Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, irreversible form of interstitial lung disease of unknown cause. Although IPF is a rare disease, it is not a rare cause of death, and the incidence of IPF is increasing. Early diagnosis of IPF is crucial to starting treatment in a timely manner to slow disease progression. The first step in the diagnosis of IPF is the exclusion of the possible causes of interstitial lung disease (ILD). Crucial tools for the diagnosis of IPF are high-resolution computed tomography (HRCT), pulmonary function tests -- mainly used to assess disease severity -- and histopathology. Recent guidelines list HRCT and histopathology patterns of fibrosis that should be considered when assessing a patient suspected of having IPF, these include: usual interstitial pneumonia (UIP), probable UIP, indeterminate for UIP, and alternative diagnosis. Guidelines also recommend that a multidisciplinary discussion (MDD); including the clinician, radiologist, and pathologist) should take place after HRCT and before bronchioalveolar lavage (BAL) and/or surgical lung biopsy is performed. Additional MDD is recommended after BAL/surgical lung biopsy in those patients undergoing BAL/surgical lung biopsy. The multidisciplinary team is important in establishing a working diagnosis of IPF in some patients; it’s key as it has implications for treatment management. Another significant fact is the availability of antifibrotic, antiproliferative, and monoclonal antibody treatment that significantly delays the decline in lung function, in a pathology in which the only previous viable option was lung transplantation. Therapeutic advances, including individualized care and interventions that reduce the deposition of extracellular matrix proteins (ECM) in the lung, could turn this pathology into a long-lasting chronic disease.
Idiopathic pulmonary fibrosis, High-resolution computed tomography, Pulmonary function test, Multidisciplinary discussion, Interstitial lung disease, Bronchiolaveolar lavage
IPF: Idiopathic pulmonary fibrosis; HRCT: High-resolution computed tomography; PFT: Pulmonary function test; MDD: Multidisciplinary discussion; ILD: Interstitial lung disease; BAL: Bronchiolaveolar lavage
IPF is a serious, relatively rare lung disease of unknown etiology, characterized by irreversible, progressive fibrosis of the lungs (Figure 1). This leads of a worsening of lung function, progressing to respiratory failure and ultimately death [1]. Main symptoms include unexplained exertional dyspnea, chronic dry cough, inspiratory Velcro-like crackles, and finger clubbing. Exertional dyspnea typically progresses over a period of months to years. In practice, patients with ILD often initially receive a diagnosis of heart failure or chronic obstructive lung disease (COPD), suggesting that clinicians frequently fail to consider ILD in patients with dyspnea. Nearly two-thirds of patients are smokers or ex-smokers. IPF has a poor prognosis. The average survival is 50% at 3 years and 20% at 5 years. The prevalence in Europe is 50 / 100,000 inhabitants, in the USA 10-60 / 100,000 inhabitants and in Canada 115 / 100,000 inhabitants. However a recent study in Sweden showed a stable incidence, but an increase in prevalence [2]. The older age (>60 years) and male sex were associated with a higher incidence of disease [3]. The annual cost per capita in the US is $20,000 and is also increasing. Patients with IPF have a much lower health related-quality of life (HR-QoL) than the general population, and they have marked deterioration in HR-QoL in the last 2 years of life [4]. Dyspnea, cough, depression and anxiety are important drivers of QoL in IPF [5]. There are several evolutionary patterns. A subgroup evolves slowly in the loss of lung function and it is those that have a greater survival at 5 years. Others have a progressive and rapid deterioration, with a one-year survival, and another group presents, after the first year of survival, an acute exacerbation, with an average survival of three years [6]. Acute exacerbation leads to a mortality rate up to 80% to 90% [7]. However, for a patient given the progression of IPF is unpredictable [8]. IPF prognosis is worse than for many cancers [9] and in the United States, IPF kills 40.000 patients per year-similar to breast cancer [10]. This monograph aims to give the clinician a simple tool to approach the diagnosis and treatment of IPF.
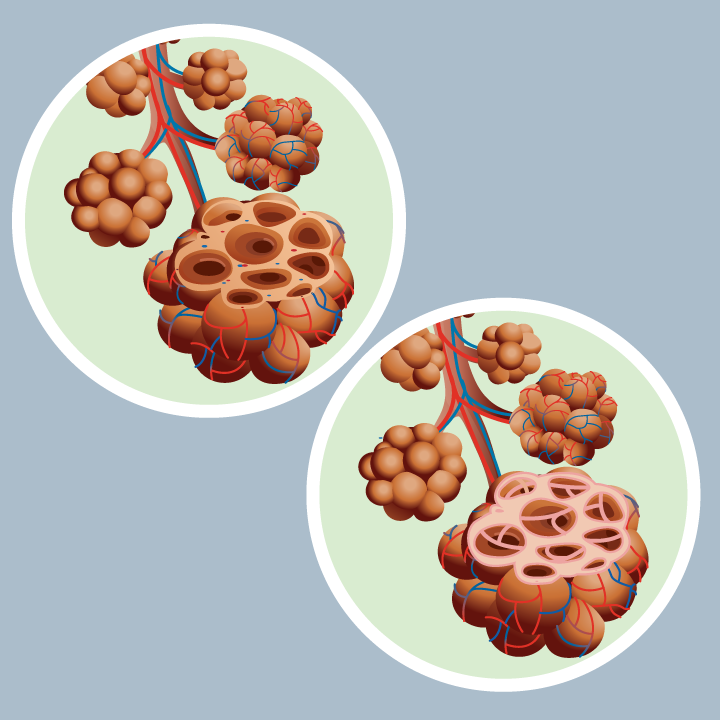
Figure 1. Interstitial Pulmonary fibrosis
See the left lung and normal alveoli. On the right see alveoli in pulmonary fibrosis. Stand out irregular thickening of tissue between alveoli; irregular, abnormal air spaces and large areas of scarring.
Consequences of missing a diagnosis of IPF are that burdensome symptoms and lung function worsening due to progressive nature of the disease, negative impact on quality of life, risk of acute exacerbation and increased mortality [11-14]. Early diagnosis of IPF is important to improve long-term treatment outcomes. Disease-modifying therapies such as nintedanib and pirfenidone can approximately halve the relative decline in FVC (forced vital capacity) vs placebo, therefore early diagnosis is vital [15]. Future considerations for identifying early IPF are more disease awareness, more active screening (currently difficult to identify clinically interstitial abnormalities) and identification of patients with minimal symptoms or non-symptomatic. Communication with patients is key to set expectations that lung function will decline less with therapy, but will not stop.
