Abbreviations
AD: Alzheimer’s disease; CNS: central nervous system; CRE: cyclic AMP response element; CREB: CRE binding protein; DMEM: Dulbecco’s modified Eagle medium; DTPA: diethylenetriamine-N,N,N’,N”,N”-pentaacetic acid; Ex: exendin; FBS: fetal bovine serum; GLP-1: glucagon-like peptide-1; GSH: glutathione; HKR: HEPES-buffered Krebs Ringer solution; HPX: hypoxanthine; HRP: horseradish peroxidase; MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide; NBT: nitrotetrazolium blue chloride; NF-kB: nuclear factor-kappa B; O2–: superoxide anion; PBS: phosphate-buffered saline; PD: Parkinson’s disease; ROS: reactive oxygen species; RT-PCR: reverse transcription polymerase chain reaction; SOD: superoxide dismutase; WST-1: water soluble tetrazolium-1; XOD: xanthine oxidase.
Abstract
Under some pathological conditions in brain, a large amount of superoxide anion (O2–) is produced, causing various cellular damages. Among three isozymes of superoxide dismutase (SOD), extracellular (EC)-SOD should play a role to detoxify O2– in extracellular space; however, a little is known about EC-SOD in brain. Exendin 4 (Ex-4), an analogue of glucagon-like peptide-1 (GLP-1), binds to GLP-1 receptor to potentiate insulin secretion in pancreatic b cells and is used extensively as a drug for type 2 diabetes mellitus. It was reported that Ex-4 might represent neuroprotective effects; however, the details of mechanisms and the effects on glial cells were unclear. In the present study, we examined the effects of Ex-4 on EC-SOD expression in cultured rat cortical astrocytes. By means of RT-PCR, EC-SOD mRNA was increased by Ex-4 exposure in a time-dependent manner. The expression of EC-SOD protein was also increased by Ex-4 exposure for 24 h dose-dependently, and exendin (9-39), an antagonist of GLP-1 receptor, inhibited Ex-4-increased EC-SOD protein expression. Moreover, the cell-surface SOD activity in astrocytes and the activity of SOD released in the medium were not significantly affected by incubation of Ex-4 and/or exendin (9-39) for 24 h. These results suggest that Ex-4 might increase EC-SOD expression via binding to GLP-1 receptor although EC-SOD activities in astrocytes and in the medium were not affected. The regulation of EC-SOD in astrocytes may contribute to the defensive mechanism against oxidative stress in brain.
Keywords
astrocyte, extracellular superoxide dismutase, exendin, GLP-1
Introduction
Exendin (Ex)-4 is a peptide found from saliva of Gila monster (Heloderma suspectum) consisting of 39 amino acids, an analog of glucagon-like peptide-1 (GLP-1), and an agonist of GLP-1 receptor [1,2]. GLP-1 binds to GLP-1 receptor in pancreatic b cells and potentiates glucose-dependent insulin secretion to decrease blood glucose levels [3]. Ex-4 has 53% amino acids homologous to GLP-1, has higher affinity to GLP-1 receptor than GLP-1, and is less degraded in serum by dipeptidyl peptidase [1]. Therefore, Ex-4 recently has been used as a drug for type 2 diabetes mellitus.
It has been reported that GLP-1 is produced also in brain and that GLP-1 receptor expresses in neurons and glial cells widely in brain [4-8]. In vitro experiments, GLP-1 was reported to protect neurons against glutamate-induced excitotoxicity and against amyloid b-induced oxidative stress and cell death, and to suppress lipopolysaccharide-induced inflammatory cytokines in astrocytes [9-11]. In vivo experiments, GLP-1 was reported to protect neurons in rodent models of Alzheimer’s disease (AD) and Parkinson’s disease (PD) [12-14].
It was reported that Ex-4 permeated blood brain barrier [15]. In model mice of diabetes mellitus, Ex-4 injection could protect neurons through upregulation of glutamate transporters and neurotrophic factors [16,17]. In rodent models of brain ischemia, AD, and PD, Ex-4 could protect neurons from oxidative stress-induced impairment [18-21]. It was known that hyperglycemia in diabetes mellitus might elevate oxidative stress and induce mitochondrial impairment to cause neuronal toxicity [22,23]. Also in human ischemia, AD and PD, oxidative stress and metabolic disturbance might cause neuronal dysfunction [24-26].
