Hypertensive disorders of pregnancy (HDP), characterized by hypertension after 20 weeks of gestation, are considered a major complication during pregnancy. When HDP worsen, both the mother and fetus can experience life-threatening conditions, such as stroke, eclampsia, fetal growth restriction, intrauterine fetal death, and placental abruption. Despite numerous studies thus far, the pathogenesis of HDP remains unclear. Recently, a 2-step theory has been advocated and widely accepted. The first step is impaired placentation during the early stage of pregnancy, while the second step is overexpression of antiangiogenic factors derived from the hypoxic placenta, leading to endothelial dysfunction, including hypertension and proteinuria. To elucidate the pathogenesis and develop therapies for HDP, various animal models have been used. Here, we describe various rodent models based on the 2-step theory. These rodent models have contributed to partially integrate each hypothesis and demonstrate that the hypotheses can overlap. Using these animal models, research on HDP is expected to progress.
animal model, advanced maternal age, antiangiogenic factor, hypertensive disorders of pregnancy, hypoxia, immune response
Hypertensive disorders of pregnancy (HDP), characterized by hypertension after 20 weeks of gestation, are considered a major complication during pregnancy. When HDP worsen, the mother and fetus experience life-threatening conditions such as stroke, eclampsia, fetal growth restriction (FGR), intrauterine fetal death (IUFD), and placental abruption [1]. HDP are also associated with cardiovascular disease several years after delivery [2]. HDP affect approximately 5-8% of pregnancies. However, because of advances in assisted reproductive technology [3-5], the prevalence of high-risk pregnancies, including HDP, has increased in the past few decades [4].
While many studies have actively investigated the pathophysiology of HDP, the mechanism is unclear. Various hypotheses have been advocated [6], among which the 2-step theory is widely accepted. This hypothesis comprises 2 steps: First, mal placentation occurs during early-stage pregnancy due to an imbalance of fetomaternal immune response and maternal pathologic conditions. Second, a severely hypoxic placenta develops after mid-gestation, leading to the production of antiangiogenic factors such as soluble vascular endothelial growth factor (VEGF) receptor-1, soluble fms-like tyrosine kinase-1 (sFlt-1), and soluble endoglin (sEng), which cause maternal endothelial dysfunction systemically, resulting in hypertension and proteinuria.
Based on the 2-step theory, the pathogenesis of HDP can be categorized as follows: (1) maternal immune response to the allogenic fetus; (2) placental oxygen dysregulation; (3) uteroplacental dysfunction, including decreased placental perfusion and placental hypoxia; (4) imbalance between angiogenic and antiangiogenic factors; and (5) insufficient trophoblast invasion. To elucidate the pathogenesis and develop therapies for HDP, various animal models have been used. In this mini-review, we discuss the mouse and rat models of HDP and categorize them by the 5 hypotheses stated above.
Immune response-induced model
One of the most important pathogeneses of HDP is immune response abnormality of fetomaternal tolerance. It has been considered that HDP are associated with an imbalance of Th1 and Th2. While a Th2-immunotolerant state is predominant in normal pregnancy, a Th1-proinflammatory environment is predominant in patients with HDP [7,8]. The Th2-predominant state contributes to prevent fetal rejection.
Hayakawa et al. developed a Th1/Th2-activated mouse model by injecting splenocytes treated with IL-4 and/or IL-12 during the late gestational period. This model demonstrated hypertension, FGR, IUFD, and glomerular abnormality, but not proteinuria [9]. Zenclussen et al. established a different mouse model by injecting Th1-activated splenocytes during mid-pregnancy. Their model demonstrated human HDP-like symptoms, including hypertension, IUFD, proteinuria, and glomerular abnormality [10]. Interestingly, when the same treatment was given to nonpregnant female mice, the model did not show any HDP-like phenotypes. These results indicate that deficiency of the fetomaternal immune system during pregnancy is associated with the development of HDP.
Aoki et al. reported another role of immunologic abnormality, autophagy deficiency, in the pathogenesis of HDP. They established a model of trophoblast-specific autophagy-related gene 7 (Atg7) knockout mice using lentiviral vector. Atg7 is an autophagy-related gene essential for autophagosome biogenesis [11]. This model demonstrated hypertension and placental abnormality in the late gestational period. These results showed that autophagy deficiency of the placenta has adverse effects on fetal and maternal outcomes [12].