Diagnosis of IPF starts with excluding other forms of ILDs. ILDs are a large and heterogeneous group of more than 200 rare parenchymal lung disorders with overlapping clinical present and pattern of lung injury [16,17]. The simplest way to classify them into idiopathic interstitial pneumonias (IIPs) and ILDs related to know causes. The IIPs are currently made up of 9 identifiable groups as: IPF, idiopathic nonspecific interstitial pneumonia (NSIP), respiratory bronchiolitis-interstitial lung disease (RB-ILD), desquamative interstitial pneumonia (DIP), cryptogenic organizing pneumonia (COP), acute interstitial pneumonia (AIP), idiopathic lymphoid interstitial pneumonia, idiopathic pleuroparenchymal fibroelastosis and unclassificable IPPs [18].. The ILDs related to know causes include: ILDs with autoimmune features (rheumatoid arthritis, Sjögren’s syndrome, systemic lupus erythematous, polymyositis and dermatomyositis, mixed connective tissue disease [MCTD], systemic sclerosis, and other connective tissue diseases), hypersensitive pneumonitis (HP), sarcoidosis and other ILDs (lymphangioleiomyomatosis, Langerhans’ cell histiocytosis), drug-associated ILD, ILDs related to other occupational exposures, vasculitis/granulomatosis and other rare ILDs). However, there are data for an ILD that can facilitate diagnostic work such as searching signs of systemic autoimmune disease and other organ involvement, exposure to antigens (to rule out hypersensitivity pneumonitis), sign of occupational or environmental lung disease, and genetic components: any family history or evidence of premature graying, cryptogenic cirrhosis, aplastic anemia, macrocytosis, thrombocytopenia. ILDs related to know causes are more common in younger and middle-age adults The majority of patients with interstitial fibrosis ultimately receive a diagnosis of chronic hypersensitivity pneumonitis (an ILD characterized by peribronchiolar and interstitial chronic inflammatory infiltrates and interstitial fibrosis with or without poorly formed granulomas due to dampness, mold or bird exposure), pulmonary sarcoidosis, an underlying autoimmune disease, drugs, environmental exposure (e.g., silica or asbestos), or if no cause is identifies, IIP (e.g., IPF). A diagnostic, necessarily requires an excellent clinical history and a good semiology and not get lost in a sea of laboratory tests ex-officio (antinuclear antibodies, rheumatoid factor, anti-cyclic citrullinated peptide antibodies, antibodies against Scl-70, Ro, La, UI-RNP, and Jo-1; creatine-kinase, myoglobin, and antisynthetase antibodies). They should be guided sequentially by clinical evaluation [19]. Many ILDs are associated with a risk of developing a progressive fibrosis phenotype. Signs and symptoms of progressive fibrosing ILD includes worsening symptoms (cough, dyspnea), deterioration of HR-QoL, increased fibrosis, (HRCT), decline on lung function (FVC and gas exchange), and acute respiratory exacerbation [17]. Commons characteristics of progressive fibrosing ILDs are common pathogenic mechanism, self-sustaining fibrosis, progression despite treatment and early mortality. Management of the fibrosis phase is the similar, regardless of etiology [18].
In the pathogenesis of PF-ILD (different interstitial lung diseases associated with pulmonary fibrosis) the triggers may vary (Figure 2). Alveolar epithelial injury and fibroblastic proliferation leading to fibro-proliferative disorders (IPF), pathogenic shift to a fibro-proliferative pathway leading to inflammatory disorders and variables degree of inflammation causing scarring and fibrosis (others ILDs). In IPF a conceptual model of pathogenesis is that the recurrent subclinical epithelial injury is superimposed on an accelerated epithelial aging, which leads to an aberrant repair of the injured alveoli and deposition of ECM by myofibroblasts. Apparently senescence of epithelial cells is a central phenotype that favors pulmonary fibrosis [20]. Shortened telomeres, oxidative stress, proteostatic dysregulation (abnormalities in the synthesis, trafficking, folding, or turnover of intracellular proteins), endoplasmic reticulum stress, and mitochondrial dysfunction decrease alveolar cell proliferation and increase the secretion of profibrotic mediators [21,22]. Prevotella, Veillonella and Escherichia coli in BAL, and abundance of streptococcus and staphylococcus have been associated with an increased risk of disease progression [22-24]. A number of non-genetic risk factors for IPF have been identified. Older age (>50 years of age), male sex, and cigarette smoking are considered risk factors for IPF [25].
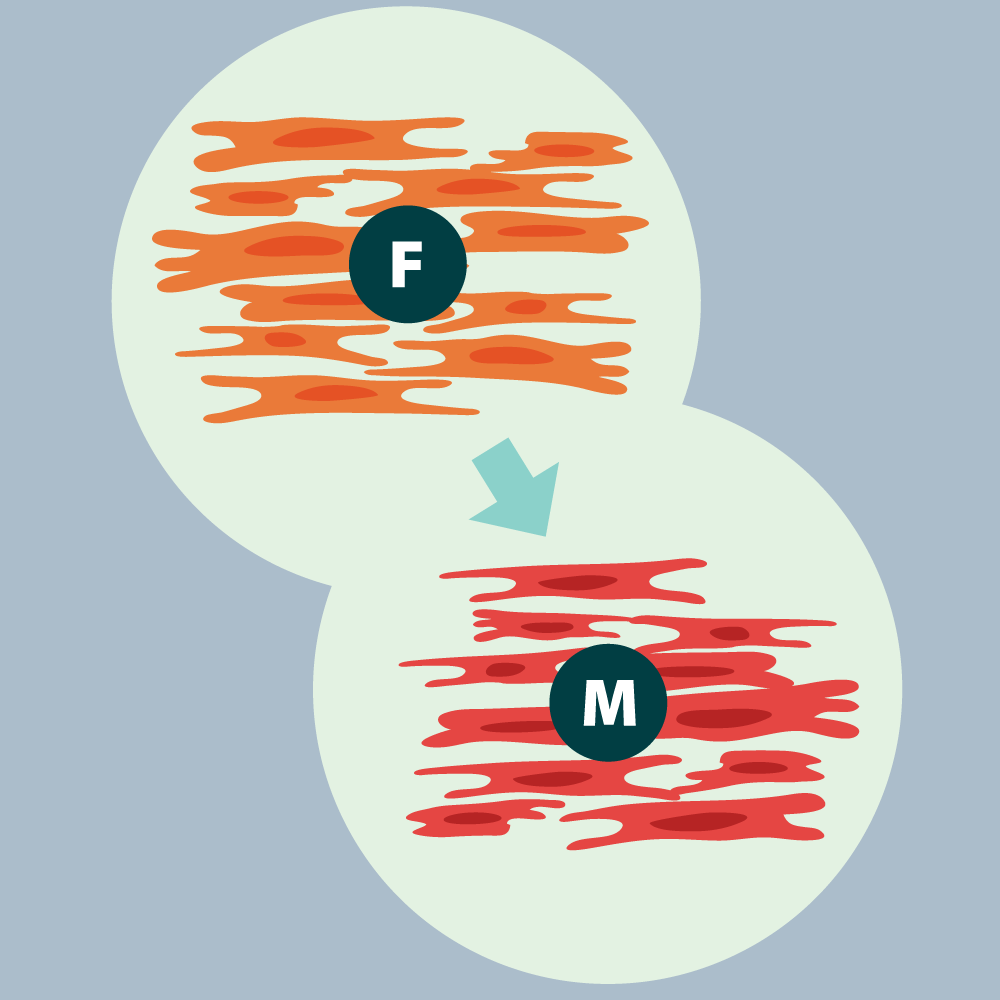
Figure 2. Pathogenesis of PF-ILD: Progressive Fibrosis
Fibroblasts clusters at site of injury and differentiate into myofibroblasts. Myofibroblasts secrete and release ECM that forms the fibrotic scar. Macrophages and lymphocytes (not shown on the figure) are recruited to the site and release profibrotic mediators and cytokines and contribute to increase inflammation. Stiffness lung tissue activates fibroblasts, creating a self-sustaining process.