Superoxide dismutase (SOD) is a unique enzyme that converts O2– into H2O2 and O2 by dismutating reaction [27] and known to be classified into three isozymes: cytosolic SOD (Cyt-SOD, Cu/Zn-SOD or SOD1) that contains Cu2+ and Zn2+ as enzyme cofactors, mitochondrial SOD (Mt-SOD, Mn-SOD or SOD2) in its matrix that contains Mn2+, and extracellular SOD (EC-SOD or SOD3) that contains Cu2+ and Zn2+ and has the enzymatic activity in extracellular space.
In the central nervous system (CNS), SOD isozymes are expressed and there are many reports suggesting that SOD plays important roles in defense against hypoxia-induced brain injury and neurodegenerative diseases such as PD [28-35]. However, most of these reports regard Cyt-SOD; not so much information on EC-SOD is known in CNS.
Although the expression level of EC-SOD in brain is considered to be much less than blood vessel or lung [36], it is reported that a proteolytic product of EC-SOD at C-terminal region is secreted in cerebrospinal fluid [37] and EC-SOD mRNA expression level increases in the brain after ischemia [38]. Furthermore, there are reports that mice overexpressing EC-SOD shows the increased resistance to focal cerebral ischemia [39,40], conversely EC-SOD deficiency in mice worsens outcome from focal cerebral ischemia [41]. Further, the beneficial function of EC-SOD is demonstrated in hyperoxia-induced brain injury in neonatal mice [42]. EC-SOD should play crucial roles in brain.
In addition, we previously reported that the expression level of EC-SOD in cultured astrocytes was higher than neurons and microglia by semi-quantitative RT-PCR and that lipopolysaccharide-stimulation increased SOD activity in the medium [43]. We also previously reported that dopamine incorporated into the cells thorough dopamine transporter triggered the EC-SOD induction via nuclear factor-kappa B (NF-kB) activation in cultured astrocytes [44]. Astrocytes play various important roles in CNS, such as maintenance of blood brain barrier, scavenging some neurotransmitters, control of ionic balance in brain parenchyma [45-47]. These functions of astrocytes serve the maintenance of brain homeostasis. Furthermore, it is reported that astrocytes protect endothelia from oxidative stress [48] and supply glutathione (GSH) to neurons and that astrocytes regulate the metabolism of ascorbic acid to protect neurons [49,50].
In the present study, we examined the effects of Ex-4 on EC-SOD expression and activity in cultured rat brain astrocytes. We found that exposure to Ex-4 increased the EC-SOD expressions in mRNA and protein, and SOD activities on the cell-surface and in the medium were not significantly affected by Ex-4.
Experimental procedures
Materials: Deoxyribonuclease I (DNase I; DN-25), trypsin, SOD (from bovine liver), xanthine oxidase (XOD; from milk), nitrotetrazolium blue chloride (NBT), anti-b-actin antibody and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (whole molecule) antibody were purchased from Sigma Chemical Co. (St Louis, MO, U.S.A.). Dulbecco’s modified Eagle medium (DMEM) and horse serum were obtained from Gibco BRL (Grand Island, NY, U.S.A.). Fetal bovine serum was obtained from Nichirei Biosciences Inc. (Tokyo, Japan). HRP-conjugated goat anti-mouse IgG (H+L) antibody was purchased from Bio-Rad Laboratories Inc. (Hercules, CA, U.S.A.). Hypoxanthine (HPX) was from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Water soluble tetrazolium-1 (WST-1, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), diethylenetriamine-N,N,N’,N’’,N’’-pentaacetic acid (DTPA) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide (MTT) were obtained from Dojindo (Kumamoto, Japan). Anti-EC-SOD antibody was purchased from Enzo Life Sciences, Inc. (Plymouth Meeting, PA, U.S.A.). FavorPrepTM Tissue Total RNA Purification Mini Kit was from Favorgen (Ping-Tung, Taiwan). Omniscript Reverse Transcription kit was from Qiagen (Hilden, Germany). SYBR® Green Realtime PCR Master Mix was from Toyobo (Osaka, Japan). EC-SOD primer and 18S rRNA primer were obtained from Operon Biotechnologies (Tokyo, Japan).