An imbalance of angiogenic and antiangiogenic factors is suggested in the pathogenesis of HDP [13]. Karumanchi and Maynard et al. revealed that antiangiogenic factors, in particular, sFlt-1 and placental growth factor (PlGF), play an important role in such pathogenesis [13-18]. Many researchers have reported that serum sFlt-1 and sEng levels are increased in patients with HDP and are associated with the severity of disease [13]. Thus, an imbalance of angiogenic and antiangiogenic factors, including PlGF and VEGF, is suggested to lead to HDP.
Our group developed a mouse model of placenta-specific human sFlt-1 overexpression using lentiviral vector. The model demonstrated human HDP-like symptoms, including hypertension, proteinuria (high albumin/creatinine ratio), FGR, high serum sFlt-1, low serum PlGF and VEGF, placental abnormality, and glomerular endotheliosis. Moreover, these phenotypes were ameliorated by pravastatin treatment [19]. Consequently, several clinical trials of pravastatin in patients with HDP have begun in some countries. So far, it has been reported that there are no complications during pregnancy and that pravastatin treatment has the effect of activating endothelial nitric oxide synthase (eNOS) and releasing anti-inflammatory factors, resulting in the protection of endothelial cells and prevention of HDP [20].
sEng-overexpression model
Vaenkatesha et al. reported that the increased levels of sEng in patients with HDP are correlated with the severity of clinical disease. They established a rat model by intravenously injecting recombinant sEng with or without sFlt-1. This model demonstrated not only hypertension in the late gestational period but also severe features of HDP, including HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome-like phenotypes and FGR [21].
RAS has been suggested as one of the most important pathogenetic factors of HDP. However, even if renin, angiotensin, angiotensin II, and aldosterone increase during pregnancy, the effect of these factors on the sensitivity of vasoconstriction is not high in normal pregnancy. On the other hand, their effect in patients with HDP is strikingly high. It also has been hypothesized that the expression of angiotensin II type 1 (AT1) receptor is increased in patients with HDP. There is evidence that AT1 agonistic autoantibody (AT1-AA) is highly detected in patients with HDP. Moreover, some investigations have revealed RAS components expressed in the placenta. According to such evidence, Falcao et al. developed a model of human angiotensin-renin double-transgenic mice and analyzed the phenotypes in pregnancy. While male mice demonstrated hypertension, female mice did not. However, this model already induced human renin and angiotensin, researchers observed their phenotype. Finally, pregnant transgenic mice demonstrated hypertension, proteinuria, FGR, decreased litter size, placental abnormality, high sFlt-1 expression, low PlGF expression, and cardiac hypertrophy [22].
AT1-AA-induced model
A previous investigation indicated that the serum of patients with HDP contains high levels of AT1 receptor [23]. Based on this, Zhou et al. established a mouse model by injecting AT1-AA at embryonic day 13.5 and analyzed the phenotypes. The model demonstrated HDP-like symptoms, including hypertension, proteinuria, glomerular endotheliosis, FGR, decreased litter size, placental abnormality, and high HIF-1α expression. Moreover, these phenotypes were suppressed by losartan treatment, an AT1 receptor blocker. These results indicate that AT1-AA can affect HIF-1α expression in the placenta [24].
Uteroplacental perfusion-deficiency model
Uteroplacental perfusion deficiency exacerbates the hypoxic environment of the placenta, increases the production of antiangiogenic factors, and results in HDP. The reduced uterine perfusion pressure model is a technique for reducing uteroplacental perfusion by clipping the uterine and ovarian arteries. After clipping the ovarian and/or uterine arteries at embryonic day 14.5, the model demonstrated HDP-like symptoms, including hypertension, FGR, IUFD, decreased litter size, high sFlt-1 expression, low PlGF expression, abnormal placenta, and high HIF-1α expression in the placenta. However, the FGR rate in the ovarian artery-clipping group was significantly higher than that in the uterine artery-clipping group, indicating that the anatomical characteristics of experimental animals can lead to unique results compared with humans [25].
