediated toxicity
Legend: a) Smouldering toxicities characterized by off-target T cell activation that may wax and wane over time. Examples include rheumatoid arthritis-like inflammation of the joints. Such effects often resolve on treatment withdrawal and/or steroids. b, c) Burnout toxicities characterized by irreversible damage to the relevant cells, typified by immune-checkpoint inhibitor-mediated endocrinopathies. Examples include destruction of the hormone-secreting cells of the pancreas (b) or thyroid (c). Such toxicities are usually irreversible and require permanent hormone-replacement therapy (from Johnson).
Basically, preclinical studies support the role of TIM-3, TIGITI and LAG-3 checkpoints in tumor growth. The over-expression of the three receptors in T cells promotes said growth and its blockade with ICIs reduces it. At least 9 clinical trials inhibiting TIM-3 in solid tumors have shown promising results, especially associated with PD-1 blockade [25]. TIGITI is involved in attenuating T-cell activation and anti-tumor effector cell differentiation and its blockade with ICIs is being investigated in various types of cancer. LAG-3 synergizes with PD-1 to promote tumor growth and is widely expressed on tumor-infiltrating lymphocytes in TME in various malignancies. The 2 ligands of LAG-3 (galectin-3 and fibrinogen-like protein 1) are involved in immune evasion mechanisms, so their blockade in T cells is a short-term objective in immunotherapy.
Manipulating T cell depletion was initiated in 2010 by blocking CTLA-4 in metastatic melanoma. A year later it was approved by the FDA and then in 2014 the blockade of PD-1 was approved. It must be said in all fairness that the response rate to this therapy (which opened a new horizon in malignant diseases and a vast field of basic and clinical research) remains modest. The ORR (objective response rate) is 10-16% with ipilimumab (anti-CTLA-4), and 30-40% with nivolumab (anti-PD-1). Together the two ICIS reach levels around 60% and can reach 90% in metastatic melanoma. But some tumors have been refractory to this therapy or have generated resistance, or patients have developed irAEs (immune-related adverse effects), which has counteracted the expected clinical success and the initial and optimistic projections. Multiple clinical trials are under study and scrutiny, either using monotherapy or optimized combination therapies (ICIs mixed with chemotherapy/radiotherapy or vaccines directed against tumor antigens). Very recently Forde PM, et al. (04/2022) have shown that the use of neoadjuvant treatment (preoperative) with Nivolumab in patients with stage IB to IIIA resectable NSCLC (non–small-cell lung cancer) associated with platinum-based chemotherapy, resulted in significantly longer event-free survival and a higher percentage of patients with a pathological complete response than chemotherapy alone. The addition of nivolumab to neoadjuvant chemotherapy did not increase the incidence of adverse events or impede the feasibility of surgery [26].
Side effects (irAEs)
ICIs can cause immune cells to become stimulated and attack healthy cells, triggering a series of side effects. As these effects are related to immunity, they are known as immune-related adverse events (irAEs). There are several mechanisms responsible for them. ICIs can aggravate or activate a previously silent autoimmune disease. A new autoimmune inflammatory disorder can be generated in each patient by the breakdown of immune tolerance, by the breakdown of tissue immune homeostasis, or by the patient's own tissue damage outside the tumor. Undesirable reactions can also appear when checkpoints are blocked in non-T cells. The microbiome can affect the response in cancer. Intestinal dysbiosis due to antibiotics can negatively impact the results in anticancer immunotherapy. This implies that irAEs originate in factors related to the tumor and in other factors not related to the tumor. A specific risk score for each immunotherapy can give important perspectives to clinicians to improve immunotherapy by limiting adverse effects [10]. The most frequently reported side effects are in organs with more extensive interfaces such as skin, respiratory and digestive tract. In skin rash and itching are the most prominent. Cough, chest pain and dyspnea at the respiratory level. Nausea, diarrhea, and hepatitis, in the digestive tract. At the endocrinological level, diabetes, hypothyroidism and hyperthyroidism, and as non-specific symptoms myalgia and numbness. Fatal toxicity occurs in 0.4-1.2% of patients. Persistent toxicity may be more common than initially reported, reaching up to 40%, mainly endocrinological and rheumatological.
Approximately 50% of patients with metastatic disease in developed countries are eligible to receive ICIs with 8 approved agents in at least 17 different malignancies (December 2021). This has led to an increase in its use in different neoadjuvant and maintenance regimens. ICIs are also used in combination regimens with cytotoxic, biological, or targeted chemotherapy. Durable responses, even in metastatic disease, have been increasing and therefore so have the chronic complications of immunotherapy [27]. CTLA-4 blockade has a higher incidence of irAEs (which is dose dependent) than blockade of the PD-1/PD-L1 axis (independent dose). Ipilimumab (anti-CTLA-4) varies from 38.6-57.9% which contrasts with only 10-15% of the PD-1/PD-L1 axis. CTLA-4 inhibition, except for metastatic melanoma, has limited activity as monotherapy. In contrast, inhibitors of the PD axis have clinical activity in various types of solid tumors. The inhibition of both checkpoints increases the effectiveness but also the irAEs [28]. irAEs generally appear in the first 3 months of treatment and if severe, the ICI(s) should be discontinued, and steroids given. The use of high doses of steroids does not appear to interfere with the antitumor response. Cancer patients generally receive steroids as symptomatic treatment for worsening of systemic symptoms and symptoms due to tumor progression. They are also used for their antiemetic effect in platinum-based chemotherapy, and with ICIs they suppress the immune responses induced by IL-2, CD8+-T, and Treg increase. However clearly defining the role of steroids given concomitantly with ICIS is another future challenge in lung cancer.