Preparation of astrocytes culture: This study was carried out in compliance with the Guideline for Animal Experimentation at Osaka Prefecture University, with an effort to minimize the number of animals used and their suffering. Astrocytes were prepared as described previously [51]. In brief, cortex from 20-day-old embryos, which were taken out from pregnant Wistar rats deeply anesthetized, were cleared of meninges, cut into about 1 mm3 blocks, and treated with 0.25% trypsin in Ca2+, Mg2+-free phosphate-buffered saline containing 5.5 mM glucose for 20 min at 37ºC with gentle shaking. An equal volume of horse serum supplemented with 0.1 mg/ml of DNase I was added to the medium to inactivate the trypsin. Then, the tissues were centrifuged at 350 x g for 5 min. The tissue sediments were triturated through a pipette with DMEM containing 10% fetal bovine serum, 100 µg/ml streptomycin and 50 unit/ml penicillin. After filtering cell suspensions through a lens-cleaning paper (Fujifilm Co., Tokyo, Japan), the cells were plated on polyethyleneimine-coated 100 mm-diameter plastic dishes (Iwaki, Asahi Glass Co., Tokyo, Japan) at a density of 0.8–1.3 x 105 cells/cm2. Cultures were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37ºC with changing medium every 3 days. After one week, astrocytes were replated to remove neurons. On days 12–14, they were replated onto 96-well plates (MS-8096F; for tissue culture, Sumitomo, Tokyo, Japan), 12-well plates (Iwaki), 35 mm-diameter plastic dishes (Thermo Fisher Scientific Inc., Waltham, MA, U.S.A.), or 60 mm-diameter plastic dishes (Thermo Fisher Scientific) using an ordinary trypsin-treatment technique at a density of 1.2 x 105 cells/cm2 and stabilized for 1 day, then we used for experiments.
More than 90% of the cells were immunoreactively positive to glial fibrillary acidic protein (GFAP) using the antibody (Sigma). Less than 10% of the cells were positive to Iba-1 using the antibody (Wako).
Cell viability: To evaluate cell viability, we measured total mitochondrial activity with so-called MTT assay. In brief, after the cells were stimulated, the medium was changed with a fresh one and one-tenth volume of 5 mg/ml MTT solution was added. The cells were incubated for 1 h at 37ºC and the formazan generated by total mitochondrial activity was dissolved in dimethylsulfoxide, and then the color development was measured at 585 nm with a microplate reader (ARVO 1420 Multilabel counter, Wallac, Turuk, Finland). When we observed cell morphology under a phase-contrast microscope, the remaining cell number is almost consistent with the results of MTT assay.
Reverse transcription-polymerase chain reaction (RT-PCR): Cultured astrocytes were washed with PBS, followed by extraction of mRNA using FavorPrepTM Tissue Total RNA Purification Mini Kit and subsequent synthesis of complementary DNA with oligo dT primers, mixture of dNTP (deoxyribonucleotide triphosphate), RNase inhibitor, Buffer RT and Omniscript Reverse Transcriptase (Omniscript Reverse Transcription Kit; Qiagen). Reverse transcriptase reaction was run at 37ºC for 60 min, followed by inactivation of the enzyme at 94ºC for 5 min, and an aliquot of synthesized complementary DNA was used for Realtime PCR.
Realtime PCR was performed in buffer containing SYBR® Green Realtime PCR Master Mix (Toyobo) and each primer for the corresponding EC-SOD and 18S rRNA. PCR was performed with primers specific for each EC-SOD and 18S rRNA described below. The conditions of each PCR cycles for these primers were as follows: denaturation at 95ºC for 15 sec; annealing at 60ºC for 15 sec; and extension at 60ºC for 30 sec. The results were analyzed using Realtime PCR System (StepOne™; Applied Biosystems).
Specific primers:
EC-SOD: sense 5’-ATGGTGGCCTTCTTGTTCTGC-3’
antisense 5’-CCAGATCTCCAGGTCTTTGGA-3’
18S rRNA: sense 5’- AGGTCTGTGATGCCCTTAGA-3’
antisense 5’- CCATCCAATCGGTAGTAGCG-3’
Western blotting: Cultured astrocytes were homogenized in 20 mM Tris-HCl (pH 7.5) buffer containing 1 mM EDTA and protease inhibitor cocktail (Sigma P8340). Each homogenate was added at a volume ratio of 4:1 to 50 mM Tris-HCl buffer (pH 6.8) containing 50% glycerol, 10% sodium dodecyl sulfate, 0.05% bromophenol blue and 25% 2-mercaptoethanol, followed by mixing and boiling at 100ºC for 5 min. Each aliquot in a certain amount of protein was loaded on a 10% polyacrylamide gel for electrophoresis at a constant voltage of 120 V for 2 h at room temperature and subsequent blotting to a polyvinylidene fluoride membrane previously treated with 100% methanol. After blocking by 5% skimmed milk dissolved in 20 mM Tris-HCl buffer (pH 7.5) containing 137 mM NaCl and 0.05% Tween 20, the membrane was reacted with antibodies against EC-SOD or b-actin followed by a reaction with anti-rabbit or anti-mouse IgG antibody conjugated with peroxidase. Proteins reactive with those antibodies were detected with the aid of chemiluminescence detection reagents (Immobilon™ Western HRP Substrate; Millipore Corporation, Billerica, MA, U.S.A.) and analyzed with lumino-image-analyzer (LAS-4000, Fujifilm). The graphs showed EC-SOD/b-actin ratio of the density of detection bands.