The three key assessments for diagnosis of IPF are PFT, HRCT and histopathology. PFT help to evaluate the severity of impairment. The three typical functional findings are reduced DLCO, hypoxemia at rest or desaturation with exertion, and a normal (early stages of ILD or when emphysema is also present) or low FVC. Serial monitor FVC and DLCO every 3-6 months is recommended. Conventional radiography may be normal or show nonspecific changes in early disease. In more established disease, bilateral reticular infiltrates, hazy opacities, and reduced inspiratory lung volumes on chest radiography should prompt consideration of interstitial lung disease. In IPF, bilateral reticulation is often predominant in lower lung zones. HRCT is as a “gatekeeper” in initial patient evaluation (supine, inspiratory and expiratory decubitus and with thin reconstruction [<1.25mm], a moderately sharp reconstruction kernel, and small reconstruction field of view). Reconstruction kernel is the algorithm used to generate CT imagen. Higher (sharper) kernels provide better spatial resolution and are preferred for high-resolution CT imaging. The pattern of fibrosis observed with HRCT affects decision regarding to need for future, potentially invasive investigations. For example, with pattern of UIP (or probably UIP), guideline-based diagnosis can be made without further testing, such as surgical lung biopsy, in selected cases. But if indeterminate for UIP or alternative diagnosis is the pattern, further tests are needed for the diagnosis of IPF (e.g., surgical lung biopsy) [26] (Figure 3). The UIP and the probable UIP has many similarities. Both has subpleural and basal predominant, distribution often heterogeneous, peripheral traction bronchiectasis or bronchiolectasis and probable UIP may have mild ground-glass opacities (hazy areas of increased lung attenuation on CT imaging) (GGO), but the most important key distinction is the presence (UIP) or absence (probably UIP) of honeycombing (thick-walled, linear clusters of cysts on CT, which are often subpleural). Honeycombing represents lung fibrosis (26). >90% of confident first-choice diagnoses based on HRCT (definitive UIP) are accurate compared with histology (Figure 4). However, less than 50% of IPF patients present with a typical UIP pattern [27]. The indeterminate for UIP pattern is subpleural and basal predominant, subtle reticulation (short, irregular, linear opacities on CT). Reticulation typically represent fibrosis and may have GGO or distortion (“Early UIP pattern”), CT features and/or distribution of lung fibrosis that not suggest any specific etiology (“Truly indeterminate for UIP”) or fibrotic lung disease or distribution features on CT that is not typical for an IPF diagnosis, but not obvious enough to confidently confirm an alternative diagnosis (Figure 5). In the alternative diagnosis pattern the findings suggestive of another diagnosis including: CT features (cyst, nodules, predominant GGO, etc.) with predominant distribution peribronchovascular or perylimphatic or another findings as pleural plaques, dilated esophagus, etc. [26]. The new HRCT category created, probable UIP, is subpleural and basal predominat, with peripheral traction bronchiectasis and no honeycombing. It is highly predictive of IPF histology where there is a clinical suspicion of IPF, and expands the range of HRCT patterns that allow for a diagnosis of IPF, without surgical lung biopsy. The HRCT category removed, inconsistent with UIP, is replaced by two categories: indeterminate for UIP and alternative diagnosis. Many patterns previously considered “inconsistent with UIP”, are occasionally observed in atypical UIP cases and the presence of these patterns does not rule out IPF. Identifying these features can be difficult, and referral to a center that specializes in interstitial lung disease may help establish an accurate diagnosis without lung biopsy.
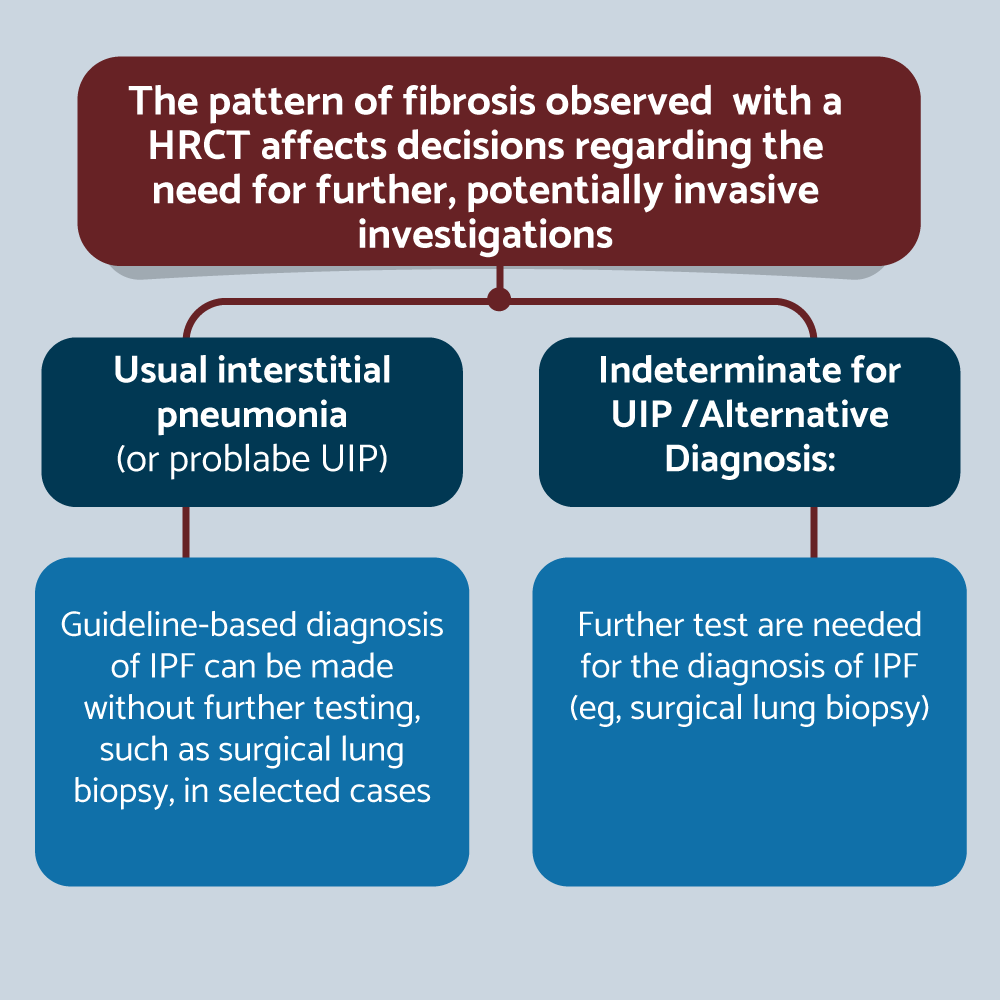
Figure 3. IPF diagnosis: HRCT as a “gatekeeper” in initial patient evaluation
Surgical decision based on radiological pattern.
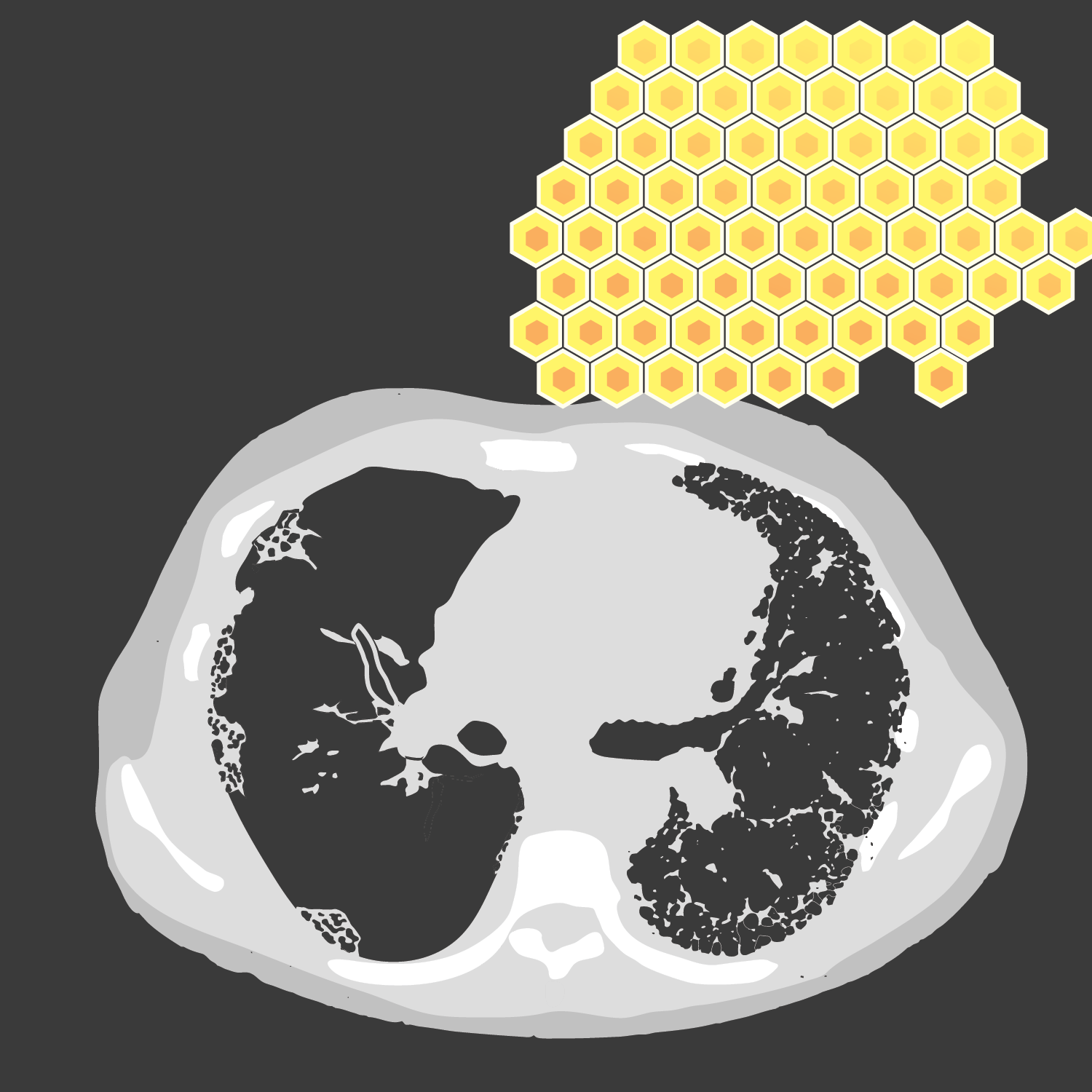
Figure 4. UIP pattern
CT images demonstrating areas of honeycombing, reticulation and subpleural predominance.