Delayed irAEs are those that appear up to one year after ICI therapy and occur in 5.3% of patients. The most frequent are rash, colitis and pneumonitis and the resolution time is not clear [29]. Chronic irAEs are defined as those adverse effects that persist after 12 weeks of discontinuation of ICI therapy. They appear in 43.2% of patients with ICIs directed against PD-1/PD-L1 (retrospective studies). The most common are hypothyroidism, type 1 DM, and arthritis [30,31] (Figure 5). Some details of irAEs induced by checkpoints deserve to be highlighted. For example, steroids are not used as treatment for endocrine effects, because you do not respond to them, and the inflammatory pathogenic mechanism is not clear. They require lifelong hormone replacement, whether hypothyroidism (the most common), hypophysitis, type 1 DM, or adrenal insufficiency (rare) [32,33]. Rheumatologic irAEs are not surprising, although lupus erythematous and mixed connective tissue disease are rare, but not rheumatoid arthritis (RA). There are some differences with the primary disorders. For example, in RA (irAE) it is generally seronegative and the response to steroids is suboptimal. The use of DMARDs (disease-modulating antirheumatic drugs) does not worsen oncological results and it is not necessary to suspend ICIs regularly. At the gastroenterological level, colitis, hepatitis, celiac disease, and pancreatic insufficiency are the most frequent (especially in regimens containing ipilimumab). They improve with steroids and immunomodulators (infliximab) [34]. At the respiratory level pneumonitis (anti-PD axis) responds to steroids and second line immunomodulators. Sarcoidosis also responds well to steroids. The bidirectional relationship between ICIs and tuberculosis is not clear. Within the cardiovascular irAEs, the picture can range from an asymptomatic elevation of troponin 1, pericarditis and acute vasculitis, fulminant myocarditis to residual cardiomyopathy. At the neurological level, they appear fundamentally with ipilimumab (5%) and the range includes alterations of the neuromuscular junction (myasthenia gravis and Eaton-Lambert), peripheral motor neuropathy and meningoencephalitis. In the skin, the most frequent are inflammatory dermatitis, vitiligo (melanoma), Stephen-Johnson and bullous pemphigus [35].
Fatal irEAs are refractory to steroids and other immunosuppressants. They occur in 0.4% with anti-PD-1/PD-L1 and in 1-2% with combined anti-CTLA-4 and anti-PD axis. Myocarditis can be fatal in 30-50% of patients, pneumonitis in 10-15%, and colitis and hepatitis are less common. It generally appears early with the use of ICIs (15-40 days) and in older patients. When combined therapy is used, clinical and laboratory monitoring is recommended for the first 3-4 weeks.
Patients with AID (autoimmune disease) have an inappropriate or dysregulated activation of the immune system, which attacks the body's own organs. and represent around 80 diseases that share a common pathogenesis. Because ICIs can exacerbate autoimmunity (break immune tolerance), AID patients are generally excluded from prospective, controlled, randomized clinical trials of cancer treatment with ICIs. However, many cancer patients have background AID (25%). A review of 17 studies of the use of ICIs in this population suggests that the efficacy of ICIs in this population to treat malignancy is comparable to that in patients with malignancy and without AIDs, and that the incidence of irAEs is higher but manageable. Inflammation and tumor lysis induced by immunotherapy generate numerous antigens that can be presented by APC as autoantigens, triggering autoimmunity. The preventive strategy proposes that in patients with pre-existing IDA, if worsening of autoimmune symptoms occurs (28%), they be managed with steroids or steroids and additional disease-modifying therapies [36]. The presence of AID should not exclude patients from ICIs, but 3 conditions must be maintained. Very close monitoring, careful risk/benefit balance and the patient must be managed by a multidisciplinary team [37].
Toward tumor immunity without autoimmunity
Despite the impressive efficacy of immune checkpoint blockade in the treatment of some cancers, the manifestation of autoimmune disease-like irAEs has become a critical limitation for the applicability of these drugs in clinic. Additionally, many patients fail to respond to current checkpoint blockade therapies. Current and future efforts in the field are directed toward promoting tumor-specific immunity with greater efficacy in more clinical settings with more tolerable side effects (Figure 4). There are certain characteristics shared between cells tumor-infiltrating exhausted T and proinflammatory helper (CD4+) cells. At the same time, depleted T cells and pathogenic TH17 are implicated in many autoimmune diseases. Identifying genes that share depleted Th1, Th17 and T signatures could allow the creation of molecules that aim to promote antitumor immunity without activating autoimmune processes. The use of specific techniques (RNA-sequencing, ATAC-sequencing) combined with computerized and functional systems could precisely define signatures or genes that encode exhausted CD8+ and thus create specific molecules against these target genes without triggering autoimmunity. There are subpopulations of exhausted CD8+ progenitor cells that respond better to anti-PD-1 therapy (melanoma). Another possible strategy is to specifically block only the tumor checkpoints, thus limiting the appearance of irAEs in non-tumor sites, or to take advantage of drugs used concomitantly with ICIs to block these irAEs. For example, blocking TNF (mediator of many autoimmune diseases) can reduce irAEs such as colitis induced by anti-TCLA-4 or anti-PD-1 therapy. It is necessary to improve the delivery of ICIs in the TEM, improve the specificity of these molecules, design molecular shields that restrict the action of ICIs to local tumor activity and investigate the option of vaccines directed against tumor antigens. The use of metabolic enzyme inhibitors should also be explored from a therapeutic point of view since there seems to be an interrelationship between metabolic rewiring in the promotion of cancer metastasis [10].