Protein concentrations were determined by the method of Bradford using CBB color solution (Nacalai Tesque, Kyoto, Japan), according to the manufacturer’s protocol, with bovine serum albumin as the standard.
Measurement of SOD activity on cell surface: SOD activity on cell surface was measured by the assay as described previously [43]. After the cells were rinsed with HEPES-buffered Krebs Ringer solution (HKR; 130 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 50 mM HEPES, 1 mM NaH2PO4, 5.6 mM glucose, 1.8 mM CaCl2, pH 7.4) twice, 90 µl of 0.3 mM HPX, 0.3 mM DTPA and 33 µM WST-1 in HKR was added into each well. Then the reaction was started with the addition of 20 µl of 46 mU/ml XOD and the absorbance (Abs) at 450 nm was immediately measured as blank, using a microplate reader. The plate was incubated in a CO2 incubator for 30 min, and then the absorbance was measured. For standard, various concentrations of SOD enzyme (Sigma S-8160 from bovine liver) were added into cell-free wells in the same plate; then, the absorbance was measured simultaneously.
The standard curve was established as follows. The absorbance at 30 min was used; at first, the blank values (time 0) were subtracted. The difference between each value and 0 SOD was plotted against the standard SOD concentrations: the central linear part of the sigmoid curve was used for the calculation of SOD activity. This procedure is based on the inhibition of O2– detection with an artificial O2– generator; therefore, the procedure is not applicable when the cells themselves have O2– generating activity. We examined the color development of WST-1 added on the astrocytes without addition of XOD: the absorbance change was less than 0.002 Abs/h and revealed that astrocytes did generate almost no O2–.
Measurement of SOD activity in medium: We had attempted to measure the SOD activity in the medium released from the cell similarly as described above; however, the measurement was disturbed by some components of the DMEM. Therefore, in order to measure the SOD activity released into the medium, we used HKR for the drug-stimulation reaction instead of DMEM. It is supplemented that cell viability in HKR was not different from that in DMEM with and without LPS (data not shown).
Data analysis: For statistical analysis of the data, one-way ANOVA followed by Tukey-Kramer multiple comparison procedure or Student’s t-test was used. Differences between treatments were considered statistically significant when p < 0.05.
Results
Expression of EC-SOD mRNA increased in astrocytes after Ex-4 stimulation: Cultured astrocytes were stimulated with 10 nM Ex-4 for indicated time, and then the mRNA expressions EC-SOD were analyzed by realtime RT-PCR. The expression of EC-SOD mRNA was significantly increased by 10 nM Ex-4 for 3 h exposure, it was remarkably increased at 6 h and the increased level was sustained up to 24 h (Figure 1).
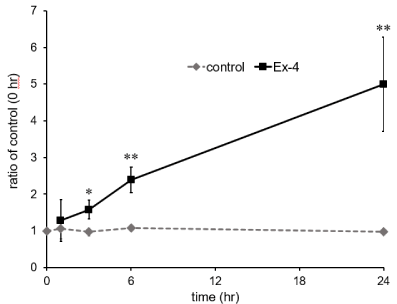
Figure 1. Time course of EC-SOD mRNA expression in cultured astrocytes after Ex-4
exposure. Cultured astrocytes were stimulated by 10 nM Ex-4 for 1, 3, 6, 24 h. The
expression of mRNA of EC-SOD was assessed by realtime RT-PCR. Data are mean ±
S.D. of three samples from different cell preparations. *P < 0.05, **P < 0.01, significantly
different from control.
Effect of Ex-4 and/or exendin (9-39) on the EC-SOD protein: In addition to the mRNA level, we examined the level of EC-SOD protein. The cells were stimulated with various concentrations of Ex-4 for 24 h, and then the expression of EC-SOD protein was assessed by western blotting. The expression level of EC-SOD protein increased in a dose-dependent manner and with 10 nM Ex-4, significantly (Figure 2A).
