Inhaled bronchodilators with or without inhaled steroids (IGCs) are the mainstay of pharmacological treatment for stable obstructive airway diseases, including beta2-agonists and muscarinic antagonists. Long-acting muscarinic antagonist bronchodilators (LAMA) and long-acting β2-agonists (LABA) are the treatment of choice for moderate to severe chronic obstructive pulmonary disease (COPD). Also, some studies have demonstrated the benefit of LAMA in the treatment of difficult-to-control asthma and the potential use in asthma-COPD overlap syndrome (ACOS). Control of airway tone in health and disease states, cholinergic and adrenergic receptors, signaling, molecular biology of mediators, and airway muscle contraction / relaxation mechanisms are reviewed concisely and as they may be handled pharmacologically. Subsequently, the evidence of the role of the LABA / LAMA combination in the aforementioned entities, their complementary mechanisms of action and other alternative mechanisms of action as well as potential deleterious effects is discussed.
COPD, bronchial asthma, LAMA/LABA combination
There are two major systems of control in the human economy, the nervous system and the endocrine system. Both control systems can be manipulated by therapeutic drugs, which may resemble or block the usual action of the control system, to produce or inhibit a physiological effect. The general organization of the nervous system can be delineated as follows: central nervous system (brain and spinal cord); the peripheral nervous system with sensory (afferent), motor (somatic or efferent) neurons and the autonomic nervous system with parasympathetic and sympathetic branches [1]. The autonomic nervous system is an unconscious and involuntary control mechanism that regulates visceral or vegetative functions. The parasympathetic system is finely regulated and controls day-to-day respiratory, cardiac, gastrointestinal and urinary bladder functions. Some of these functions have been called SLUD syndrome (salivation, lacrimation, urination and defecation). In contrast, the sympathetic system is a “fight or flight” alarm system that prepares the body for maximum acute bodily function, and affects bronchial smooth muscle (bronchodilation) as well as blood flow (increased heart rate and blood pressure) [2].
The parasympathetic nerve fibers originate from the craniosacral regions of the spinal cord, with the vagus nerve originating in the brain stem as the tenth cranial nerve and a small parasympathetic innervation leaving the sacral portion of the spinal cord. The vagus (wandered) innervates many of the organs of the thorax and abdomen, and is involved in the parasympathetic innervation of the airway. The preganglionic fibers are long, and in the thorax they form a local synapse in the autonomic pulmonary plexus around the airway wall, whereas the postganglionic fibers are very short innervating the smooth muscle, and the mucous glands of said wall. The neurotransmitter of the parasympathetic branch is acetylcholine (Ach) which is concentrated in the presynaptic neurons (both in the ganglia and at the effector site). Ach is synthesized from acetyl CoA and choline by the enzyme choline acetyltransferase. Ach is stored in vesicles (1000 to 50,000 molecules per vesicles). When a nervous impulse (action potential) reaches the presynaptic neuron, an influx of calcium into the neuron causes exocytosis of the vesicles containing the neurotransmitter at the end of nervous termination, joining the receptors in the postsynaptic membrane initiating its effect on the tissue or organ. Ach is then inactivated through hydrolysis by cholinesterase, which catabolize it in Acetyl CoA and choline and acetate, ending with stimulation in the postsynaptic membrane [1]. Non-neuronal cholinergic system is widely expressed in epithelial cells, eosinophils, submucosal glands, smooth muscle cells and a variety of immune cells, including lymphocytes, macrophages and mast cells of the airway. This suggests that non-neuronal cholinergic signals could play an important role in the pathophysiology of COPD and bronchial asthma [3]. Therefore, the use of anticholinergics may be beneficial in obstructive airway diseases, to block cholinergic bronchoconstriction not only neuronal but also non-neuronal. The normal airways have a small degree of cholinergic vagal tone, but as the airways are patent this effect is not perceptible and airflow reduction is minimal, but when the airways are irreversibly obstructed in COPD, the cholinergic vagal tone has greater effect on airway resistance for geometric reasons. When this increased constriction is relieved by anticholinergic drugs, a noticeable improvement in airflow occurs.
Sympathetic nerve fibers originate from the thoracolumbar portions of the spinal cord. The first 4 roots (T1-T4) generate sympathetic innervation of the airway. The short preganglionic fibers very soon make synapses within the ganglia of the right and left sympathetic trunks that move vertically, lateral to the medulla and posterior in the thoracic and abdominal wall. The long postganglionic sympathetic fibers are mixed with the parasympathetic vagal fibers to form the autonomic pulmonary plexus. Branches of this plexus accompany the bronchi and bronchioles. In the presynaptic neuron, tyrosine is converted to dopa, dopamine and by means of dopamine β-hydroxylase to norepinephrine, accumulating in the deposit vesicles. The opening of calcium channels by action potentials allows the influx of calcium which produces exocytosis of the vesicles with the neurotransmitter which binds to the receptor in the postsynaptic membrane. As the adrenal medulla releases noradrenaline (norepinephrine US) and adrenaline (epinephrine US), circulating catecholamine can stimulate adrenergic receptors throughout the body without necessarily having nerve fibers present. Noradrenaline molecules in the synaptic space may experience re-uptake by the presynaptic neuron for further reuse and / or be captured by postsynaptic muscle termination and metabolized by monoamine oxidase and catechol-ortho-methyl transferase (MAO and COMT respectively) [1,4].