HIF-1α-overexpression model
Increased expression of antiangiogenic factors alone cannot fully explain the pathogenesis of HDP. There are some cases in which even normal levels of these factors can result in HDP [26]. Thus, it has been hypothesized that a hypoxic environment exists in the placenta of patients with HDP.
Clinically, there is evidence that levels of catechol-O-methyltransferase (COMT) and 2-methoxyestradiol (2-ME) are low in patients with HDP. COMT is expressed in human placenta and decidua. The level of 2-ME increases during pregnancy, which inhibits the effect of HIF-1α and regulates the expression of HIF-1-associated genes [27,28].
Kanasaki et al. developed a model of COMT-knockout mice, which demonstrated HDP-like symptoms under the absence of 2-ME. Consequently, they noted that 2-ME treatment attenuated such HDP-like phenotypes and improved placental hypoxia, including high HIF-1α expression and serum sFlt-1 levels [29].
HIF-1α-overexpression model
HIF-1α is a transcription factor associated with placental development, and its expression is increased in the placenta of patients with HDP. Therefore, excessive expression of HIF-1α can lead to HDP. Tal et al. developed a mouse model of HIF-1α overexpression using cytomegalovirus. Their model demonstrated many human HDP-like symptoms, including HELLP syndrome, and elevated antiangiogenic factors [30].
HIF-1α-knockdown model
Several reports have advocated that HIF-1α expression is associated with the development of HDP. In particular, prolonged elevated HIF-1α expression induces the production of antiangiogenic factors, including sFlt-1, sEng, and sET-1. AT1-AA and tumor necrosis factor induce HIF-1α expression in the vascular smooth muscle, kidney, and hepatocyte [24]. Induction of these factors by intravenous injection results in HDP-like symptoms.
Iriyama et al. reported a mouse model of SiRNA knockdown of HIF-1α. SiRNA improved the HDP-like phenotypes, including hypertension, proteinuria, kidney injury, abnormality of placental vasculature, and high serum sFlt-1. Next, they found that HIF-1α expression increased with the injection of an inflammatory agent. From these results, HIF-1α expression seems to play a role in the development of HDP.
L-nitro-arginine methyles (L-NAME) inhibits eNOS and induces HDP-like symptoms. Yallampalli et al. developed a rat model of eNOS inhibition by subcutaneously injecting L-NAME. This model demonstrated HDP-like symptoms, including hypertension, FGR, and IUFD. Moreover, nitroglycerine treatment ameliorated these phenotypes [31].
HDP-associated gene-induced model
STOX-1 has been identified as the first gene associated with HDP [32]. In vivo experiments of STOX-1 overexpression have demonstrated HDP-like phenotypes. Doridot et al. established a model of STOX-1 transgenic mice, which showed HDP-like symptoms, including hypertension, proteinuria, increased serum sFlt-1 and sEng levels, and placenta and kidney abnormalities. Consequently, these phenotypes were attenuated or prevented by aspirin treatment [33].
Pre-existing hypertension model (BPH/5 mice)
BPH/5 mice are known as an inbred mouse strain with pre-existing mild hypertension [34]. Davisson et al. attempted to mate these mice and observed whether HDP developed. In their model, pregnant BPH/5 mice demonstrated elevated blood pressure in the late gestational period, which recovered to baseline postpartum. This model also showed other HDP-like phenotypes, including proteinuria, glomerular abnormality, and FGR [35].
Advanced maternal age (AMA) model
Recently, AMA has increased because of late marriage and advances in assisted reproductive technology, which has also increased the prevalence of HDP [3,5]. We established an AMA mouse model using pregnant mice aged over 6 months. This model demonstrated the phenotypes and pregnancy complications of human AMA, including maternal obesity, declining fertility, FGR, higher IUFD rate, and smaller fetal/placental weight ratio. The model also demonstrated hypertension and lower serum PlGF, indicating placental dysfunction, in the late gestational period. Thus, these results indicate not only the complications of AMA but also the development of HDP. Urinary protein was not significantly different between AMA and control (young pregnant) mice. The conclusions of our investigation are as follows: (1) AMA mice developed HDP; (2) serum sFlt-1 levels in extremely AMA (over 40 years of age) mice were significantly lower than those in young mice; and (3) while the AMA model mice and human AMA complicated with HDP, serum sFlt-1 levels demonstrated a low level. We focused on the vessels and placental senescence in the cause of hypertension with low sFlt-1. There was evidence of placental and systemic senescence in AMA mice in the late gestational period [36].