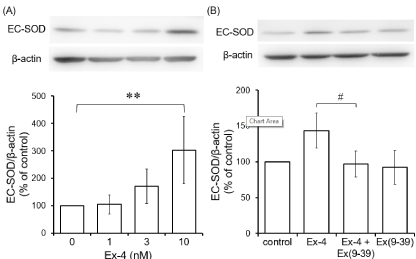
Figure 2. Effects of Ex-4 and Ex (9-39) on expression of EC-SOD protein in cultured
astrocytes. (A) Cultured astrocytes were stimulated by various concentrations of Ex-4 for
24 h. (B) The cells were preincubated with 100 nM Ex (9-39) for 30 min and stimulated by
10 nM Ex-4 for 24 h. The expression of EC-SOD protein was detected by western blotting.
Typical bands of western blotting for EC-SOD and β-actin proteins are shown in the
photograph. The graph shows EC-SOD/β-actin ratio of the density of detection bands. Data
are mean ± S.D. of five samples from different cell preparations. **P < 0.01, significantly
different from control. #
P < 0.05, significantly different from Ex-4.
The cells were preincubated with 100 nM exendin (9-39), an antagonist of GLP-1 receptor, for 30 min and stimulated by 10 nM Ex-4 for 24 h, and then the expression of EC-SOD protein was assessed by western blotting. Preincubation with 100 nM exendin (9-39) inhibited Ex-4-increased EC-SOD expression (Figure 2B).
Effect of Ex-4 on cell-surface SOD activity and the cell viability: We examined the effect of Ex-4 on cell-surface SOD activity in cultured astrocytes. The cells were incubated with various concentrations of Ex-4 for 24 h, and washed with HKR. Then, SOD activity on the cell-surface was assayed. The activity was not significantly affected (Figure 3A).
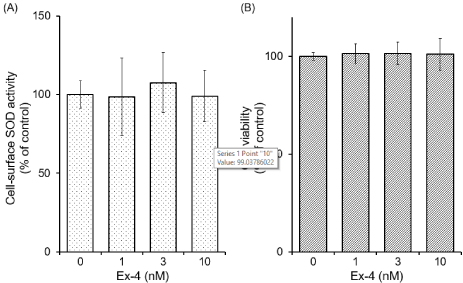
Figure 3. Effects of Ex-4 on the cell-surface SOD activity and the cell viability in cultured
astrocytes. Cultured astrocytes were stimulated by various concentrations of Ex-4 for 24 h.
(A) After the stimulation, the cells were washed with HKR. Then, SOD activity remained
on the cell-surface was assayed. (B) The cell viability was assessed by MTT assay. Data are
mean ± S.D. of four samples from different cell preparations.
We also examined the effect of Ex-4 on cell viability in cultured astrocytes. The cells were incubated with various concentrations of Ex-4 for 24 h, and cell viability was assessed by MTT assay. Cell viability was not affected by Ex-4 used in the present study (Figure 3B).
Effects of Ex-4 and/or exendin (9-39) on SOD activity in the medium: We examined the effect of Ex-4 on SOD activity in the medium. Cultured astrocytes were preincubated with 100 nM exendin (9-39) for 30 min and stimulated by 10 nM Ex-4 for 24 h in HKR, and then SOD activity in the medium was assayed. The activity was affected by neither Ex-4 nor exendin (9-39) (Figure 4).
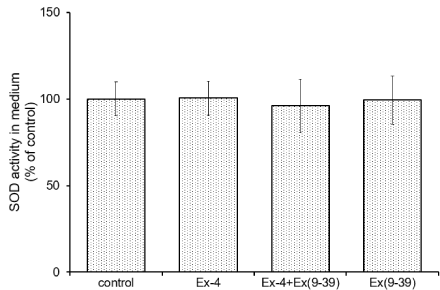
Figure 4. Effects of Ex-4 and Ex (9-39) on activity of the released SOD in the medium.
Cultured astrocytes were preincubated with 100 nM Ex (9-39) for 30 min and stimulated by
10 nM Ex-4 for 24 h in HKR. The medium was assayed for the released SOD activity. Data
are mean ± S.D. of four samples from different cell preparations.
Discussion
In the present study, we demonstrated that the expressions of EC-SOD, both of mRNA and protein, were induced when the cells were stimulated by Ex-4 in cultured rat cortex astrocytes. However, the activities of SOD on the cell-surface and in the medium were not significantly affected by Ex-4. The Ex-4-increased EC-SOD expression was inhibited by an antagonist of GLP-1 receptor. These results suggest that Ex-4 increases the expressions of EC-SOD mRNA and protein via binding to GLP-1 receptor.