Since the appearance of short-acting bronchodilators (SABA), which stimulate the β2 receptor and bronchodilators that block muscarinic receptors (SAMA), large-scale studies with pressurized metered-dose inhalers (pMDI) were designed that demonstrated the superiority of the combination of ipatropium with salbutamol (Combivent ®) on the individual elements (ipatropium bromide alone and salbutamol alone) and the benefits of the nebulized combination were also identified [5-9]. With the design of long-acting bronchodilator molecules (LAMAs, LABAs and ULTRA-LABAs) and devices that allow them to be used together and dispensed every 12-24 hours, the combination has become a useful and frequent tool in the treatment of obstructive airways diseases. A condensate of the latest works and evidence of his indications will be presented in this article.
The plant ma huang (Ephedra sinica) has been used in traditional Chinese medicine for the treatment of asthma for over 5000 years [10]. Ephedra is a shrub-like plant that grows in many desert regions, although Ephedra sinica is found especially in northern China and Mongolia. The crude powdered stem of Ephedra sinica is rich in ephedrine, a plant-derived compound with adrenaline-like properties and is ingested as tea. It was not until 1900 that Solis-Cohen used "adrenal substance" to treat asthma, with the successful use of adrenaline (epinephrine) for asthma attacks reported in 1903. Chemically synthesized β-selective isoprenaline was introduced in 1948, while β2-selective agonist, salbutamol (albuterol) was introduced as a bronchodilator in 1969, followed by terbutaline in 1974. LABAs, salmeterol and formoterol became available as inhaled agents in clinical practice in the 1990s, to be used every 12 hours [11]. ULTRA-LABAs (LABAs for once-daily use) such as indacaterol, vilanterol, and olodaterol have recently been developed and approved [12].
Bronchodilator therapy is based on the effects of drugs on the autonomic (involuntary) nervous system. In 1948, Ahlquist distinguished the sympathetic alpha (α) and beta (β) receptors based on different responses to adrenergic drugs [13], and in 1967 Lands differentiated the β receptors into β1 and β2 [14]. Β1 receptors are found primarily in cardiac muscle and β2 in other muscles including skeletal muscle and vascular and bronchial smooth muscle and in another variety of cells as indicated in the introduction. A third type of β-receptor, β3 is found in fat cells and its stimulus produces lipolysis.
The stimulation of the β2-adrenergic receptors (β2-AR) by agonists causes bronchodilation by activation of the β2-AR found on the surface of the smooth muscle cell membrane of the airway. These receptors are distributed from the trachea to the terminal bronchioles and the density increases as the diameter becomes smaller. β2-Agonists therefore, cause relaxation of small airways (<2 mm in diameter) [15]. They are calculated between 30-40,000 β2-AR in each smooth muscle cell. β2-AR is a serpiginous receptor, member of a seven-domain transmembrane receptor family (TMD), which are seven α-helices that cross the plasma membrane, and is composed of 413 amino acids [16]. The terminal amino group is outside the cell, while the terminal carboxyl group is in the cellular cytoplasm.
Occupation of β2-AR by neurotransmitters (catecholamine or by β2-agonists) results in the activation of adenylyl cyclase, vía the stimulatory G-protein (Gs), which catalyzes the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate cAMP [17]. This second messenger (cAMP) within the smooth muscle cell activates protein kinase A (PKA), which phosphorylates several target proteins within the cell. When PKA phosphorylates myosin regulatory light chain kinases (MLCK), the affinity for calmodulin / calcium decreases, and muscle relaxation occurs (Figure 1). The activity of MLCK and calmodulin / calcium is essential for the formation of bridges between actin and myosin and for muscle contraction. This interaction is diminished or inhibited by the occupation of β2-AR. In addition, the inositol triphosphate receptor (IP3R) in the endoplasmic reticulum (ER) is phosphorylated by PKA, decreasing the release of calcium from the ER to the cytosol. This sequestration of calcium within the intracellular organelles further decreases the availability of calcium in the cytoplasm, favoring and amplifying the decreased calmodulin / calcium interaction with MLCK. In addition, PKA can phosphorylate the potassium channels favoring the outflow of the same out of the cell, depolarization and relaxation of smooth muscle. It is obvious that these mechanisms, via cAMP / PKA, act in concert to produce muscle relaxation of the airway [18].
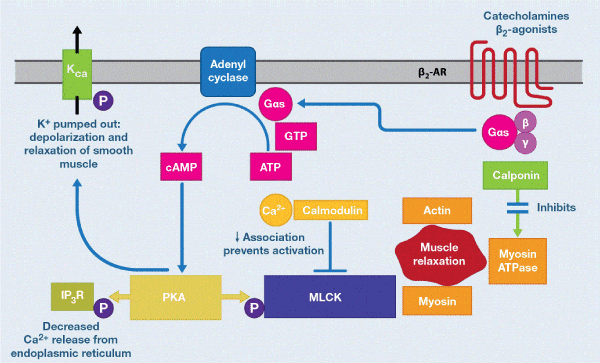
Figure 1. Β2-adrenoreceptor-mediated smooth muscle relaxation: cell signaling. Smooth muscle relaxation is caused by binding of agonists (catecholamine, SABA, LABA or ULTRA-LABA) to the β2-adrenoreceptor. This is a Gαs-coupled receptor that stimulates adenylyl, cyclase, resulting in the conversion of ATP to cAMP. Increased cAMP levels activate protein kinase A (PKA) which phosphorylates myosin regulatory light chain kinase (MLCK). This decreases its association with calcium/calmodulin. In addition, inositol triphosphate receptors (IP3R) on the endoplasmic reticulum (ER) are phosphorylated by PKA; this decreases Ca2+ release from the ER into the cytosol. The two pathways result in depolarization of the smooth muscle. Adapted from Figure 4A of Jhonson EN.