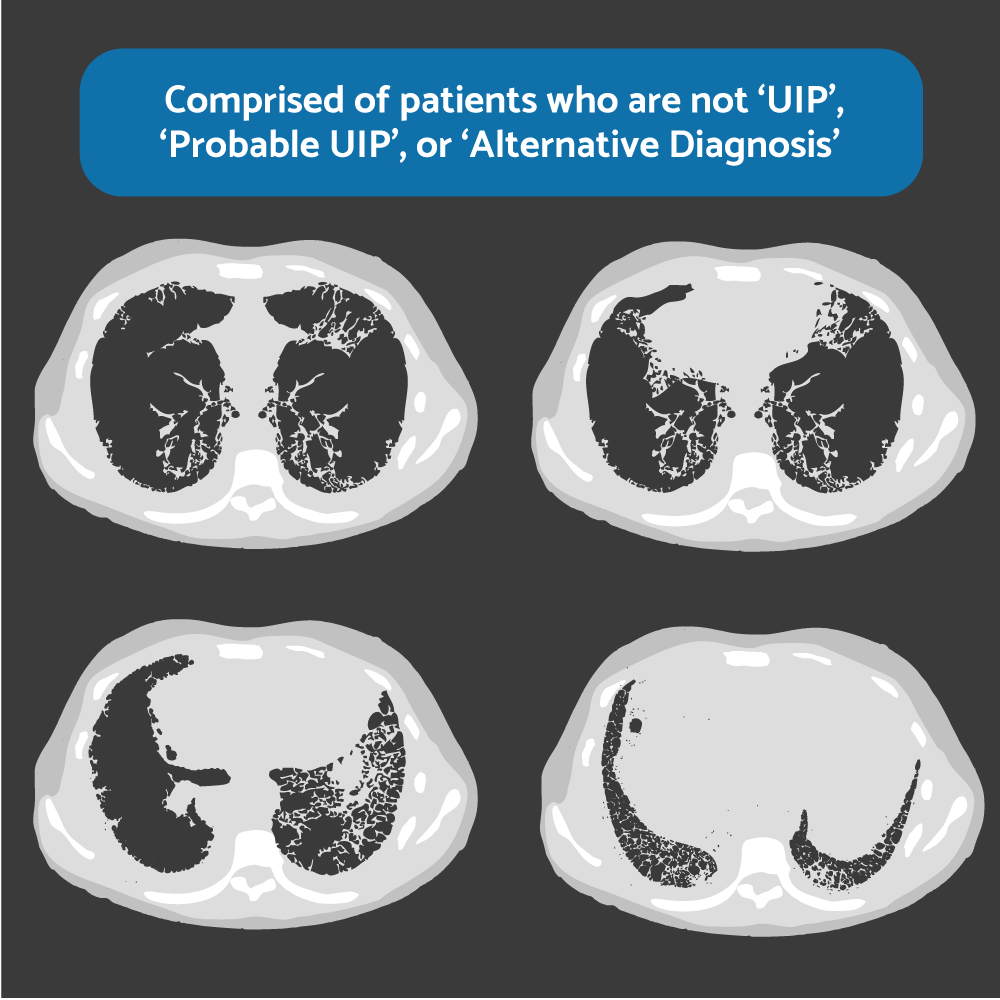
Figure 5. HRCT. “Indeterminate for UIP”
No honeycombing, no traction bronchiectasis, no features suggestive of alternative diagnosis (eg, CHP or fibrotic NSIP). It is an Indication for lung biopsy.
The decision to use histopathology (thoracoscopic lung biopsy) requires a MDD discussion including the patient, as the procedure carries some risks. In a patient with newly detected ILD or clinical suspicion of IPF, the HRCT is critical. If it´s UIP pattern histopathology (BAL, surgical biopsy) is not recommended, but if it´s probable UIP (some patients), indeterminate for UIP, or alternative diagnosis pattern, histopathology (BAL, surgical biopsy) is recommended (Figure 6). Factors that may preclude use of surgical lung biopsy include: advanced age, poor lung function, especially low DLCO level (<25% after correction for hematocrit) or severe hypoxemia at rest, presence of comorbidity (severe pulmonary hypertension), rapid progressing disease or frailty. If these factors allow for a surgical lung biopsy then bee should performed in multiples lobes to diagnose IPF in patients in whom HRCT scan is indeterminate [28]. Transbronchial lung cryobiopsy has poor concordance with surgical lung biopsy. For patients with newly detected ILD of unknow cause and clinical suspicion of IPF, guidelines do not recommended cryobiopsy if HRCT pattern is UIP. No recommendations made for patients with probable UIP, indeterminate for UIP, or alternative diagnosis [29,30]. Surgical biopsy remains the standard procedure for obtaining lung tissue to establish a diagnosis of ILD. Histopathologic patterns for IPF diagnosis have the same acronyms as the HRCT patterns. UIP has dense fibrosis with architecture distortion (ie, destructive scarring and/or honeycombing), predominant subpleural and/or paraseptal distribution of fibrosis, patchy involvement of lung parenchyma, fibroblast foci and, absence of features to suggest an alternative diagnosis. In probable UIP some histological features of UIP are present but to an extent that preclude a definitive diagnosis of UIP/IPF, and absence of features to suggest an alternative diagnosis. Indeterminate for UIP pattern has fibrosis with or without architecture distorsion, with features favoring either a pattern other than UIP or features favoring UIP secondary to another cause; or some histological features of UIP, but with other features suggesting an alternative diagnosis. Alternative diagnosis pattern has features of other histological patterns of IIPs (eg, absence of fibroblast foci or loose fibrosis) in all biopsies or histological findings indicative of other diseases (eg, hypersensitivity pneumonitis, Langerhans cell histiocytosis, sarcoidosis, LAM [lymphangioleiomyomatsis]). The concordance or discordant of the HRCT and histopathology patterns allow to “refine” the diagnostic work in IPF. If IPF is suspected, and the HRCT pattern is UIP; the histopathology patterns of UIP, probable UIP, and indeterminate for UIP strongly suggests IPF. Only alternative diagnosis does not suggest it. Unlike, if IPF is suspected, and the HRCT pattern is alternative diagnosis, the histopathology pattern UIP, maybe suggest IPF, but the other histopathology patterns suggest Non-IPF diagnosis. Ideally, the matching should be done by the MMD (Figure 7).