In cultured astrocytes, 10 nM Ex-4 increased EC-SOD mRNA time-dependently for 1-24 h, and 1-10 nM Ex-4 exposure for 24 h increased EC-SOD protein in a dose-dependent manner, significantly with 10 nM. The increased expression of EC-SOD protein by Ex-4 was blocked by exendin (9-39), an antagonist of GLP-1 receptor. GLP-1 receptor is a G-protein coupled receptor, and binding of agonists to it causes activation of adenylate cyclase to increase cAMP and activate protein kinase A [52]. Ex-4 treatment reported to induce the elevation of intracellular cAMP levels in astrocytes [52]. The promoter region of EC-SOD contains various regulatory elements including cAMP response element (CRE) [53]. Therefore, Ex-4 in the present study might increase EC-SOD mRNA via intracellular cAMP production. Moreover, the promoter region of EC-SOD contains various regulatory elements other than CRE, including antioxidant response element, activator protein-1 binding sites, NF-kB motifs, and xenobiotic response elements [53]. Further investigation is necessary to elucidate the total mechanisms of the EC-SOD induction by Ex-4 stimulation, inclusive of other transcriptional factors as well as CRE-binding protein (CREB).
It was reported that Ex-4 administration protected neurons through the upregulation of antioxidants in ischemic rat brain [18]. However, in the present study, the activities of SOD on the cell-surface and in the medium were not significantly affected by Ex-4 treatment for 24 h. The effects of Ex-4 on mRNA and protein only in EC-SOD among three isozymes of SOD were assessed in the present study; therefore, the effects of Ex-4 on the expressions of other two SODs, Cu/Zn-SOD and Mn-SOD should be further investigated. Moreover, it is likely that Ex-4 might affect the utilization efficiency of Cu2+ and Zn2+ which are contained as enzyme cofactors and needed for enzymatic activity of EC-SOD.
EC-SOD is a unique enzyme having the enzymatic activity in extracellular space and at least two steps are known to be occurred for secretion [27,54]. It was reported that N-glycosylation was essential for the transport of intracellular EC-SOD protein to cellular membrane [55]. Further, EC-SOD is known to be released through the proteolytic processing at binding region in various cells including glial cell line U1169 CG [56]. The C-terminal proteolytic processing of EC-SOD was reported to be regulated by redox state [57]. In the present study, these post-translational modifications might not be stimulated by Ex-4 treatment in cultured astrocytes although Ex-4 increased the expressions of EC-SOD mRNA and protein. Further investigation is needed to elucidate the effects of Ex-4 on SOD activity in more detail; however, the induced EC-SOD in astrocytes by Ex-4 exposure might play a role in neuronal protection.
Conclusion
In the present study, Ex-4 triggered the EC-SOD induction via binding to GLP-1 receptor in cultured astrocytes. EC-SOD induced by Ex-4 in astrocytes may play a role in extracellular anti-oxidative defense for surrounding neurons. The regulation of EC-SOD expression in astrocytes should be an important target in treatment of CNS diseases.
Acknowledgement
This work was supported in part by JSPS KAKENHI Grant Number JP15J12259 to K.K., JP26850209 to K.T., JP26450447 to M.M., and JP15K07768 to Y.N.
References
- 1. Eng J (1992) Exendin peptides. Mt Sinai J Med 59: 147-149.
- 2. Goke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, et al. (1993) Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting beta-cells. J Biol Chem 268: 19650-19655.
- 3. Gefel D, Hendrick GK, Mojsov S, Habener J, Weir GC (1990) Glucagon-like peptide-I analogs, effects on insulin secretion and adenosine 3′,5′-monophosphate formation. Endocrinology 126: 2164-2168.
- 4. Campos RV, Lee YC, Drucker DJ (1994) Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 134: 2156-2164.
- 5. Calvo JC, Yusta B, Mora F, Blázquez E (1995) Structural characterization by affinity cross-linking of glucagon-like peptide‐1 (7–36) amide receptor in rat brain. J Neurochem 64: 299-306.
- 6. Mora F, Expösito I, Sanz B, Blazquez E (1992) Selective release of glutamine and glutamic acid produced by perfusion of GLP-1 (7–36) amide in the basal ganglia of the conscious rat. Brain Res Bull 29: 359-361.
- 7. Satoh F, Beak SA, Small CJ, Falzon M, Ghatei MA, et al. (2000) Characterization of human and rat glucagon-like peptide-1 receptors in the neurointermediate lobe, lack of coupling to either stimulation or inhibition of adenylyl cyclase 1. Endocrinology 141: 1301-1309.