Stimulated β2-AR can also bind to Kca channels via Gs, and relaxation of airway smooth muscle can occur independently of the intracellular increase of cAMP. Also, and independent of cAMP, the direct stimulation of β2-AR can activate calponin which inhibits the myosin ATPase, which in turn blocks the actin / myosin interaction producing relaxation. Some other actions of β2-agonists are mediated via other cAMP-regulated proteins, such as EPAC (exchange protein activated by cAMP) [19]. For example, inhibition of airway smooth muscle proliferation by β2-agonists appears to be dependent on EPAC rather than PKA. The smooth muscle of the airways shows resistance to desensitization of β2-AR, probably because of the large reserve of receptors and the low levels of expression of the GRK2 (G-protein receptor kinase-2) enzyme, which phosphorylates and inactivates receptors β2 occupied [20]. However, it is unclear how far abnormal signals occur in the smooth muscle of the airways in bronchial asthma and COPD.
Β2-agonists may have additional effects on airways, as β2-receptors are localized to several different cells types in the airways. They may therefore cause bronchodilation in vivo not only by a direct action on airway smooth muscle but also indirectly by inhibiting the release of bronchoconstrictors mediators from inflammatory cells and of neurotransmitters from airway nerves. For example, β2-agonists inhibit mediator release of mast cell through closing an intermediate conductance Ca2+-activated K channel (KCa3.1) coupled to Gs [21]. Stem cell factor, which is secreted by epithelial cells in asthmatic patients, is an important factor in keeping mast cells at the airway surface in asthma and counteracts this effect of β2-agonists [22]. The anti-inflammatory effects of β2-agonists in asthma are controversial. The inhibitory effect on mast cell mediator release and microvascular leakage are clearly anti-inflammatory, suggesting modification in acute inflammation, but they do not have significant inhibitory effect on the chronic inflammation of asthmatic airways, in contrast to the resolution of inflammation that occurs with inhaled IGCs [23]. This is like to be related to the fact that β2-agonists effects on macrophages, eosinophils and lymphocytes are rapidly desensitized due to the low density of β2-AR on these cells and high expression of GRK2 [20], contrary to what occurs in smooth muscle cells of the airway. Indeed, exposure to LABA increases the expression of GRK2 and GRK5 in human peripheral lung [24].
In another order, it is now recognized that, although β2-AR are coupled through Gs to relax airway smooth muscle, they may activate alternative signaling pathways that may have deleterious effects, such as increasing inflammation. When β2-overexpressing occurs as in asthma, Gq coupled to phospholipase Cβ1 (PLCβ1), is activated, resulting in enhanced bronchoconstrictor response to mediators such as ACh and histamine, that signal through Gq what favors the inflammatory process [25]. β2-AR also interacts with β-arrestin-2, which in turn interacts with p38MAPK and PI3K which are pro-inflammatory mediators with deleterious effects on asthma as they activate genes encoding inflammatory protein synthesis (Figure 2). This may contribute to the reported adverse effects of LABA [26,27]. The fundamental detail of these findings is that β2-agonists should not be used as maintenance monotherapy in asthma and should almost always be associated with IGCs. Synthesizing β2-agonists that favor Gs signals rather than the β-arrestin recruitment may prove to be more effective bronchodilators therapy in the future [28].
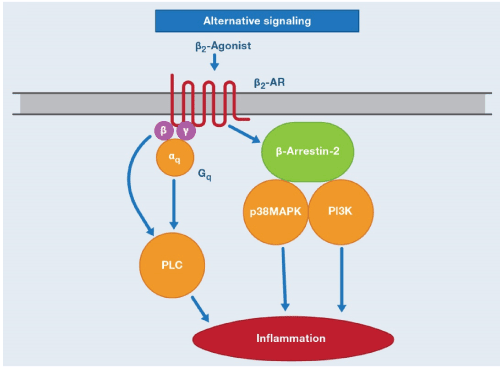
Figure 2. β2-Agonist alternative signaling pathway. Alternative signaling by β2-AR may activate βγ-subunits of Gq and via Gαq, resulting in activation of PLC (phospholipase C). β2-AR also interact with β-arrestin-2, which interacts with p38 MAPK (p38 mitogen-activated protein kinase) and PI3K (phosphatidylinositol 3 kinase). These alternatives signaling pathways may increase the expression on inflammatory proteins and therefore may have a deleterious effect in asthma [18].
There are several single-nucleotide polymorphisms (SNPs) and haplotypes of the human β2-AR gene (ADRβ2) that may affect the β2-AR function. As early as 2000, a total of 17 SNPs within ADRβ2 and their regulatory regions were described [29]. The common variants are G16R and Q27E, which have in vitro effects on receptor desensitization, but clinical studies have shown inconsistent effects on the bronchodilator responses to SABAs and LABAs [30]. Some studies have shown that patients with the common homozygous Arg16/Arg variant have more frequent adverse effects and a poorer response to β2-agonists than heterozygotes or the Gly16/Gly homozygotes [31]. However, these differences are small, and there appears to be no clinical value in measuring ADRβ2 genotype. No differences have been found with responses to LABAs between these genotypes [32].