In conclusion, we reviewed various experimental rodent models of HDP (Table 1) (Figure 1). These rodent models have contributed to partially integrate each hypothesis and demonstrate that the hypotheses can overlap. Using these animal models, research on HDP is expected to progress.
Table 1. Various rodent models of HDP. HDP models are categorized by the following concepts: (1) immune response-induced models, (2) hypoxia-induced models, (3) RAS models, and (4) other models (including pregnant BPH/5 mice, STOX-1 transgenic mice, and advanced maternal age mice)
Abbreviations: ADA: adenosine deaminase; FGR: fetal growth restriction; HDP: hypertensive disorders of pregnancy; HELLP: hemolysis elevated liver enzymes and low platelets; IUFD: intrauterine fetal death; PlGF: placental growth factor; RAS: renin-angiotensin system; RUPP: reduced uteroplacental perfusion; sEng: soluble endoglin; sFlt-1: soluble fms-like tyrosine kinase-1
Classifications |
Pathophysiology of HDP |
Name of models |
Increasing
blood pressure |
Proteinuria |
IUGR |
Other complications and novel findings |
Investigators |
Immune response induced models |
Immune response induced model |
Th1 & Th2 activated splenocytes induced pregnant mice |
+ |
- |
+ |
Glomerular abnormality |
Hayakawa, et al. (2000) |
| |
|
Th1 cell injected pregnant mice |
+ |
+ |
+ |
IUFD and glomerular abnormality |
Zenclussen, et al. (2004) |
| |
Autophagy deficiency model |
Placenta-specific Atg7 conditional knokcout mice |
+ |
- |
+ |
Placental abnormality |
Aoki, et al. (2018) |
Anti-angiogenetic factors induced models |
Anti-angiogenic factor overexpression model |
Placental sFlt-1 overexpression mice |
+ |
+ |
+ |
IUFD, decline of fetal/placental weight, and glomerular abnormality. Pravastatin ameliorates HDP-like phenotypes. |
Kumasawa, et al. (2010) |
| |
Anti-angiogenic factor injected model |
sFlt-1 injected rat |
+ |
+ |
+ |
IUFD, glomerular abnormality |
Maynard, et al. (2003) |
| |
sEng injected rat |
+ |
+ |
+ |
Increasing sFlt-1 levels, and HELLP syndrome |
Vaenkatesha, et al. (2006) |
Hypoxia induced models |
Hypoxia induced model |
HIF-1α overexpression mice |
+ |
+ |
+ |
Decline of litter size, Increasing of sFlt-1 and sEng levels, decreasing of PlGF levels, placental abnormality, glomerular abnormality, and HELLP syndrome |
Tal, et al. (2010) |
| |
|
AT1-AA and inflammatory agent (LIGHT) treatment
with HIF-1α knockdown mice |
+ |
+ |
+ |
Decline of litter size, increasing of IUFD, increasing of sFlt-1 levels and HIF-1α expression, and placental abnormality |
Iriyama, et al. (2015) |
| |
|
ADA knockout mice |
+ |
+ |
+ |
Increasing of sFlt-1 levels and HIF-1α expression, glomerular abnormality, placental abnormality, and high expression of Adora2b gene Adora2b deletion ameliorates these complications. |
Iriyama, et al. (2015) |
| |
|
COMT knockout mice |
+ |
+ |
+ |
Decline of litter size, increasing of IUFD and sFlt-1 levels, decreasing of PlGF levels and placental abnormality |
Kanasaki, et al. (2008) |
| |
Reduced uteroplacental perfusion model |
RUPP mice |
+ |
+ |
+ |
Increasing of IUFD and sFlt-1 levels, decreasing of PlGF levels and placental abnormality, and higher expression of HIF1-α |
Fushima, et al. (2012) |
Renin-angiotensin system (RAS) models |
Renin-angiotensin system (RAS)
overexpression model |
Renin and angiotensin double transgenic model mice |
+ |
+ |
+ |
Decline of litter size, Increasing of sFlt-1 levels, decreasing of PlGF levels, and placental abnormality, and glomerular abnormality |
Falcao, et al. (2009) |
| |
AT1-AA induced