- 8. Uttenthal LO, Toledano A, Blázquez E (1992) Autoradiographic localization of receptors for glucagon-like peptide-1 (7–36) amide in rat brain. Neuropeptides 21: 143-146.
- 9. Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J (2006) Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci Res 55: 352-360.
- 10. Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH (2002) Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Experiment Therapeut 302: 881-888.
- 11. Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, et al. (2003) Glucagon‐like peptide‐1 decreases endogenous amyloid‐b peptide (Ab) levels and protects hippocampal neurons from death induced by Ab and iron. J Neurosci Res 72: 603-612.
- 12. Harkavyi A, Whitton PS (2010) Glucagon‐like peptide 1 receptor stimulation as a means of neuroprotection. Br J Pharmacol 159: 495-501.
- 13. Holst JJ, Burcelin R, Nathanson E (2011) Neuroprotective properties of GLP-1, theoretical and practical applications. Curr Med Res Opin 27: 547-558.
- 14. Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, et al. (2009) GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci 106: 1285-1290.
- 15. Kastin AJ, Akerstrom V (2003) Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes 27: 313-318.
- 16. Gumuslu E, Mutlu O, Celikyurt IK, Ulak G, Akar F, et al. (2016) Exenatide enhances cognitive performance and upregulates neurotrophic factor gene expression levels in diabetic mice. Fundam Clin Pharmacol 30: 376-384.
- 17. Zhang Y, Zhang J, Wang Q, Lei X, Chu Q, et al. (2011) Intravitreal injection of exendin-4 analogue protects retinal cells in early diabetic rats. Invest Ophthalmol Vis Sci 52: 278-285.
- 18. Briyal S, Gulati K, Gulati A (2012) Repeated administration of exendin-4 reduces focal cerebral ischemia-induced infarction in rats. Brain Res 1427: 23-34.
- 19. Kim S, Moon M, Park S (2009) Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson's disease. J Endocrinol 202: 431-439.
- 20. Lee CH, Yan B, Yoo KY, Choi JH, Kwon SH, et al. (2011) Ischemia‐induced changes in glucagon‐like peptide‐1 receptor and neuroprotective effect of its agonist, exendin‐4, in experimental transient cerebral ischemia. J Neurosci Res 89: 1103-1113.
- 21. Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, et al. (2010) GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. J Alzheimer's Dis 19: 1205-1219.
- 22. Piconi L, Quagliaro L, Ceriello A (2003) Oxidative stress in diabetes. Clin Chem Lab Med 41: 1144-1149.
- 23. Yang CM, Lin CC, Hsieh HL (2017) High-glucose-derived oxidative stress-dependent heme oxygenase-1 expression from astrocytes contributes to the neuronal apoptosis. Mol Neurobiol 54: 470-483.
- 24. Benzi G, Moretti A (1995) Are reactive oxygen species involved in Alzheimer's disease? Neurobiol Aging 16: 661-674.
- 25. Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson's disease, is there a causal link?. Experiment Neurol 193: 279-290.
- 26. Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, et al. (2006) Involvement of oxidative stress in Alzheimer disease. J Neuropathol Experiment Neurol 65: 631-641.
- 27. McCord JM, Fridovich I (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244: 6049-6055.
- 28. Bowling AC, Beal MF (1995) Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci 56: 1151-1171.
- 29. Chan PH, Chu L, Chen SF, Carlson EJ, Epstein CJ (1990) Reduced neurotoxicity in transgenic mice overexpressing human copper-zinc-superoxide dismutase. Stroke 21: 80-82.
- 30. Kawase M, Murakami K, Fujimura M, Morita-Fujimura Y, Gasche Y, et al. (1999) Exacerbation of delayed cell injury after transient global ischemia in mutant mice with CuZn superoxide dismutase deficiency. Stroke 30: 1962-1968.
- 31. Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen H-C, et al. (1998) Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci 18: 687-697.
- 32. Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, et al. (1997) Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci 17: 4180-4189.
- 33. Li Y, Copin J-C, Reola LF, Calagui B, Gobbel GT, et al. (1998) Reduced mitochondrial manganese-superoxide dismutase activity exacerbates glutamate toxicity in cultured mouse cortical neurons. Brain Res 814: 164-170.
- 34. Matsuyama T, Michishita H, Nakamura H, Tsuchiyama M, Shimizu S, et al. (1993) Induction of copper-zinc superoxide dismutase in gerbil hippocampus after ischemia. J Cereb Blood Flow Met 13: 135-144.
- 35. Murakami K, Kondo T, Kawase M, Li Y, Sato S, et al. (1998) Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci 18: 205-213.