Functional antagonism occurs when two drugs each produce an effect, and the two effects cancel each other. For example, methacholine can stimulate parasympathetic (muscarinic) receptors in the airways, causing bronchoconstriction, epinephrine can stimulate β2 receptors in the airways, causing bronchodilation, but each one is an agonist of its own receptor.
All β2-agonists share the molecular and genetic mechanisms discussed above. All are functional antagonists, but of course there are differences between them in duration of action (SABAs, LABAs and ULTR-LABAs) but also in terms of solubility. For example, salbutamol is hydrophilic and therefore interacts directly with the receptor of the aqueous phase, at the active site located in the transmembrane domains 3,5 and 6, hence its onset of rapid and short action so it is of choice for rescue and treatment of crisis or exacerbations of obstructive airway diseases [28,33]. Salmeterol is lipophilic and is associated initially and predominantly with the lipid phase, followed by membrane translocation. This indirect binding to the active site in the aqueous phase causes a delay in onset of action. In fact, the translocation takes about 30 minutes [15]. Formoterol is partially hydrophilic and forms a deposit in the membrane. Its onset of action is faster than salmeterol. These molecular mechanisms carry clinical differences. For example, both LABAs cause bronchodilation for at least 12 hours after a single administration but the onset of action of salmeterol is delayed for at least 15 minutes while with formoterol is almost immediate. Therefore, formoterol, but not salmeterol, can be used "as required" or "PRN" (pro re nata=for necessary reason), and the dose of formoterol is more flexible than salmeterol, which cannot be increased because of long-lasting systemic side-effects. Salmeterol is a partial agonist with lesser efficacy in isolated smooth muscle than the full agonist formoterol. However, while it has been speculated that full agonism may in certain cases lead to therapeutic advantages, there is also the theoretical possibility of increasing tolerance and adverse effects [2].
Medications obtained from herbs have been used since ancient times to treat human diseases and some of these plant extracts contain anticholinergic agents [33-36]. Ayurvedic medicine source from India at around AD450 describes the use of thorn-apple or jimsonweed extracts for their bronchial relaxant effects (stramonium).
In the 19th century, in the early 1800s, the British Army medical officers serving in India introduced anticholinergics in Western medicine [37]. The leaves and roots of Datura stramonium (source of stramonium), Hyoscyamus niger (source of hyoscine or scopolamine) and Atropa belladonna (which contains the alkaloid atropine), had all their place in most pharmacopoeias [38]. Given the fat solubility of these natural anticholinergics, they are easily absorbed through the oral and respiratory mucosae. High doses used as bronchodilators generate side effects such as decreased salivation, dry mouth, mydriasis, blurred vision, urinary retention, decreased bowel motility, nausea, tachycardia and decreased salivation. The discovery of β2-agonists in 1920s displaced these agents as the first-line treatment in asthma and emphysema [2].
Since the 1970s, there has been renewed interest in the use of anticholinergics. This is due to several facts, such as the increase in the prevalence, morbidity and mortality of asthma and COPD, the need to develop alternatives to therapy with β2-agonists, a better understanding of the cholinergic mechanisms controlling airways caliber in health and disease states and the development of synthetic atropine analogues [39]. The new anticholinergics are water-soluble, poorly absorbed quaternary ammonium compounds, causing only mild systemic effects when administered by inhalation or nebulization. These agents include: ipatropium, thiazinamium, oxitropium, glycopyrronium, aclidinium (umeclidinium) and tiotropium bromide [40].
Muscarinic receptors have been classified into five subtypes, initially based on drug selectivity and subsequently confirmed by molecular cloning. M1, M2 and M3 receptors are found in human airways [2]. M4 and M5 are in the central nervous system [1]. M1 receptors are found in alveolar walls and in the parasympathetic ganglia, their blockade reduces the bronchoconstriction response. M2 receptors are located on postganglionic cholinergic nerve endings, and these auto-receptors limit the magnitude of vagally induced bronchoconstriction. When the rate of release and transport of Ach is increased (favoring bronchospasm), these receptors activated by said Ach inhibit the release of Ach to exercise a kind of pattern in neurotransmitter trafficking. If these receptors are dysfunctional, this mechanism of servo-control of the vagal tone is lost, perpetuating the spasm of the airway. M3 receptors are located in the airway smooth muscle and submucosal glands, where they mediate bronchoconstriction and mucus secretion, stimulated by Ach or cholinergic agonists (eg, methacholine) (Figure 3) [10]. The cholinergic fibers in the lung are dense in the hilar region and decrease towards the peripheral airways. Muscarinic cholinergic receptors also decrease their density in the distal airways (mainly M3 receptors), unlike β2-AR (Figure 4) [1].