model mice |
+ |
+ |
+ |
Decline of litter size, Increasing of sFlt-1 levels, decreasing of PlGF levels, and placental abnormality, and glomerular abnormality |
Zhou, et al. (2008) |
| |
eNOs inhibition model |
L-NAME induced model rat |
+ |
- |
+ |
Increasing of IUFD |
Yallampalli, et al. (1993) |
| |
Nitroglycerine treatment ameliorates HDP-like phenotypes. |
Others |
HDP-associated gene (STOX-1)
induced model |
STOX-1 transgenic mice |
+ |
+ |
+ |
Decline of litter size, Increasing of sFlt-1 levels, decreasing of PlGF levels and placental abnormality, and glomerular abnormality. Preventing these complications treated with aspirin. |
Doridot, et al. (2012) |
| |
Mouse strain with preexisting hypertension
(mild hypertension) |
Pregnant BPH/5 mice |
+ |
+ |
+ |
Glomerular abnormality |
Davisson, et al. (2002)
Dokras, et al. (2006) |
| |
Placental and systemic senescence model |
Advanced maternal age (AMA) model mice |
+ |
- |
+ |
Resembled the phenotypes of human AMA, decline of litter size, lower pregnancy rate, decreasing of PlGF levels and evidences of placental senescence. AMA represent relatively lower levels of sFlt-1 compared with young individuals under placental dysfunction. |
Furuya, et al. (2019) |
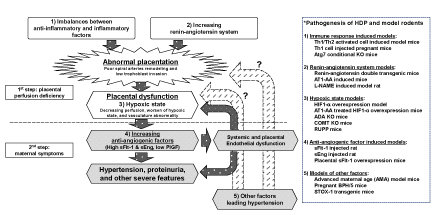
Figure 1. Relationship between the pathogenesis of HDP and model rodents. An arrow with a white dot on a black background indicates the mechanisms that have been already elucidated. A broken line arrow on a shaded line background indicates the theory that has partially or not completely elucidated
Abbreviations: sFlt-1: serum soluble fms-like tyrosine kinase-1; PlGF: placental growth factor; sEng: soluble Endoglin; AT1-AA: angiotensin II Type 1 receptor agonistic autoantibody; COMT: catechol-O-methyl transferase; L-NAME: L-nitro-arginine methyl ester; ADA: adenosine deaminase; RUPP: reduced uterine perfusion pressure
The authors declare that they have no competing interests.
This work was supported by JSPS KAKENHI JP17K11232.
No ethical approval was required.
- American college of obstetricians and gynaecologists, Task force on hypertension in pregnancy (2013) Hypertension in pregnancy. report of the american college of obstetricians and Gynecologists’ task force on hypertension in pregnancy. Obstet Gynecol 122: 1122-1131. [Crossref]
- Jim B, Karumanchi SA (2017) Preeclampsia: pathogenesis, prevention, and long-term complications. Semin Nephrol 37: 386-397. [Crossref]
- Jacobsson B, Ladfors L, Milsom I (2004) Advanced maternal age and adverse perinatal outcome. Obstet Gynecol 104: 727-733. [Crossref]
- Krieg SA, Henne MB, Westphal LM (2008) Obstetric outcomes in donor oocyte pregnancies compared with advanced maternal age in in vitro fertilization pregnancies. Fertil Steril 90: 65-70. [Crossref]
- Yogev Y, Melamed N, Bardin R, Tenenbaum-Gavish K, Ben-Shitrit G, et al. (2010) Pregnancy outcome at extremely advanced maternal age. Am J Obstet Gynecol 203: 558. [Crossref]
- Broughton Pipkin F, Rubin PC (1994) Pre-eclampsia--the 'disease of theories'. Br Med Bull 50: 381-396. [Crossref]
- Saito S (2010) Th17 cells and regulatory T cells: new light on pathophysiology of preeclampsia. Immunol Cell Biol 88: 615-617. [Crossref]
- Perez-Sepulveda A, Torres MJ, Khoury M, Illanes SE (2014) Innate immune system and preeclampsia. Front Immunol 5: 244. [Crossref]
- Hayakawa S, Fujikawa T, Fukuoka H, Chisima F, Karasaki-Suzuki M, et al. (2000) Murine fetal resorption and experimental pre-eclampsia are induced by both excessive Th1 and Th2 activation. J Reprod Immunol 47: 121-138.