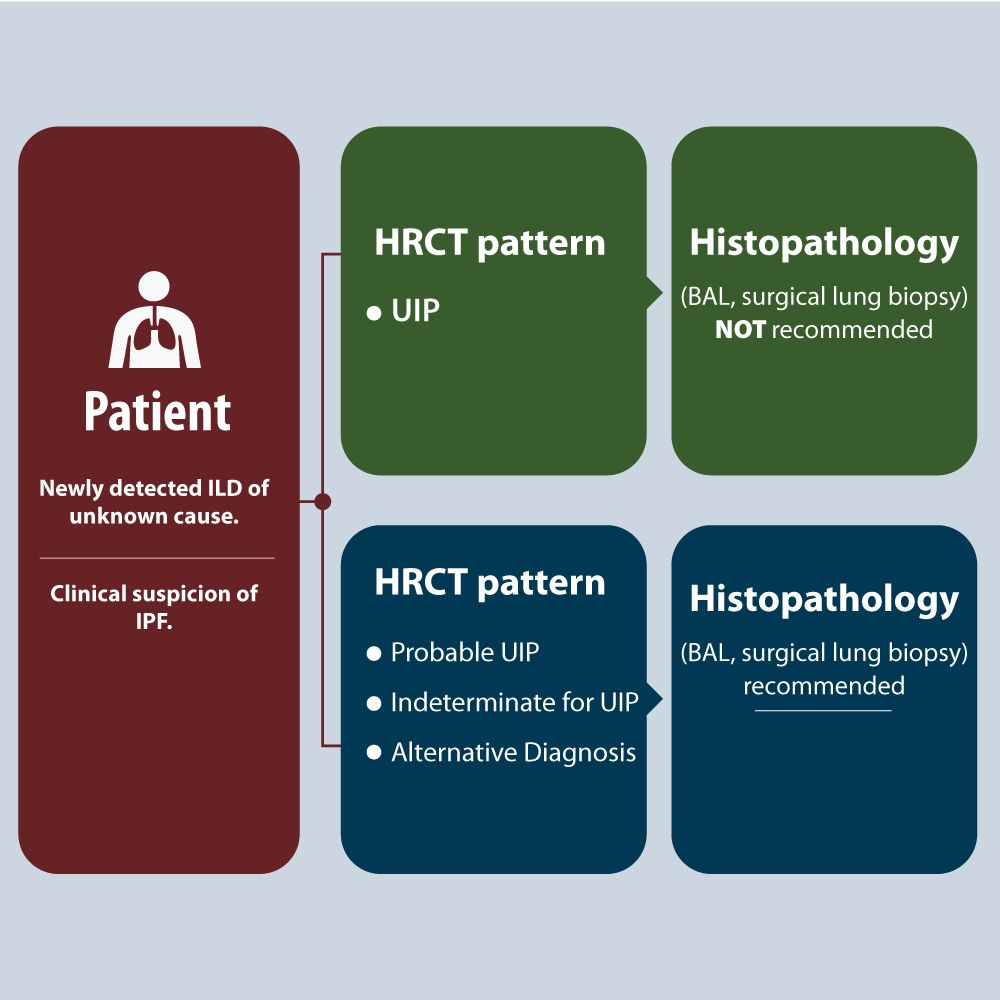
Figure 6. When is histopathology necessary?
The decision to use histopathology requires MDD discussion including the patient, as the procedure carries some risks.
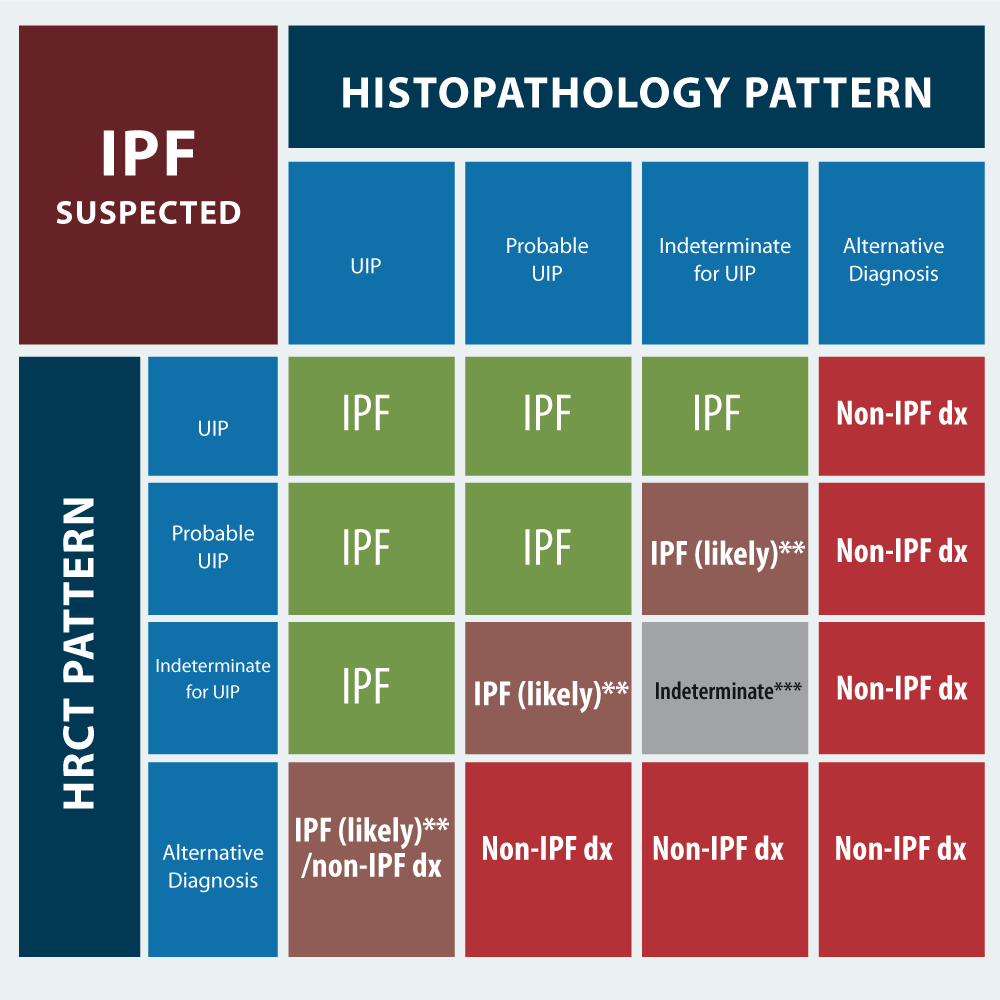
Figure 7. Diagnosis based in HCRT and histopathology patterns
If IPF is suspected, the combination of histological and HRCT patterns, makes it possible to predict the most likely diagnosis (IPF vs. Non-IPF).
There are key challenges in the diagnosis of IPF. Challenge 1 is excluder other causes of pulmonary fibrosis. Challenge 2 is adequately performing and assessing HRCT (used volumetric rather interspaced imaging). Patients who should have but are unable to undergo lung biopsy may be categorized as unclassifiable ILD or a multidisciplinary team may establish a working diagnosis of IPF [19]. Misdiagnosis and diagnosis retard delay poses a burden on patients and healthcare services [7]. Agreement among MDD teams is good and (non-significantly) better than the agreement among individual specialties [31]. Benefits of MDD are greatest when HRCT pattern is probable UIP, indeterminate for UIP, alternative diagnosis or, when, clinical, radiological, and/or histological data are discordant. Median agreement between single-discipline decision-making (SDD) and MDD was 70%. It means that for every 100 patients, SDD and MDD would agree on diagnosis in 70 patients and disagree in 30 patients. Thus, 30 patients could potentially undergo delayed or incorrect therapy or unnecessary diagnostic testing with SDD [26].