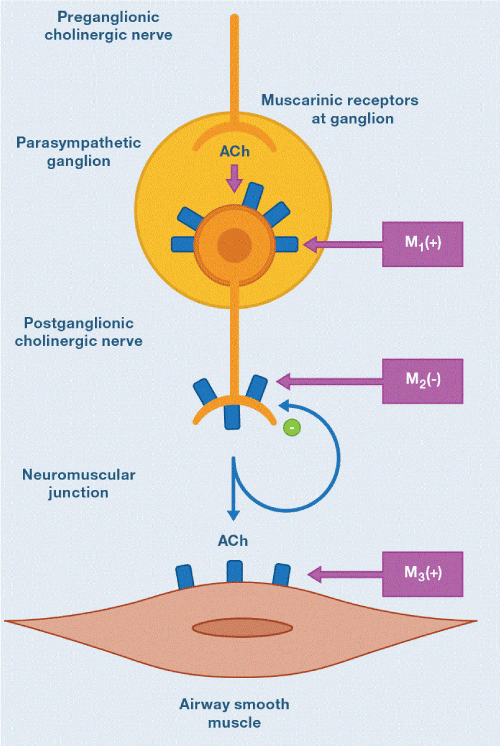
Figure 3. Muscarinic receptor subtypes. M1 receptors are localized in parasympathetic ganglia and augment ganglionic transmission. M2 receptors are at cholinergic nerve terminals and inhibit acetylcholine (ACh) release, where M3 receptors are in muscle (and mucus glands) and mediate bronchoconstriction (and mucus secretion). M2 receptors may be dysfunctional in bronchial asthma.
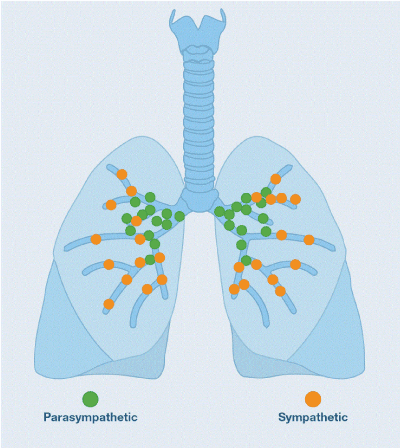
Figure 4. Airway autonomic neuronal control. Note the predominantly central distribution of cholinergic and peripheral adrenergic receptors.
Muscarinic Ach receptors are G protein-coupled receptors (GPCRs). M1 and M3 receptors are coupled to Gq proteins, while M2 are coupled to Gi/o protein [17]. Once the receptor is activated, guanosine triphosphate (GTP) binds to the corresponding G-protein subunit (for example Gq in the case of M3), which stimulates phospholipase Cβ (PLCβ). PLCβ cleaves phosphoinositol bisphosphate (PIP2) into inositol triphosphate (IP3) and diacyl glycerol (DAG) (second messengers in this system). IP3 interacts with the corresponding receptors (IP3R) in the endoplasmic reticulum (ER) and causes release of calcium into the cytoplasm, which binds to calmodulin by activating MLCK. MLCK phosphorylates the myosin facilitating the interaction between actin and myosin and muscle contraction (Figure 5). DAG in turn activates protein kinase C (PKC), which phosphorylates calponin, which prevents the inhibition it exerts on myosin-ATPase. Active myosin ATPase induces muscle contraction. All muscarinic anticholinergics occupy the cholinergic receptor blocking the Ach-mediated muscle contraction molecular processes described above and are therefore antagonists [18,41].
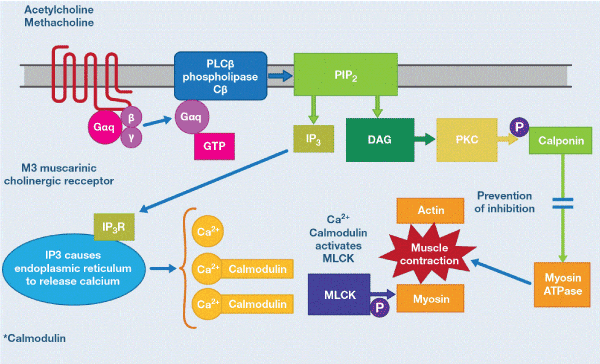
Figure 5. Cholinergic mechanisms of smoot muscle contraction. ACh binding to the M3 receptor results in guanosine triphosphate (GTP) binding to the Gαq subunit. GTP-bound Gαq stimulates phospholipase Cβ (PLCBβ), which cleaves phosphoinositol bisphosphate (PIP2) into inositol triphosphate (IP3) and diacyl glycerol (DAG). IP3 interacts with IP3 receptors (IP3R) on the ER and evokes Ca2+ release from the ER into the cytosol. Liberated Ca2+ binds calmodulin, forming a complex that activates MLCK. Phosphorylation (P) of myosin by MLCK facilitates interaction between actin and myosin and contraction. In addition, DAG activates protein kinase C (PKC), in an alternate mechanism of smooth muscle contraction. These mechanisms are blocked by muscarinic antagonists. Adapted from Figure 4B of Johnson EN.
Bronchial asthma and COPD are inflammatory diseases, and during exacerbations, a further intensified inflammatory response occurs. The three LAMAs approved by GOLD (ipatropium bromide, glycopyrronium bromide and aclidinium bromide) for maintenance treatment of COPD, reduce the frequency of acute exacerbation of COPD (AECOPD) and some increase the time to the first exacerbation [42-45]. It is known that Ach increases neutrophil chemotactic activity in COPD, and this effect is attenuated in vitro by tiotropium, suggesting a possible anti-inflammatory mechanism, although it remains controversial [46]. A British study including 142 patients, randomized either to receive tiotropium or placebo for 1 year, failed to demonstrate a significant reduction in systemic or airway inflammatory markers (IL-6, IL-8, and myeloperoxidase) with anticholinergic [47]. In vitro studies show that Ach induces the release of inflammatory mediators, such as the granulocyte-macrophage colony stimulating factor (GM-CSF), leukotriene B4 (LTB4) and prostaglandins E2 of epithelial cells. This release is mediated via muscarinic receptors that could be inhibited by tiotropium [48]. In the British study mentioned above, levels of LTB4 and / or GM-CSF were not measured, and they seem to be more relevant to evaluation cholinergic effects.