Biomarkers
A biomarker is an objectively measurable and evaluable characteristic that indicates a normal or pathogenic biological process or pharmacological response to a therapeutic intervention. Biomarkers of neoplastic cells (which translate certain genetic changes) can indicate which drugs could produce a better result. For example, the presence of MSI-H (microsatellite-instability) and/or dMMR (mismatch repair) are mutations that imply difficulty for the DNA repair, which can lead to uncontrolled cell growth. Specific ICIs have been designed for these mutations. However, patients do not necessarily need to be positive for these biomarkers to indicate immunotherapy. Positivity usually occurs in advanced cancer such as melanoma, bladder, and lung cancer. In a decade, since the introduction of ICIs, there have been more than 3,000 clinical papers evaluating T-cell modulators, which represents 66.6% of all oncology papers [38,39]. In the last 7 years, the FDA has approved 7 anti-PD-1 or anti-PD-L1 only monoclonal antibodies and more than 85 oncological indications for these ICIs. There are more than 2000 clinical papers specifically evaluating 33 monoclonal antibodies directed towards the PD-1/PD-L1 axis, which makes it impossible to discuss the biomarkers of all neoplasms [40]. Lung cancer is a neoplasm that serves to exemplify the subject for several reasons. Globally lung cancer is the deadliest: 1.6 million deaths a year. 85% of patients diagnosed with lung cancer have NSCLC (non-small cell carcinoma of the lung) which includes adenocarcinoma (the most common histologic subtype), large cell, and squamous cell carcinoma. As of 2018 and, based on molecular tests such as NGS (next generation sequence), it is possible to give a better personalize therapy. One third of patients have an oncogenic driver amenable to targeted medication, one third have an inflammatory MTE that can be treated with ICIs, and the rest are treated with chemotherapy, usually combined with other therapies [41]. Lung cancer is not only the leading cause of death worldwide, but also and even in early disease, survival is worse than in other types of cancer such as colon, prostate, and breast. Survival at 5 years is modest at best due to local, regional, or metastatic exacerbation [42-44].
Mutations with oncogenic drivers are generally exclusive, that is, the presence of one excludes the other in the same patient, allowing therapy to be personalized. The carrier of the oncogenic driver EGFR (epidermal growth factor receptor) can be treated with inhibitors of the tyrosine-kinase of the EGFR (EGFRTKIs), for example Geftinib or with Cetuximab (novel molecular-targeted agent, is an inhibitor of EGFR monoclonal humanized antibody interacting with the extracellular binding site of EGFR to block ligand stimulation. MW: 145.781 KD). 90% of EGFR mutations occur from exon 9-21 and are susceptible to EGFRTKIs. Trastuzumab (anti-human HER2) is a humanized, recombinant monoclonal antibody that binds to the extracellular domain of HER2, MW:145.53 KD. ALK (anaplastic lymphoma kinase gene) is a biomarker associated with adenocarcinoma in young people, non-smokers, and with metastatic disease in the pleura, pericardium, and brain. Crizotinib is a targeted treatment and so is Alectinib, which is more effective for Central Nervous System metastases. ROS-1 (c-ros oncogene-1) occurs in 1% of NSCLC. Entrectinib is a personalized therapy for this mutation as well as for the TRK mutation. BRAF (V-rat murine sarcoma viral oncogene homolog B1) is another mutation in NSCLC. 2-3% of advanced NSCLC have the BRAF gene mutation and 50% of them have the V 600 E mutation. FDA has approved Dabrafenib and Trametinib for both. MET (mutation in exon 14), RET fusion (rearranged during transfection) and NTRK (tropomyosin receptor kinase) are other marker genetic mutations that are targets of targeted and personalized immunotherapy [45]. While it is true that the efficacy and prolongation of life or response to therapy occurs, the effect on overall survival is more debatable.
NSCLCs can generate mechanisms of acquired resistance to targeted therapy. For example, modifications of the oncogenic driver may occur, or secondary mutations may appear. TKIs (tyrosine kinase inhibitors) act by interrupting the binding of ATP or inducing suppressive conformational changes, but secondary mutations in the gene that encodes tyrosine kinase can counteract this inhibition by having a greater affinity for ATP, generating a steric hindrance for the tyrosine kinase-TKI binding. This occurs in all three generations of TKIs. At least 50-60% of NSCLCs with mutated EGFR develop new mutations for the TKIs and it also occurs in 20-30% of ALKTKI NSCLC patients. Identifying secondary mutations that create post-treatment resistance may allow sequential therapies to be produced. Amplification of the oncogenic driver can also occur, which means that secondary mutations are amplified, that is, the original mutation passes to another driver [46]. Many pathways of oncogenic mutations occur through already established and known primary cellular signals (JAK/STAT, PI3K/AKT, etc.), but in therapy with TKIs alternative or downstream pathways may emerge that avoid the inhibitions produced by TKIs (parallel bypass). MET (hepatocyte growth factor receptor gene) is one of those avenues of resistance. One obvious result is that primary or target blockade is not sufficient for long-term control of NSCLC. A practical aspect is to use combination therapy to overcome the primary targets and acquired resistance. For example, dispense Geftinib in NSCLC (inhibit mutated EGFR) plus Campnatinib (inhibit MET). Although multi kinase inhibitors can inhibit the initial activation of a pathway, downstream protein modifications can independently maintain cell signaling [47]. Phenotypic transformation has also been described as a resistance mechanism. For example, in an NSCLC treated with EGFRTKI and ALKTKI, which becomes resistant, MET has been detected facilitating tumor migration and invasion. ctDNA (circulating tumor DNA) has been approved by the FDA for detection of active EGFR mutations in NSCLC in clinical trials and is likely to help guide therapy. It is a biomarker with high specificity and moderate sensitivity to detect EGFR mutations [48].