In probable UIP, indeterminate for UIP or alternative diagnosis in HRCT patterns, the MDD can choose BAL as an alternative to surgical biopsy particularly if patients unable or unwilling to undergo surgical lung biopsy. Almost every patients with suspected IPF should undergo BAL. BAL has low risk, is cost-effective, and can provide diagnostic information. The limitation is BAL should be performed and interpreted in centers with expertise. In BAL, neutrophil inflammatory pattern seems to be the most common profile in patients with IPF (>10%) [32]. Although BAL neutrophil levels may provide prognostic information in IPF, BAL neutrophilia is, in reality, highly nonspecific; it is merely a marker of more extensive fibrosis, whether in IPF or in other fibrosing disorders such as CTD-ILD. The true diagnostic value of BAL lies in the presence or absence of major lymphocytosis (>30%). Lymphocytosis content is similar in IPF and healthy control subject, and when increased in IPF (diagnosed historically) is associated with moderate to severe alveolar septal inflammation, raising the possibility of an alternative form of UIP, such as HP. By contrast, a major BAL lymphocytosis is often present in HP, NSIP, and CTD-ILD [33]. The utility of BAL lies not only in immediate diagnostic evaluation but also in the optimal selection of patients to undergo a diagnostic surgical biopsy procedure.
The ultimate goal is to help patients live as well as possible for a long as possible, alleviating the symptoms burden. In addition to the available pharmacologic therapies, nonpharmacologic options and palliative care aim to prevent and relieve suffering by controlling symptoms and to provide other support to patients and families in order to maintain and improve their quality of living.
The lung presents special challenges with regard to therapy targeting fibrosis On the one hand, the lung has easily measured clinical features that allow for assessment of lung function, a surrogate for fibrosis. One the other hand, pulmonary fibrosis appears to be less dynamic than fibrosis occurring in other organ system. Non-pharmacologic management strategies help patients with IPF live heathier, more normal lives, and the importance of these approaches cannot be overemphasized. Smoking cessation should be a priority for patients who are actively using tobacco products (conventional or vaping) [34]. Influenza, pneumococcal, and other age-appropriate vaccines should be administered [35]. Clinical practice guidelines strongly recommended supplemental oxygen for patients with IPF. Oxygen reduces exertional dyspnea and improves exercise tolerance [36]. In other pathologies, pulse oximetry should not be used as a sole criterion to indicate long term oxygen therapy (LTOT), but to justify an arterial gas (ABG). ABG should be indicated (8 weeks after stabilization of an acute exacerbation, eg., AECOPD), and should be repeated at 3 weeks (British guidelines). If both samples define described criteria, LTOT is considered [37], but on IPF, an oxyhemoglobin saturation of 88% or less at rest, during exertion, or during sleep should prompt initiation of home oxygen therapy [35]. Pulmonary rehabilitation, an exercise structure programed designed for adult with advanced lung disease, has been shown to improve exercise capacity and HR-QoL for patients with IPF [38].
Only a minority of patients with IPF receive a transplant [39]. Lung transplantation can prolong survival and improve quality of life for highly selected candidates [40,41]; however, only 66% of transplant recipients with IPF survival for more than 3 years after transplantation and only 53% survive for more than 5 years [39]. Common complications include primary graft dysfunction, acute and chronic forms allograft rejection, cytomegalovirus and other infections, and cancer. IPF has not been show to recur in the allograft. Referral to a transplantation center should be made at the time of diagnosis, since the evaluation process and waiting time can last for month or years [42]. Common contraindications include recent cancer, advanced non-pulmonary organ failure, and lack of a reliable social support system. Poverty, meager health budget and little experience of health personnel with this tool threaten the possibility of implementing them successfully in third world countries [43].
Treatment guidelines for IPF include a strong recommendation against the use of prednisone in combination with azathioprine and oral N-acetylcysteine, a regimen associated with an increase in mortality by a factor of 9, as compared with placebo [44]. Interferon-γ [45], endothelin antagonists [46], and warfarin [47], are ineffective or harmful in patients with IPF. The Food and Drug Administration has appropriately warned consumers against various unapproved stem-cell “therapies” advertise for the treatment of IPF [48]. Although current guidelines recommended the use of antacid therapy to treat IPF, there are not data from clinical trials to support this recommendation [49]. More recent data suggest that antacid therapy to treat IPF may increase risk of respiratory infections in patients with IPF [50].
In fibrosis (pulmonary or other organs), inflammation or other stimuli (unknown in IPF) damages the epithelial and endothelial cells leading to the production and release of inflammatory mediators (cytokines, chemokines and others) that recruit a wide range of inflammatory cells (PMNs, eosinophils, basophils, mastocells, T lymphocytes, platelet, and macrophages). These cells, in turn, release profibrotic mediators (eg, transforming growth factor beta (TGF-β), platelet-derivate growth factor (PDGF), fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), IL-1, Il-4, IL-6, IL-10, TNF-α, angiotensin II, endothelin I, and integrins). These factors will stimulate cells to release ECM. These cells are fibroblasts, myofibroblasts, cells derived from bone marrow, fibrocytes, epithelial cells in transition to mesothelial cells (EMT), endothelial cells in transition to mesothelial cells (EndMT), and mesothelial cells in transition to mesenchymal cells (MMT). For example, fibroblasts stimulated by these factors become myofibroblasts, much more biologically active than their parents and release ECM. These profibrotic factors use co-stimulatory molecules to accelerate the signal at the intracellular level and / or enzymes, such as tyrosine kinases to stimulate the corresponding receptor in the effector cell (eg, myofibroblasts). ECM that form the fibrotic scar are collagen I and II (synthetized from pro-collagen), cellular fibronectin, basement membrane protein such laminin, proteoglycans and aggrecan [51]. Therefore, a logical treatment, which aims to alter the dynamics and plasticity of the fibrotic scar, involves inhibiting profibrotic factors or enzymes that make the signal to release ECM effective in receptors. Two pharmacological alternatives today meet these objectives in IPF.
Two medications, nintedanib and pirfenidone, has been shown to be safe and effective in the treatment of IPF; both are recommended for use in patients with IPF [49]. In placebo-controlled, randomized trials, each drug has been show to slow the rate of FVC decline by approximately 50% over the course of 1 year (INPULSIS-1 and ASCEND respectively) [52,53]. Both have shown some efficacy in reducing severe respiratory events, such as acute exacerbations, and hospitalization for respiratory events [54,55]. Pooled data and meta-analysis suggest that these agents may reduce mortality [56,57]. The cost of each medication is estimated to exceed $100.000 annually. Again, in low-income countries it is very difficult to implement these therapies on a regular basis for patients with IPF due its high cost, especially, that they are of indefinite use.
Nintedanib is an intracellular inhibitor of tyrosine kinase that targets growth factor pathways, including those downstream VEFG receptors 1,2, and 3, FGF receptors 1,2, and 3, and PDGF receptor, by occupying their intracellular ATP-binding pockets. By blocking this signaling, it is believed that profibrotic processes including fibroblast proliferation, migration and differentiation, and the secretion of ECM proteins in the interstitial space are curbed. Patients should initially be prescribed 150 mg of nintedanib, to be taken by mouth twice daily. The dose can be decreased to 100 mg twice daily if unmanageable side effects occur. The medication should be taken with food and can be continued indefinitely. Patients taking nintedanib commonly have diarrhea, which can often be managed with antidiarrheal agents [52]. Cases of drug-induced liver injury have been reported. Liver function should be tested at baseline, monitored monthly for the first 3 months, and then monitored as clinically indicated. Since nintedanib is associated with a small increase in the risk of bleeding, this agent should be used with great caution, if at all, in patients receiving full-dose anticoagulant therapy. Atheroembolic events, including myocardial infarction, have also been reported with nintedanib. Caution should be used when treating patients with cardiovascular risk factors, including those with who have coronary artery disease. In a recent work, Flaherty, et al. enrolled patients with IPF and specific effort were made to enroll patients with a progressive fibrotic phenotype other than IPF (eg, chronic hypersensitive pneumonitis and autoimmune interstitial lung diseases). The patients who received nintedanib had significantly slower rate of progression of interstitial lung disease than those who received placebo, assessed over a 52-week period, independent of the fibrotic pattern (IPF or others ILDs). This change in physiological outcomes was not accompanied by meaningful changes in measures of HR-QoL, although nintedanib was associated with higher frequency of diarrhea, nausea, and vomiting [58]. The results of the SENSCIS trials showed that nintedanib has a beneficial effect by reducing the rate of decline in FVC in patients with ILD associated with systemic sclerosis (SSc-ILD) over a 1-year period. No other clinical benefit was observed [59]. Nintedanib has been approved by the FDA for the management of adult whit SSc-ILD and as of September 9, 2019 is under review for this indication in Europe.