In the murine model of COPD the use of glycopyrronium bromide significantly reduced the expression of cigarette-induced pro-inflammatory mediators in bronchoalveolar lavage (BALF) [49]. Recently, aclidinium has been shown to inhibit the transition of human fibroblasts to myofibroblasts, and reduces the proliferation and migration of them, which could impact airway remodeling [50,51]. Therefore, there is evidence that anticholinergics could prevent contraction and proliferation of smooth muscle airway cells and fibroblast proliferation. These findings support the hypothesis that the cholinergic system plays a role in the pro-fibrotic process of airway remodeling. Peribronchiolar fibrosis may be a key event in the progressive FEV1 decline in COPD [52]. Even this evidence suggests that LAMAs may offer benefits as maintenance therapy in bronchial asthma, particularly in those who fail to gain control with the LAMA-IGCs association or in those with frequent exacerbations [53].
From another perspective, M2 receptors are highly expressed smooth muscle cells in the airways and inhibit the adenylyl cyclase pathway Gi, thus counteracting the bronchodilator effect of β2-agonists. M2 receptors also counteract β2-agonists by inhibit KCa in tracheal smooth muscle cells via the Gβγ subunit of G protein [54]. There is evidence that pre-synaptic M2 receptors are dysfunctional in patients with bronchial asthma [55].
Thus, as with β2-agonists, the counter-effects on inflammation of anticholinergics leave a conceptual vacuum around the anti-inflammatory mechanism of these molecules in obstructive airway diseases.
Overall
The combination of β2-adrenergics with anticholinergics in the treatment of obstructive airway diseases rests on several facts. At the level of mechanisms of action, the molecular combination has complementary effects. The predominantly hilar distribution of cholinergic receptors facilitates the central bronchodilation of anticholinergics and the access of β2-agonists to the periphery where β2-AR are most abundant. This is a mechanism in which a bronchodilator facilitates access and action of the other. A common conceptual error is to believe that anticholinergics and β2-adrenergics have additive effects. Additivity occurs when two drugs act on the same receptor and the combined effect is simply the linear sum of the two drugs, to obtain a maximum effect. The action of the two classes of bronchodilators on two different receptors supports the concept of synergism. This mechanism occurs when both drugs act on the same target tissue (in this scenario, smooth muscle) and the combined effect is greater than the simple sum of the separate effects of the two drugs [1]. What happens is a capitalization or amplification of the effect, in this case, bronchodilation. For example, β2-adrenergic when forming cAMP and it activates PKA (which phosphorylates the IP3R) inhibits the release of calcium from the ER. Concomitantly, the anticholinergic, by blocking the formation of PLCβ (and inhibiting the formation of IP3 which is the second messenger that stimulates the IP3R) blocks the release of calcium from the ER. (Figures 1 and 4). It is obvious that acting through different metabolic pathways of each receptor, the end result is the reduced availability of calcium in the cytosol, which is fundamental for the concerted action of actin and myosin for contraction. Another example is the action of both bronchodilators on calponin. The β2-adrenergic stimulus increases its concentration in the cytosol and the anticholinergic prevents it being inactivated by PKC. The end result is increased availability of the enzyme that favors the inhibition of myosin ATPase and thus relaxation [2]. It is also important to consider that sympathetic activity occurs predominantly during the day, whereas the parasympathetic system is more active at night, therefore controlling both systems can provide beneficial effects [3].
Although this monograph deals with the combined therapy of bronchodilators as a maintenance strategy, it is fair to say that it can also occupy a place in the management of crisis or exacerbations and as rescue therapy. GINA states that for the treatment of acute or sub-acute asthma exacerbations in primary care, the only bronchodilator is SABA [56]. For mild to moderate exacerbations the doses of salbutamol (SABA) is 4-10 puffs every 20 minutes the first hour and then 4-10 puffs every 3-4 hours (each puff has 100 micrograms =100 µg salbutamol). The most cost-effective route of delivery is pMDI with spacer [57]. Delivery of SABA pMDI with a spacer or DPI (dry powder inhaler) leads to a similar improvement in lung function than nebulized delivery [58,59]. Due to the static load on the plastic spacers, they must be pre-washed with detergent and air-dried prior to immediate use. If the pre-treated spacer is not available, a new spacer should be primed with at least 20 puffs of salbutamol before use [60]. In severe or near-fatal asthma the evidence of cost-effectiveness of pMDI use with respect to nebulization is less robust and if conflicting. A recent review found reduced hospitalizations and better lung function with continuous nebulization compared to intermittent, particularly in patients with poor lung function [61]. A reasonable approach is to use the continuous medication initially, followed by intermittent, on-demand therapy in the hospitalized patient. For nebulization, 2.5-5 milligrams (0.5-1 cc) of the salbutamol nebulizer solution are used for each nebulization, diluted with 3.5-3 cc of saline every 20 minutes the first hour (number 3) and then once every 4 hours. Intravenous β2-agonists should not be used in patients with severe asthma. Adrenaline (epinephrine US) intramuscular (0.5-1 milligram) can be added to standard therapy for acute asthma associated with anaphylaxis and angioedema. It is not indicated for other asthmatic exacerbations [56].