Engaging the immune system to treat cancer appears to be an effective strategy. ICIs, by reversing T cell depletion, particularly mediated by PD-1(which is a biomarker in itself), have raised expectations in NSCLC [49]. Chemotherapy plus Pembrolizumab is superior to platinum-based chemotherapy. This has improved the therapeutic landscape for NSCLC; for example, 5-year survival is 32% higher with ICIs than with Docetaxel in NSCLC, but the impact has not been as successful for this type of cancer associated with driver mutations, for example EGFR. These mutations have low expression of PD-1, which explains the relative lack of efficacy of anti-PD-1 ICIs [50]. Additionally, the addition of TKIs plus immunotherapy adds toxicity to the regimens [51-53]. Since 2016, ICIs have been widely used as first and second line in both NSCLc and SCLC (small cell lung cancer). More studies of combined therapy with ICIs in SCLC are required. In general, SCLC have less expression of PD-1 and the results with ICIs are less good than in NSCLC.
Other indications for ICIs
In the same way that the list of checkpoint receptors, their ligands and the ICIs under investigation has been increasing, other indications have also emerged [54]. A point that is not clear and that should be investigated is the use of ICIs in cancer patients and certain populations, such as post-organ transplant autoimmune disorders, patients with chronic viral infection, or concomitant use of immunosuppressants, patients with dysfunction of organs, patients with brain metastases, or use in extreme ages, pregnancy or altered functional status [55]. Emerging data suggest that the immune checkpoint axis is dysregulated in COPD. Lung T cells from these patients express PD-1 and have loss of cytotoxic function. Excessive T-cell inflammation may be a consequence of acute infections, which in turn may contribute to future acute exacerbations of COPD. Studying the expression of PD-L1 and exposure to cigarette smoke could define in the future a connection between COPD and lung cancer, and it should also be defined if the check points in patients with COPD could be future therapeutic targets [56]. Other works do not find evidence of alterations in the expression of immune checkpoints associated with cigarette use and COPD, therefore this is a fertile field of research to define whether there is such an association and the potential benefit of ICIs [57]. More than 25 new types of autoantibodies have been recently described in neurological diseases, which are related to autoimmune diseases and clinical syndromes. This is related to immune checkpoint dysregulation, pathogenicity by autoantibodies, and possibly will lead to targeted immune-specific therapy [58]. B lymphocytes contribute to the pathogenesis of many rheumatic diseases. Directing or using the checkpoints that control and activate their effector functions as therapeutic targets is a promising option. Rituximab (anti-CD20) is a chimeric anti-CD20 mAb that binds the CD20 antigen on B cells with a binding affinity of 5 nM, MW: 143.86 KD [59].
Checkpoint agonists
The basic idea is to activate checkpoint receptors with agonists (as opposed to inhibiting them with ICIs) in autoimmune diseases to restore lost immune tolerance and modulatory function of these receptors. In review, PD-1 and CTLA-4 recognize ligands expressed on the surface of self-tissues and act as buffers against unwanted immune activation (autoimmunity) by those ligands. This is immune tolerance. If an autoreactive T cell (which has these PD-1 and CTLA-4 receptors, that is, TCR) escapes this tolerance, it can encounter an antigen from its own tissue and harm it [60]. Checkpoints are designed to avoid this contingency. But if the checkpoints are dysfunctional, autoimmune inflammatory infiltrate will occur in multiple organs. For example, cardiomyopathies, extensive immune dysregulation, SLE, RA, Atopy, Multiple Sclerosis, Hepatitis, Ankylosing Spondylitis, Psoriasis, EAE (experimental autoimmune encephalomyelitis) and, CIA (collagen-induced arthritis. The basic idea is to induce signals originating from these receptors that turn off deleterious immune responses and again, regain immune tolerance that is lost in autoimmune diseases. The T cell would be the cellular target of the agonist checkpoint to exert autoimmunity immunosuppression. The matter is not as simple as it seems. When a checkpoint agonist acts, it depends not only on receptor binding but also on the ability to deliver a signal. For example, the action with TNFR agonists (which acts at the CD40 receptor), depends in part on the receptor for the Fc fraction (crystallizable fraction of the agonist antibody) to bind to the lymphocyte scaffolding (FCjRIIB) [61]. Agonism can be improved by using a human IgG2 isoform that makes the structure more compact and rigid, making aggregation more efficient. And the quality of the signal depends on the epitope on which it acts (antigenic determinant or part of a macromolecule that is recognized by the immune system. Specifically, the sequence to which the antibodies that are the receptors of B or T cells in soluble state bind). Work is being done on checkpoint agonism with viral proteins that improve receptor stimulation. The promising results have been demonstrated mainly in mice and the therapeutic use in human autoimmune diseases is still uncertain [62].