- 36. Marklund SL (1984) Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest 74: 1398-1403.
- 37. Ohe Y, Ishikawa K, Itoh Z, Tatemoto K (1996) Cultured leptomeningeal cells secrete cerebrospinal fluid proteins. J Neurochem 67: 964-971.
- 38. Fukui S, Ookawara T, Nawashiro H, Suzuki K, Shima K (2002) Post-ischemic transcriptional and translational responses of ec-sod in mouse brain and serum. Free Radical Biol Med 32: 289-298.
- 39. Sheng H, Bart RD, Oury TD, Pearlstein RD, Crapo JD, et al. (1999a) Mice overexpressing extracellular superoxide dismutase have increased resistance to focal cerebral ischemia. Neuroscience 88: 185-191.
- 40. Zaghloul N, Patel H, Codipilly C, Marambaud P, Dewey S, et al. (2014) Overexpression of extracellular superoxide dismutase protects against brain injury induced by chronic hypoxia. PLoS One 9: e108168.
- 41. Sheng H, Brady TC, Pearlstein RD, Crapo JD, Warner DS (1999b) Extracellular superoxide dismutase deficiency worsens outcome from focal cerebral ischemia in the mouse. Neurosci Lett 267: 13-16.
- 42. Zaghloul N, Nasim M, Patel H, Codipilly C, Marambaud P, et al. (2012) Overexpression of extracellular superoxide dismutase has a protective role against hyperoxia-induced brain injury in neonatal mice. FEBS J 279: 871-881.
- 43. Iitsuka I, Motoyoshi-Yamashiro A, Moriyama M, Kannan-Hayashi Y, Fujimoto Y, et al. (2012) Extracellular superoxide dismutase in cultured astrocytes: Decrease in cell-surface activity and increase in medium activity by lipopolysaccharide-stimulation. Neurochem Res 37: 2108-2116.
- 44. Takano K, Tanaka N, Kawabe K, Moriyama M, Nakamura Y (2013) Extracellular superoxide dismutase induced by dopamine in cultured Astrocytes. Neurochem Res 38: 32-41.
- 45. Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier, an overview, structure, regulation, and clinical implications. Neurobiol Dis 16: 1-13.
- 46. Merrill JE, Jonakait GM (1995) Interactions of the nervous and immune systems in development, normal brain homeostasis, and disease. FASEB J 9: 611-618.
- 47. Pierre K, Pellerin L (2005) Monocarboxylate transporters in the central nervous system, distribution, regulation and function. J Neurochem 94: 1-14.
- 48. Schroeter ML, Mertsch K, Giese H, Müller S, Sporbert A, et al. (1999) Astrocytes enhance radical defence in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett 449: 241-244.
- 49. Stewart VC, Stone R, Gegg ME, Sharpe MA, Hurst RD, et al. (2002) Preservation of extracellular glutathione by an astrocyte derived factor with properties comparable to extracellular superoxide dismutase. J Neurochem 83: 984-991.
- 50. Wilson JX (1997) Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol 75: 1149-1163.
- 51. Takano K, Sugita K, Moriyama M, Hashida K, Hibino S, et al. (2011) A dibenzoylmethane derivative protects against hydrogen peroxide-induced cell death and inhibits lipopolysaccharide-induced nitric oxide production in cultured rat astrocytes. J Neurosci Res 89: 955-965.
- 52. Reiner DJ, Mietlicki-Baase EG, McGrath LE, Zimmer DJ, Bence KK, et al. (2016) Astrocytes regulate GLP-1 receptor-mediated effects on energy balance. J Neurosci 36: 3531-3540.
- 53. Fattman CL, Schaefer LM, Oury TD (2003) Extracellular superoxide dismutase in biology and medicine. Free Radical Biol Med 35: 236-256.
- 54. James DC, Enghild JJ (2004) The intracellular proteolytic processing of extracellular superoxide dismutase (EC-SOD) is a two-step event. J Biol Chem 279: 22152-22157.
- 55. Ota F, Kizuka Y, Kitazume S, Adachi T, Taniguchi N (2016) N‐Glycosylation is essential for the secretion of extracellular superoxide dismutase. FEBS Lett 590: 3357-3367.
- 56. Marklund SL (1990) Expression of extracellular superoxide dismutase by human cell lines. Biochem J 266: 213-219.
- 57. Gottfredsen RH, Tran SMH, Larsen UG, Madsen P, Nielsen MS, et al. (2012) The C-terminal proteolytic processing of extracellular superoxide dismutase is redox regulated. Free Radic Biol Med 52: 191-197.