For the patient who becomes worse in primary care and is considered to be transferred to a hospital and for adults and children presenting in an emergency with moderate to severe asthma attacks, a cycle of ipatropium bromide (SAMA) may be added. This tool is associated with reduction in hospitalizations and great improvement in FEV1 and PEF compared to salbutamol alone [62,63]. GINA states that ipatropium should be added basically in severe crises [56]. If inhaled ipatropium is to be used, 4 puffs (80 µg) should be used every 20 minutes the first hour (along with inhaled salbutamol) and then 2 inhalations every 4 hours (40 µg) (together with inhaled salbutamol). As in severe crisis (where SAMA is indicated), nebulization may be more effective, salbutamol (0.5-1 cc) may be mixed with 2 cc ipatropium (500 µg) with the same dosage regimen; three continuous the first hour and then a nebulization of the mixture every 4 hours. From 1-17 years the dose is halved (1cc = 250 µg). The solution prepared to nebulize the mixture of salbutamol with ipatropium (Combivent® which by nebulizing ampoule has 2.5 cc with 2.5 milligrams of salbutamol + 0.5 milligrams of ipatropium) is available in the market, making the scheme very easy as a nebulization ampoule diluted with 1.5 cc of saline solution. One drop of the mixture has 10 µg of Ipatropium and 50 µg of salbutamol. As the dose per kilogram of weight in children is 150 µg of salbutamol and 30 µg of Ipatropium, it is also very easy to dosage the mixture in Pediatrics: 3 drops per kilogram of weight by nebulization up to 17 kg. With the upper weights, the entire blister is used [39]. Whether separate solutions are used for misting or mixing, the total volume of nebulization should be 4 cc, therefore this final volume should be adjusted with physiological saline. As a rescue medication in asthma GINA definitely recommends SABAs in all 5 steps, although in 3,4 and 5 as an alternative it suggests low dose IGCs with formoterol. It does not routinely suggest anticholinergics basically for a slower onset of action than SABAs, but neither does it contraindicate them and there are studies that show that when used together (SAMA / SABA) rescue, anticholinergic adds significant improvement in lung function, and are also an alternative for patients with SABA who experience tachycardia, arrhythmia and tremor [3,16].
In AECOPD, as in an asthmatic crisis, the use of SAMA is elective and not mandatory. The use of SABA with or without anticholinergics is the initial bronchodilator for treatment [64]. There are no significant differences in FEV1 between using pMDI (with or without a spacer) or nebulization to deliver the drug [65]. However, in patients with moderate to severe AECOPD and the elderly, nebulization may be the most appropriate method for delivery. If you use pMDI in the first hour you should receive 2-3 puffs and then one every 2-4 hours. If the nebulized route is used 0.5 cc (2.5 milligrams) of the salbutamol solution should be given to nebulize followed alone the first hour with or without 1cc (250 µg) of the solution to nebulizer of ipatropium bromide + 2.5 physiological serum (3 doses continuous). Then the patient can receive this same dose every 4 hours, based on the response. If the mixture is available, an ampoule diluted with 1.5 cc of saline may be used for each nebulization. In patients with hypertrophic cardiomyopathy, tachyarrhythmia, myocardial ischemia, recent infarction (last 3 months), hyperthyroidism or pheochromocytoma, anticholinergic therapy alone is the treatment of choice [66]. It is recommended that the patient continue the maintenance treatment he had with LABA and / or LAMA, or combined and with or without IGCs, or if he did not have it prescribed, to initiate it before his discharge from the hospital. Short-acting bronchodilators “PRN” are only indicated in group A of the EPOC classification (GOLD) as an alternative to long-acting bronchodilators. From group B to D, the long-acting bronchodilators are superior to the short-acting bronchodilators used when necessary (prn) and are therefore recommended [66].
LABA / LAMA in COPD
COPD is a highly prevalent disease, with prevalence in people ≥ 40 years of 10.1% worldwide [67]. COPD has become the third leading cause of death worldwide and will become the seventh largest burden in 2030 [68]. It is estimated that in 2010 the number of COPD cases globally amounted to 384 million, with 3 million deaths annually. With the increase in smoking in developing countries, and with an older population in high-income countries, the prevalence of COPD is expected to increase over the next 30 years and to exceed 4.5 million deaths annually in both COPD and related conditions [69]. In the United States (US) COPD is the second cause of death [70]. It is obvious that optimizing treatment is vital in this entity.
Apart from the bronchodilator effects that can be obtained from the synergism LABA / LAMA and the availability of devices that allow the joint delivery of both, it is the fact that they can be dispensed once a day which facilitates adherence to treatment, reduce doses of each molecule and thus the side effects associated with high doses of a single drug. Dual therapy is superior to monotherapy and they recognize it and recommend previous GOLD trials [71].