Pirfenidone has a number of anti-inflammatory and anti-fibrotic effects, including inhibition of collagen synthesis, down-regulation of TGF-β, and TNF-α, a reduction in fibroblast proliferation, and an antioxidant effect. The mechanism is not fully established [60]. Pirfenidone is prescribed in an escalating-dose fashion over 14-day period: 267 mg (one capsule) by mouth three times daily for 1 week, 534 mg (two capsules) three times daily for 1 week, and 801 mg (three capsules) three times daily thereafter. Patients can subsequently be transitioned to an 801-mg tablet three times daily. Pirfenidone must be taken with food and can be continued indefinitely. Common side effects, such anorexia, nausea, vomiting, can often be ameliorated by judicious use of antacid and antiemetic agents. In some cases, side effects are severe enough to require a lower total daily dose (six to eight capsules daily). A photosensitive rash can occur. Liver function should be monitored periodically [53]. The treatment is generally safe and tolerable, the side effects usually mild to moderate and reversible, and infrequently led to discontinuation of treatment [61,62]. Pirfenidone has also been investigated in other fibrous ILDs that are not IPF, with promising results [63], and others, waiting future results [64,65].
Pirfenidone and nintedanib provide similar benefits [66,67]. Recent data on treatment that combines these agents suggest clinically significant gastrointestinal effects in patients with IPF [68]. It is difficult to recommend one agent over the other.
There are several possible approaches for the management of cough in IPF. Thalidomide, gefapixant (the P2X3 antagonist) and inhaled cromolyn preparation were shown to ameliorate cough in patients with IPF [69-72].
IPF remains a fatal disease, but there are now pharmacologic treatments that slow the disease progression and prolong survival.
Awareness of IPF is low, leading to delays in diagnosis and treatment.
Pharmacologic and non-pharmacologic therapies provides the opportunity to reduce these delays and improve survival and QoL.
Patients should be treated in accordance with their needs and wishes, to prevent or alleviate suffering and ensure a dignified death.
A team effort is need to provide support to patients and their families.
Future research in fibrosing lung disease (IPF and non-IPF) should be directed in two ways: first in an early molecular diagnosis of fibrogenesis. There are studies with diagnostic molecular markers with circulating matrix metalloproteinases, combination of circulating proteins and peripheral blood transcriptomic signature; and gene-expression signature developed with the use of a machine-learning biopsy approach has been applied to surgical lung-biopsy samples to enhance the identification of a UIP pattern [35]. But it remains unclear how such molecular approaches will be integrated into clinical practice, and their use cannot be recommended until further data are available. At the time, these tools are not reliable enough to identify progressive fibrosing ILD with confidence. The most promising is UIP pattern on HRCT. Second, by knowing better the central mechanisms that underlie the fibrogenic process, it is possible to develop new and safer therapeutic tools that have a better impact on the natural evolution of fibrosis and can offer these patients a better life expectancy than they have today.
This work was only carried out by the author. Author AA contributed in the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
No.
No.
- Diamantopoulous A, Wright E, Vlahopoulou K, Comic L, School N, et al (2018) The burden pf Illness of Idiopathic Pulmonary Fibrosis: a comprehensive evidence review. Pharmacoeconomics 36: 779-807. [Crossref]
- Ferrra G, Dahlstrom L, Janson C, Kirchgässler KU, Levine A, et (2019). Epidemiology of pulmonary fibrosis: a cohort study using health care data in Sweden. Pulm Ther 5: 55-68. [Crossref]
- Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR (2016) Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18-64 years old. Eur Respir J 48: 179-186. [Crossref]
- Rajala K, Lehto JT, Sutinen E, Kautiainen H, Myllärniemi M, et al (2018) Marked deterioration in the quality of life of patients with idiopathic pulmonary fibrosis during the last two years of life. BMC Pulm Med 18: 172. [Crossref]
- Glaspole IN, Chapman SA, Cooper WA, Ellis SJ, Goh NS, et al (2017) Health-related quality of life in idiopathic pulmonary fibrosis. Respirology 22: 950-956. [Crossref]
- Ley B, Collard HR, King TE (2011) Clinical course of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183: 431-440. [Crossref]
- Cosgrove GP, Bianchi P, Danese S, Lederer DJ (2018) Barriers to timely diagnosis of interstitial lung disease. BMC Pulm Med18: 9. [Crossref]
- King TE jr, Pardo A, Selman M (2011) Idiopathic pulmonary fibrosis. Lancet 378: 1949-1961. [Crossref]
- Vancheri C, Faila M, Crimi N, Raghu G (2010) Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J 35: 496-504. [Crossref]
- Siegel RL, Miller KD, Jemal A (2019) Cancers statistics. Ca Cancer J Clin 69: 7-34. [Crossref]
- Fernández Pérez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, et al (2010) Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 137: 129-137. [Crossref]
- Rush B, Wiskar K, Berger L, Griesdale D (2016) The use of mechanical ventilation in patients with idiopathic pulmonary fibrosis in the United States: a nation-wide retrospective cohort analysis. Respir Med 111: 72-76. [Crossref]
- Silva Santos P, Cruz-Esquina AM (2016) Mechanical ventilation in idiopathic pulmonary fibrosis: unresolved dilemma. Respir Med 117: 283. [Crossref]
- Kamiya H, Panlaqui OM (2019) Prognostic factors for acute exacerbation of idiopathic pulmonary fibrosis: protocol for a systemic review and meta-analysis. BMJ Open 9: e028226.
- Richeldi L, Collard HM, Jones MG (2017) Idiopathic pulmonary fibrosis. Lancet 389: 1941-1952. [Crossref]
- Cottin V, Hirani NA, Hotchkin DL, Nambiar AN, Ogura T, et al (2018) Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev 27: 180076. [Crossref]
- Cottin V, Wollin L Fischer A, Quaresma M (2019) Fibrosing interstitial lung diseases: knows and unknows. Eur Respir Med 28: 180100. [Crossref]
- Kolb M, Vašáková M (2019) The natural history of progressive fibrosing lung diseases. Respir Res 20: 57. [Crossref]
- Lynch DA, Sverzellati N, Travis WD, Browun KK, Colby TV, et al (2018) Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir Med 6: 138-153. [Crossref]
- Selman M, Pardo A (2014) Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med 189: 1161-1172. [Crossref]
- Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, et al (2008) Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA 105: 13051-13056. [Crossref]
- 22. Armanios MY, Chen JJ, Cogan JD, Akder JK, Ingersoll RG, et al (2007) Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356: 1317-1326. [Crossref]
- Han MK, Zhou Y, Murray S, Taylor N, Noth I, et al (2014) Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med 2: 548-556. [Crossref]
- Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russel KE, et al (2014) The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 190: 906-913. [Crossref]
- Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, et al (2000) Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol 152: 307-315. [Crossref]
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, et al (2018) Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 198: e44-e68. [Crossref]
- Hunninhake GW, Zimmerman MB, Schwartz DA, King TE Jr, Lynch J, et al (2001) Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 164: 193-196. [Crossref]
- Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB (2016) In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United State. 2000 to 2011. Am J Respir Crit Care Med 193: 1161-1167. [Crossref]
- Rogmanoli M, Colby TV, Berhet JP, Gamez AS, Mallet JP, et al (2019) Poor concordance between sequential transbronchial lung cryobiopsy and surgical biopsy in the diagnosis of diffuse interstitial lung disease. Am J Respir Crit Care Med 199: 1249-1258. [Crossref]
- Romagnoli M, Colby TV, Suehs CM, Vachier I, Molinari M, et al (2019) Cryobiopsy compared with surgical lung biopsy in ILD: reply to Maldonado et al, Froidure et al, Bendstrup et al, Agarwai et al, Richeldi et al, Rajchgot et al and Quadrelli et al. Am J Respir Crit Care Med 200: 944-946. [Crossref]
- Walsh SLF, Wells AU, Desai SR, Poletti V, Piciucchi S, et al (2016) Multi-center evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a cause-cohort study. Lancet Respir Med 4: 557-565. [Crossref]
- Aouadi S, majdoub s, ghrairi n, brahem e, gharsalli h, et al (2017) Broncoalveolar lavage in idiopathic pulmonary fibrosis. Eur Respir J 50: PA3811. [Crossref]
- Wells AU, Kokosi MA (2017) Should BAL be routinely performed in the diagnostic evaluation of idiopathic pulmonary fibrosis? Yes. Chest 152: 917-919. [Crossref]
- Bhatta DN, Glantz SA (2020) Association of E-cigarette use with respiratory disease among adults: a longitudinal analysis. Am J Prev Med 58: 182-190. [Crossref]
- Lederer DJ, Martínez FJ (2018) Idiopathic pulmonary fibrosis. N Engl J Med 378: 1811-1823. [Crossref]
- Dowman LM, McDonald CF, Bozinovski S, Vlahos R, Gillies R, et al (2017) Greater endurance capacity and improve dyspnea with acute oxygen supplementation in idiopathic pulmonary fibrosis patients without resting hypoxaemia. Respirology 22: 957-964.