T-cell transfer therapy (TCTT)
It is an immunotherapy that allows the patient's own immune cells to attack their cancer more and better. There are two types: TIL (tumor infiltrating lymphocytes) therapy and CAR-T (chimeric antigen receptors-T cell) therapy. Both consist of collecting the patient's own immune cells, growing them in the laboratory and returning them to the patient intravenously. Synonyms used for TCTT are: Immune cell therapy and, Adoptive immunotherapy. The process of growing them takes 2-8 weeks and in the meantime the patient may be receiving chemotherapy and/or radiotherapy to eliminate TME cells, which helps the transfused T cells to be more effective. In TIL, the T cells are selected (separating them from the other extracted cells) that infiltrate the tumor. This selection is made in the laboratories by means of markers. These cells are the ones that "know" the tumor cells best, which is why they are grown in large numbers and quickly in the laboratory. The basic idea of this approach is that the T lymphocytes that are inside the area of the tumor, surrounding it, are the ones with the greatest ability to recognize and inactivate tumor cells. In the patient "in vivo" there may not be enough of these lymphocytes to inactivate the tumor cells or block the signals emanating from them to block immunity. Infusing large numbers of these lymphocytes can help the patient's immune system overcome these barriers. In CAR-T, the T cells extracted from the patient, in the laboratory, undergo a process so that they can generate an artificial receptor produced by genetic engineering (CAR), that is, a protein (encoded by a gene that is introduced [transduced] into the cell’s genome), then said lymphocytes are injected into the patient and that receptor will recognize antigens of the surface of neoplastic cells allowing it to attack said cells. It is called chimeric because it is made up of different parts of antibodies and because it combines the specificity of an antibody (the extracellular part) with T cell-activating function of a T-cell receptor (the intracellular part) (63). This type of immunotherapy could be used in patients, for example with metastatic breast cancer, where other therapeutic options have failed. Neoantigen T-Cell Receptor Gene Therapy uses autologous T cells that had been genetically engineered to clonally express two allogeneic HLA-C*08:02–restricted T-cell receptors (TCRs) targeting mutant KRAS G12D (K-ras oncogene. The resultant protein acts as an “on-off” switch that instructs cells to grow and divide or mature and take on different functions) expressed by the tumors. Recently, this strategy has been used to treat metastatic pancreatic and colorectal cancer.
TCTT is still experimental. It has been used in metastatic melanoma, squamous carcinoma of the cervix, cholangiocarcinoma. In these last two fundamentally TIL. Six CART-T options have been approved by the FDA for hematologic malignancies. In solid tumor (breast and brain) it is still experimental. The increase in cytokines released by immune cells explains secondary effects such as fever, nausea, headache, rash, tachycardia, hypotension, dyspnea, and if very severe, it can cause capillary leak syndrome and cytokine storm (T-cell Transfer Therapy was originally published by the National Cancer Institute. Updated: April 1, 2022).
Epigenetic therapy
Epigenetics is the process by which mediating heritable patterns of gene expression are established without changing the sequence of DNA. Epigenetics can thus be viewed as a virtual ‘software package’ to control and utilize the information coded in the ‘hard drive’ of DNA. Epigenetic therapy modulates key regulatory features of both immune cells and tumor cells in ways that might overcome some of the current limitations of immunotherapy. For example, epigenetic drugs have the potential to reverse many processes that tumors engage to evade immune-mediated destruction. Epigenetic therapy has the potential to convert a tumor from an immune repressive (immune cold) to an immune permissive (immune hot) state through effects on several factors of the tumor microenvironment that normally impede the therapeutic activity of immune-checkpoint inhibition. Immune cold tumors are characterized by the absence of tumor-infiltrating lymphocytes, the presence of immunosuppressive cell populations, such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), and/or a lack of expression of programmed cell death 1 ligand 1 (PD-L1) by the tumor cells. Epigenetic agents can modulate the immune composition of the TME by decreasing the abundance of TAMs and MDSCs and increasing the numbers of CD8+ effector T cells and memory T cells. As well as having the potential to shift the differentiation of CD8+ tumor-infiltrating lymphocytes towards effector and/or memory phenotypes, epigenetic drugs can augment innate immune-related signaling and the expression of inflammatory proteins, such as chemokines, which aid the recruitment of T cells to the tumor. In addition, epigenetic therapy can revert key aspects of cancer immunoediting via increased expression of tumor antigens, such as cancer/testis antigens (CTAs), and restoration of the MHC class I (MHC I) antigen processing and presentation machinery (which is often dysregulated in tumor cells) thus potentiating the immune recognition of tumors. Obviously, this is an exciting field of molecular biology that is under intense scrutiny and that may generate combined therapy with ICIs (64).