- Zenclussen AC, Fest S, Joachim R, Klapp BF, Arck PC (2004) Introducing a mouse model for pre-eclampsia: adoptive transfer of activated Th1 cells leads to pre-eclampsia-like symptoms exclusively in pregnant mice. Eur J Immunol 34: 377-387. [Crossref]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, et al. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425-434. [Crossref]
- Aoki A, Nakashima A, Kusabiraki T, Ono Y, Yoshino O, et al. (2018) Trophoblast-Specific conditional Atg7 knockout mice develop gestational hypertension. Am J Pathol 188: 2474-2486. [Crossref]
- Maynard SE, Karumanchi SA (2011) Angiogenic factors and preeclampsia. Semin Nephrol 31: 33-46. [Crossref]
- Maynard SE, Min JY, Merchan J, Lim KH, Li J, et al. (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649-58.
- Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, et al. (2004) Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350: 672-683. [Crossref]
- Roberts JM, Hubel CA (1999) Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 354: 788-789. [Crossref]
- Roberts JM, Gammill HS (2005) Preeclampsia: recent insights. Hypertension 46: 1243-1249. [Crossref]
- Roberts JM, Von Versen-Hoeynck F (2007) Maternal fetal/placental interactions and abnormal pregnancy outcomes. Hypertension 49: 15-16. [Crossref]
- Kumasawa K, Ikawa M, Kidoya H, Hasuwa H, Saito-Fujita T, et al. (2011) Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc Natl Acad Sci U S A 108: 1451-1455. [Crossref]
- Costantine MM, Cleary K, Hebert MF, Ahmed MS, Brown LM, et al. (2016) Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high-risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol 214: 720.e1-720.e17. [Crossref]
- Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, et al. (2006) Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 12: 642-649. [Crossref]
- Falcao S, Stoyanova E, Cloutier G, Maurice RL, Gutkowska J, et al. (2009) Mice overexpressing both human angiotensinogen and human renin as a model of superimposed preeclampsia on chronic hypertension. Hypertension 54: 1401-1407. [Crossref]
- Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, et al. (1999) Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 103: 945-52.
- Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, et al. (2008) Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med 14: 855-862. [Crossref]
- Fushima T, Sekimoto A, Minato T, Ito T, Oe Y, et al. (2016) Reduced uterine perfusion pressure (RUPP) model of preeclampsia in mice. PLoS One 11: e0155426. [Crossref]
- Solomon CG, Seely EW (2004) Preeclampsia -- searching for the cause. N Engl J Med 350: 641-642. [Crossref]
- Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, et al. (1994) The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature 368: 237-239. [Crossref]
- Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, et al. (2003) 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3: 363-375. [Crossref]
- Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, et al. (2008) Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 453: 1117-1121. [Crossref]
- Tal R, Shaish A, Barshack I, Polak-Charcon S, Afek A, et al. (2010) Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. Am J Pathol 177: 2950-2962. [Crossref]
- Yallampalli C, Garfield RE (1993) Inhibition of nitric oxide synthesis in rats during pregnancy produces signs similar to those of preeclampsia. Am J Obstet Gynecol 169: 1316-1320. [Crossref]
- van Dijk M, Mulders J, Poutsma A, Könst AA, Lachmeijer AM, et al. (2005) Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet 37: 514-519. [Crossref]
- Doridot L, Passet B, Mehats C, Rigourd V, Barbaux S, et al. (2013) Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension 61: 662-8. [Crossref]
- Schlager G (1994) Biometrical genetic analysis of blood pressure level in the genetically hypertensive mouse. Clin Exp Hypertens 16: 809-824. [Crossref]
- Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, et al. (2002) Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension 39: 337-342. [Crossref]
- Furuya K, Kumasawa K, Nakamura H, Nishimori K, Kimura T (2019) Novel biomarker profiles in experimental aged maternal mice with hypertensive disorders of pregnancy. Hypertension Research 42: 29-39. [Crossref]