- Alvarado A (2017) Domiciliary oxygen: facts and fallacies. Clin Res Trials 3: 1-10.
- Dowman LM, McDonald CF, Hill CJ, Lee A, Baker K, et al (2017) The evidence of benefits on exercise training in interstitial lung disease: a randomized controlled trial. Thorax 72: 610-619. [Crossref]
- Valapour M, Lehr CJ, Skeans MA, Smith JM, Carrico R, et al (2018) OPTN/SRTR 2016 Annual Data Report: Lung. Am J Transplant 1: 363-433. [Crossref]
- Singer JP, Katz PP, Soong A, Shrestha P, Huang D, et al (2017) Effect pf lung transplantation on health-related quality of life in the era of lung Allocation Score: A US prospective cohort study. Am J Transplant 17: 1334-1345. [Crossref]
- Titman A, Rogers CA, Bonser RS, Banner NR, Sharples LD (2009) Disease-specific survival benefits of lung transplantation in adults: a national cohort study. Am J Transplant 9: 1640-1649. [Crossref]
- Weill D, Benden C, Corris PA, Deark JH, Davis RD, et al (2015) A consensus document for the selection of lung transplant candidates: 2014-an update from the Pulmonary Transplantation Council of the International Society for Hearth and Lung transplantation. J Hearth Lung Transplant 34: 1-15. [Crossref]
- Alvarado A (2018). Fibrosis: the sixth element. Clin Res Trials 4: 5-10.
- The Idiopathic Pulmonary Fibrosis Clinical Research Network (2012) Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366: 1968-1977. [Crossref]
- King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, et al (2009) Effect of interferon gamma-1b on survival in patients in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicenter, randomized, placebo-controlled trial. Lancet 374: 222-228. [Crossref]
- Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, et al (2013) Treatment of idiopathic pulmonary fibrosis with ambisentan: a parallel, randomized trial. Ann Intern Med 158: 641-649. [Crossref]
- Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, et al (2012) A placebo- controlled-randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186: 88-95. [Crossref]
- Marks PW, Witten CM, Califf RM (2017) Clarifying Stem-Cell therapy’s benefits and risks. N Engl J Med 376: 1007-1009. [Crossref]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788-824. [Crossref]
- Kreuter M, Wuyts W, Renzoni E, Koschel D, Maher TM, et al (2016) Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: a pooled analysis. Lancet Respir Med 4: 381-389. [Crossref]
- Rockey DC, Bell PD, Hill JA (2015) Fibrosis-a common pathway to organ injury and failure. N Engl J Med 372: 1138-1149. [Crossref]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, et al (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071-2082. [Crossref]
- King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, et al (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370: 2083-2092. [Crossref]
- Ley B, Swigris J, Day BM, Stauffer JL, Raimundo K, et al (2017) Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 196: 756-761. [Crossref]
- Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, et al (2016) Efficacy of nintedanib in idiopathic pulmonary fibrosis: across pre-specified subgroups in INPULSIS. Am J Respir Crit Care Med 193: 178-185. [Crossref]
- Nathan SD, Alberta C, Bradford WZ, Costabel HR, Glaspole I, et al (2017) Effect of pirfenidone on mortality: pooled analysis and meta-analysis of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 5:33-41. [Crossref]
- Richeldi L, Cottin V, du Bois RM, Selman M, Kamura T, et al. (2016) Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS trials. Respir Med 113: 74-79. [Crossref]
- Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, et al (2019) Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 381: 1718-1727. [Crossref]
- Distler OD, Highland KB, Gahlemann M, Azuma A, Fischer A, et al (2019) Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 380: 2518-2528. [Crossref]
- Kolb M, Bonella F, Wollin L (2017) Therapeutic targets in idiopathic pulmonary fibrosis. Respir Med 131: 74-79.
- Noble PW, Albera C, Bradford WZ, Costabel U, du Bois RM, et al (2016) Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 47: 243-253. [Crossref]
- Lancaster LH, de Andrade JA, Zibrak JD, Padilla ML, Albera C, et al (2017) Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis 26: 170057. [Crossref]
- Maher TM, Wuyts W (2019) Management of fibrosing interstitial lung disease. Adv Ther 36: 1518-1531. [Crossref]
- Behr J, Neuser P, Prasse A, Kreuter M, Rabe K, et al (2017) Exploring efficacy and safety of oral pirfenidone for progressive, non-IPF lung fibrosis (RELIEF)-a randomized, double-blind, placebo-controlled, parallel group, multicenter, phase II trial. BMC Pulm Med 17: 122. [Crossref]
- Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, et al (2018) Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomized, placebo-controlled phase II trial. BMJ Open Respir Res 5: e3000289. [Crossref]
- Vancheri C, Kreuter M Richeldi L, Ryerson CJ, Valeyre D, et al (2018) Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis: results on the INJOURNEY trial. Am J Respir Crit Care Med 187: 356-363. [Crossref]
- Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE (2016) Drug treatment of idiopathic pulmonary fibrosis: systematic review and network meta-analysis. Chest 149: 756-766. [Crossref]
- Rochwer B, Neupane B, Zhang Y (2016) Treatment of idiopathic pulmonary fibrosis: a network meta-analysis. BMC Med 14: 18. [Crossref]
- Horton MR, Santopietro V, Mathew LK, Horton KM, Polito AJ, et al (2012) Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med 157: 398-406.
- Van Manen MG, Birring SS, Vancheri C, Vindigni V, Renzoni E, et al (2017) Effect of pirfenidone on cough in patients with idiopathic pulmonary fibrosis. Eur Respir J 50: 1701157-1701157. [Crossref]
- Abdulqawi R, Dockry R, Holt K, Layton G, McCarthy BG, et al (2015) P2X3 receptor antagonist (AF-2019) in refractory chronic cough: a randomized, double-blind, placebo –controlled phase 2 study. Lancet 385: 1198-1205. [Crossref]
- Birring SS, Wijsenbeek MS, Agrawal S, van der Berg JWK, Stone H, et al (2017) A novel formulation of inhaled sodium cromoglicate (PA101) in idiopathic pulmonary fibrosis and chronic cough: a randomized, double-blind, proof-of-concept, phase 2 trail. Lancet Respir Med 5: 806-815. [Crossref]
\