Cancer, ICIs, and COVID-19
Age and receipt of immune-checkpoint inhibitor treatment remained significantly associated with COVID severity in a multivariate analysis of 563 patients with cancer [65]. But it is not clear whether cancer is an independent risk factor for severe COVID-19. Aging, obesity, metabolic syndrome, and exposure to carcinogens are predisposing factors for cancer. Aging, obesity, and metabolic syndrome also represent comorbidities that influence susceptibility to and severity of SARS-CoV-2 infection. In patients with cancer, metastatic dissemination and poor ECOG performance status also favor COVID severity. Immunosenescence and inflammaging, which are also promoted by aging and obesity, result in declining functions of the innate and adaptive immune systems, exacerbating overt inflammation and cancer dissemination and increasing vulnerability to SARS-CoV-2 infection and risk of severe COVID-19 [66]. Lymphopenia often accompanies cancer diagnosis, treatment or progression and is a side effect of chemotherapy and steroids. Radiotherapy also negatively impacts circulating lymphocyte counts. An increased number of circulating neutrophils is often combined with decreased lymphocyte counts, resulting in a marked elevation of the neutrophil-to-lymphocyte ratio. A high neutrophil-to-lymphocyte ratio is a poor prognostic marker and predicts short cancer-specific progression-free survival after blockade of programmed cell death protein 1 (PD-1), as well as severe COVID-19 [67]. Given the critical role of T effector lymphocytes in eliminating virus-infected cells, an attenuated and functionally compromised T cell pool may pave the way toward the higher incidence and severity of COVID-19 in patients with cancer. The failure of the immune system to control early viral replication and to prevent endothelial injury may lead to a marked release of chemokines, cytokines and/or alarmins and a viral sepsis initiated or maintained by pulmonary or medullary hematopoiesis. Altogether, these results imply that cancer and COVID-19 may be concomitantly aggravated by comorbidities such as aging, metabolic disorders, and innate and cognate immunosuppression. These comorbidities may also compromise the efficacy of immune-based anticancer and antiviral therapies.
An association of checkpoint inhibitor–based immunotherapy with the aggravation of COVID-19, including increased hospitalization and severe respiratory conditions, was first reported in 31 patients [68]. This negative prognostic link was independent of age, cancer type and other comorbid conditions or co-administered medications such as steroids. In this case, immune-checkpoint inhibitors may have exacerbated immune-related pneumonitis or T cell cytokine release. Given that many therapeutic actions currently used in oncology may increase the risk of severe SARS-CoV-2 infection, current guidelines related to cancer care during the COVID-19 crisis advise the postponement of all non-mandatory cancer therapies [69]. Despite their specificity, however, small-molecule inhibitors and antibody-based therapies induce both on- and off-target effects—the latter including immune-related pneumonitis and diabetes, among other conditions—that could increase the susceptibility of patients with cancer to COVID-19. Side effects of ICIs such as hypertension, cardiomyopathy, heart failure, cytokine storm, senescence, pulmonary fibrosis, and pneumonitis favor the appearance of Covid-19.
Conclusions
With a greater understanding of the molecular biology and immune response in cancer, we have moved from a "shotgun" approach to chemotherapy to a more targeted treatment paradigm with immunotherapy [70]. In some tumors (NSCLC), up to 75% of their genomic alterations are amenable to targeted therapy. These patients have a different outlook, better outcomes, and better quality of life, but subgroups of patients who show resistance to ICIs persist. To this is added the appearance of irAEs that limit the application of the treatment.
Methods of patient selection, combined therapies to increase efficacy, predictive factors of efficacy in immunotherapy must also be defined. Another problem with ICIs is that there are no comparative studies between different ICIs, "face to face", and the rapid expansion of production has been messy. These products are not biosimilars and cannot rely on prior approval of other monoclonal antibodies. They are expensive. Many sponsors have rushed to license and develop these antibodies in unapproved combination regimens. Multiple "in vitro" tests for patient selection is evidence of incoordination since there is no consensus that these tests identify the same groups of patients. Therefore, greater standardization between products and indications is required.
Anti-PD-1 and anti-PD-L1 have low response rates and randomized trials designed to verify benefit have shown inconsistent results. Initial ORR with ICIs does not predict long-term results. The FDA has advised sponsors developing new ICIs not to conduct trials of a single group of patients with refractory disease to approve the drug. There has been unbridled growth with a stampede of sponsors. Beaver calls it "The wild west of checkpoint inhibitor development" (40). With the ORDIS project, FDA intends to establish cooperation among themselves on this issue, but commercial interests are likely to prevail over cooperation.
Financial support
None.
Conflict of interest
None.
Authorship
This work was only carried out by the author. Author AA contributed on the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
References
- Parkin J, Cohen B (2001) An overview of the immune system. Lancet 357: 1777-1789. [Crossref]
- Delves PJ, Roitt IM (2000) The immune system. First of two parts. N Engl J Med 343: 37-49. [Crossref]
- Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptative immune responses. Cell Res 20: 34-50.
- Mak TW, Saunders ME (2004) Innate immunity. The immune response: basic and clinical principles. Amsterdam: Elsevier Academy Press. pp: 79-92.
- Chen L, Klies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibitors. Nat Rev Immunol 13: 227-242. [Crossref]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, et al (2002) Projection of an immunological self-shadow within the thymus by the AIRE protein. Science 298: 1395–1401. [Crossref]
- Derbinski J, Schulte A, Kyewski B, Klein L (2001) Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol 2: 1032–1039. [Crossref]
- Richards DM, Kyewski B, Feuerer M (2016) Re-examining the nature and function of self-reactive T cells. Trends Immunol 37: 114–125. [Crossref]
- Chen L, Flies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13: 227-242. [Crossref]
- Schnell A, Bod L, Madi A, Kuchroo VK (2020) The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res 30: 285-299. [Crossref]
- Xiu J (2020) Regulation of cancer immune checkpoints. Molecular and cellular mechanisms and therapy. (1st Ed) Springer Nature Switzerland AG.
- Alvarado A (2020) Programmed cell death. Review and its impact in Covid-19. Clin Res Trials 7: 1-11.
- Yang G, Elbadawi M, Efferth T (2020) Multiple cell death modalities and their key features (Review) World Acad Sci J 2: 39-48.
- Onoi K, Chihara Y, Uchino J, Shimamoto T, Marimioto Y, et al (2020) Immune checkpoint inhibitors for lung cancer treatment. A review. J Clin Med 9: 1362. [Crossref]
- Ledford H, Else H, Warren M (2018) Cancer immunologists coop medicine Nobel prize. Nature 562: 20-21. [Crossref]
- Tundo GR, Sbardella D, Lacal PM, Graziani G, Marini S (2019) On the horizon: targeting-next-generation immune checkpoints for cancer treatment. Chemotherapy 64: 62-80. [Crossref]
- Dardalhon V, Korn T, Kuchroo VK, Anderson AC (2008) Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun 31: 252-256. [Crossref]
- McKinney EF, Smith KGT (2016) T cell exhaustion and immune mediated disease-the potential for therapeutic exhaustion. Curr Opin Immunol 43: 74-80. [Crossref]
- Paluch C, Santos AM; Anzilotti C, Cornall RJ, Davis SJ (2018) Immune checkpoints as therapeutic targets in autoimmunity. Front Immunol 9: 2306. [Crossref]
- Dixon KO, Michelle S, James N, Yassaman E, Zohreh A, et al (2018) Functional anti-TIGITI antibodies regulate development of autoimmunity. J Immunol 200: 3000-3007. [Crossref]
- Wherry EJ, Ha S-J, Kaech SM, Blattman JN; Barber DL, et al (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27: 670-684. [Crossref]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczk MS, et al (2016) A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 116: 1500-1511. [Crossref]
- Huang PY, Guo SS, Zhang Y, Lu JB, Chen QY, et al. (2016) Tumor CTLA-4 over expression predicts poor survival in patients with nasopharyngeal carcinoma. Oncotarget 7: 13060-13068. [Crossref]
- Juneja VR, McGuire KA, Manguso RT, Lafleur MW, Collins N, et al. (2017) PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8+ T cell cytotoxicity. J Exp Med 214: 895-904. [Crossref]
- He Y, Ca OJ, Li X, Zhou C, Hinsch FR (2018) TIM-3, a promising target for cancer immunotherapy. Onco Target Ther 11: 7005-7009. [Crossref]
- Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, et al (2022) Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med 386: 1973-1985. [Crossref]
- Johnson DB, Nebhan CA, Moslehi JJ, Balko JM (2022) Immune checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol 19: 254-267. [Crossref]
- Wolchhok JD, Chiarion-Sileni V, González R, Rutköwski P, Grob JJ, et al (2017) Overall survival with combined nivolumab and ipilumimab in advanced melanoma. N Engl J Med 337: 1345-1356. [Crossref]
- Owen CN, Bai X, Quah T, Lo SN, Allayous C (2021) Delayed immune-related adverse events with anti-PD-1-based immunotherapy. Ann Oncol 32: 917-925. [Crossref]
- Wing JR, Kimball A, Rengarajan M (2022) Case 6-2022. A 68-year-old man with fatigue, weight, and hyperglycemia. N Engl J Med 368: 781-787. [Crossref]
- Mur CA, Clifton-Bligh RJ, Long GV, Scolyer RA, Lo SN, et al (2021) Thyroid immune-related adverse events following immune checkpoint inhibitor treatment. J Clin Endocrinol Metab 106: e3704-e3713. [Crossref]
- Filote JMK, Pen JJ, Decoster L, Vissers T, Bravenboer B, et al (2019) Immune checkpoint inhibitors and type 1 diabetes mellitus: a case report and systematic review. Eur J Endocrinol 181:363-374. [Crossref]
- Tsang VHM, McGrah RT, Clifton-Bligh R, Scolyer R, Jakrot V, et al (2019) Checkpoint inhibitor-associate autoimmune diabetes is distinct from type 1 diabetes. J Clin Endocrinol Metab 104: 5499-5506. [Crossref]
- Haslam A, Gill J, Prasad V (2020) Estimation of the percentage of US ´patients with cancer who are eligible for immune checkpoint inhibitor drugs. JAMA Netw Open 3: e200423-e200423.
- Robert C (2020) A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun 11: 3801-3801.
- Tang H, Zhou J, Bai C (2021) The efficacy and safety of immune checkpoint inhibitors in patients with cancer and preexisting autoimmune disease. Front Oncol 11: 625872. [Crossref]
- Alexander S, Swami V, Kaur A, Fatima M, Ginn MM, et al (2021) Safety of immune checkpoint inhibitors in patients with cancer and preexisting autoimmune disease. Ann Transl Med 9: 1033. [Crossref]
- Xin YJ, Hubbard-Lucy VM, Tang J (2019) Immuno-oncology drug development goes global. Nar Rev Drug Discov 18: 893-900. [Crossref]
- Robert C (2020) A decade of immune-checkpoint inhibitors in cancer therapy. Nat Communicat 11: 3801.
- Beaver JA, Pazdur R (2021) The wild west of checkpoint inhibitor development. N Engl J Med 386: 1297-1301. [Crossref]
- Paudel KR, Panth N, Pangeni R, Awasthi R, Chawla V, et al (2020) Targeting lung cancer using advanced drugs delivery system. In: Targeting chronic Inflammatory lung disease using advance drug delivery system. Elsevier-Amsterdam, The Netherlands pp: 493-516.
- Goldstraw P, Chausky K, Crowley J, Mitchel A, Bolejac KV, et al (2016) The IASLC lung cancer staging project: proposal for revision of the TNM stage grouping in the forthcoming (Eighth) edition of TNM classification for lung cancer. J Thorac Oncol 11: 39-51. [Crossref]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjoataraw I, et al (2021) Global cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca Cancer J Clin 71: 209-249. [Crossref]
- Louly CM (2022) Expanding horizons for treatment of early cancer. N Engl J Med 386: 2050-2051. [Crossref]
- Pakkala S, Ramalingtam SS (2018) Personalized therapy for lung cancer: striking a moving target. JCI Insight 3: e120858. [Crossref]
- Lim SM, Kim HR, Lee JS, Lee KH, Lee YG, et al (2017) Open-label, multicenter, phase II study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol 35: 2613-2618. [Crossref]
- Wu YL, Lim DW, Felip E, Zhang L, Liu X, et al (2016) Phase (Ph) II safety and efficacy results of a single-arm ph ib/II study of capmatinib (INC280) + gefitinib in patients (pts) with EGFR-mutated (mut), cMET-positive (cMET+) non-small cell lung cancer (NSCLC) J Clin Oncol 34(15-suppl): 9020. [Crossref]
- Mayo-de-Las-Casas C, Garzón-Ibáñez M, Jordana-Ariza N, García-Peláez B, Balada-Bel A, et al (2018) An update on liquid biopsy analysis for diagnostic and monitoring applications in non-small cell lung cancer. Expert Rev Mol Diagn18: 35-45.
- Reck M, Rodríguez-Abrev D, Robinson AG, Hui R, Csöszi T, et al (2016) Pembrolizumab versus chemotherapy for PD-1 in positive non-small-cell lung cancer. N Engl J Med 375: 1823-1833. [Crossref]
- Wang Y, Ellis P (2017) EGFR mutation positive non-small cell lung cancer: can we identify predictors of benefit from immune checkpoint inhibitors? Ann Transl Med 5:424. [Crossref]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, et al (2017) T cell stimulatory target for PD-1-mediated inhibition. Science 355: 1428-1437. [Crossref]
- Antonia SJ, Villegas A, Daniel D, Vicente D, Hui R, et al (2018) Overall survival with Durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med 379:2342-2350.
- Getinger S, Borghaei H, Brahmer J, Chow L, Burgio M, et al. (2019) Five-year outcomes from the randomized phase 3 trial check mate 017/057: Nivolumab vs Docetaxel in previously treated NSCLC. J Thorac Oncol. 14:244-245. [Crossref]
- Marin-Acevedo JF, Kimbrough EO, Lou Y (2021) Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol 14: 45. [Crossref]
- Johnson DB, Sullivan RJ, Menzies AM (2017) Immune checkpoint inhibitors in challenging populations. Cancer 123: 1904-1911. [Crossref]
- Wilkinson TMA (2017) Immune checkpoint in chronic obstructive pulmonary disease. Eur Respir Rev 26: 170045. [Crossref]
- Mark NM, Karg IJ, Busch SE, Yang GHY, Meta HE, et al (2018) Chronic obstructive pulmonary disease alters immune cell composition and immune checkpoint inhibitors efficacy in Non-Small Cell Lung Cancer. Am J Respir Crit Care Med 197: 325-336. [Crossref]
- Prüss H (2021) Autoantibodies in neurological disease. Nat Rev Immunol 21: 798-813. [Crossref]
- Rubin SJS, Bloom MS, Robinson W (2029) B cell checkpoint in autoimmune rheumatic disease. Biol Med Nat Rev Rheumatol 15: 303-315. [Crossref]
- Faje A. (2016) Immunotherapy and hypophysitis: clinical presentation, treatment, and biologic insights. Pituitary 19: 82–92. [Crossref]
- Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, et al (2017) Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 31: 383-395. [Crossref]
- McKinney EF, Smith KGT (2016) T ell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr Opin Immunol 43: 74-80. [Crossref]
- Bluestone JA, Anderson M (2020) Tolerance in the age of immunotherapy. N Engl J Med 383:1156-1166. [Crossref]
- Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB (2020) The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol 17: 75-90. [Crossref]
- Robilotti, E. V. Babady NE, Mead PA, Rolling T, Perez-Johnston R. et al (2020) Determinants of COVID-19 disease severity in patients with cancer. Nat Med 26: 1218–1223. [Crossref]
- Derosa L, Melenotte C, Griscelli F, Gachot B, Marabelle A, et al (2020) The immuno-oncological challenge of COVID-19. Nat Cancer 1: 946-964. [Crossref]
- Zhang B, Zhou X, Zhu C, Song Y, Feng F, et al (2020) Immune phenotyping based on the neutrophil-to-lymphocyte ratio and IgG level predicts disease severity and outcome for patients with COVID-19. Front Mol Biosci 7: 157.
- Vardhana S A, Wolchok J D (2020) The many faces of the anti-COVID immune response. J Exp Med 217: e20200678.
- Routy B, Derosa L, Zitvogel L, Kroemer G (2020) COVID-19: a challenge for oncology services. Oncoimmunology 9: 1760686. [Crossref]
- Phimister EG, Rubin EJ (2022) Targeting Cytotoxic T Cells to Tumor. N Engl J Med 386: 2145-2148. [Crossref]