For a decade, only LAMA tiotropium bromide was available for patients and clinicians. It is by far the anticholinergic with studies of greater statistical power and extension, as evidenced by UPFLIT, TIOSPIR and very recently paper by Zhou and collaborators of the effect of tiotropium on lung function in early stages of COPD. UPLIFT is a 4-year, randomized, placebo-controlled study, and was performed in patients with moderate to severe stages. Patients received 18 µg of inhaled tiotropium (dry powder, HandiHaler®). The study showed significant improvement in lung function, health-related quality of life, a reduction in AECOPD and hospitalizations, but there was no significant reduction in FEV1 decline rate or mortality compared to placebo (p ≤ 0.09) [42]. TIOSPIR is a randomized, double-blind study that included 17,135 patients with COPD. The study compared patients treated with tiotropium Respimat® in inhaler (daily doses of 2.5 and 5 µg) with a control group using HandiHaler® (18 µg daily). In this study, tiotropium Respimat® was not inferior to HandiHaler® with respect to the risk of death, nor was it better than HandiHaler® with respect to the time of the first AECOPD. The three doses with two different devices were comparable in terms of safety and efficacy [43]. In addition to the statistical power of the sample, one of the most important aspects of TIOSPIR is that it enrolled a substantial number of patients with cardiac conditions (1,825 with arrhythmias and 3,152 with ischemic heart disease, coronary artery disease, or heart failure). Tiotropium did not increase the risk of death or adverse effects in these patients. Zhou's work conducted in China is a randomized, double-blind, placebo-controlled study that included 841 patients with stage 1 COPD (mild) and 2 (moderate) COPD. The treatment group (419) received tiotropium HandiHaler® (18 µg) and 422 patients received placebo, for 2 years. The tiotropium group had a significantly higher FEV1 during the study than the placebo group (P ≤ 0.001) and also significantly reduced the annual decline in post-bronchodilator FEV1 compared to placebo (P = 0.006) [67]. Taking all of this evidence together, it is valid to state that tiotropium has a primordial and well documented role in the treatment not only of advanced stages to improve the quality of life related to the health of patients with COPD but also, depending on the stage in that is dispensed, may impact deterioration of lung function. It would be interesting and valuable in the future to design protocols, longitudinal and extensive to see if the use of anticholinergics in early stages of the disease, with or without LABAs, impacts the natural evolution of the same.
From 2012 the panorama changed with the approval of two new LAMAS: glycopyrronium bromide and aclidinium bromide (and umeclidinium). The three molecules are considered therapeutic options in recent GOLD updates [66,70]. Glycoyrronium bromide is a synthetic quaternary ammonium compound, recently reformulated as a dry powder for inhaled use once a day. After preclinical studies and promising clinical results, phase III studies called glycopyrronium bromide in COPD (GLOW) were conducted. These studies showed that 50 µg inhaled once daily improved FEV1, dyspnea and health status [71-73]. A reduction in the number of AECOPD was also observed [44]. GLOW 3 showed that glycopyrronium is superior to placebo over effort tolerance, with improvement in endurance exercise over a 3-week period [72]. Glycopyrronium has an acceptable safety profile, and low incidence of cardiac effects and secondary anticholinergics. Consequently, has been considered an alternative LAMA [73]. Aclidinium bromide has been approved in the European Union (EU) and in the US as maintenance treatment for COPD. In phase III studies, aclidinium bromide (compared to placebo) significantly improved lung function, dyspnea and health status in the first 24 weeks of treatment and this improvement was maintained for up to 52 weeks in subsequent studies. The frequency of AECOPD was significantly reduced compared with placebo, and systemic bioavailability was low, with a reduced tendency to produce cardiac arrhythmias, indicating an adequate tolerance profile [45]. Aclidinium (400 µg DPI, 400 µg pMDI) is administered twice daily whereas umeclidinium (62.5 µg DPI) is administered once daily [74].
LABAs, salmeterol (25-50 µg MDI and DPI) and formoterol (4.5-9 µg DPI) are used every 12 hours as single drugs in one device or mixed with steroids or LAMAs. ULTRA-LABAs (indacaterol, olodaterol and vilanterol) are the most frequently mixed with LAMAS in a single device for 24 hours, although there are formulations that dispense ULTRA-LABA as a single drug [66]. Indacaterol was the first ULTRA-LABA approved to be prescribed once daily in COPD. It was designed to have a rapid onset of action (5 minutes) and an ultra-long-acting duration (24 hours). Initially approved in the EU in 2009 and in US in 2011, it currently has approval in more than 100 countries. This drug has been shown to have a greater bronchodilator activity than LABA, formoterol (12 µg twice daily), and to work at least similarly to tiotropium [75,76]. The European Commission has authorized, and also FDA, olodaterol, a long-acting bronchodilator (2.5-5 µg SMI = soft mist inhaler), for the treatment of mild to severe COPD, as well as vilanterol (25 µg) once daily [77,78].
Adding a second bronchodilator to patients whose symptoms are under-controlled with monotherapy improves lung function, symptomatology and health status, without increasing the risk of side effects. This was demonstrated in INTRUST-1 and INTRUST-2 (indacaterol plus tiotropium versus tiotropium) and in GLOW6 (indacaterol plus glycoyrronium versus indacaterol) [79,80]. Also, administration of a fixed dose combination of 150 µg indacaterol plus 50 µg of glycopyrronium was shown to be superior to single component administration [81]. The Ultibro® Breezhaler® inhaler (indacaterol plus glycopyrronium) has been approved in the US, Japan and other countries as of 2013. The SPARK study evaluated the effect of inhaled treatment with long-acting dual molecules in severe to very severe COPD. Fixed-dose patients were randomized to a combination of 110 µg indacaterol plus 50 µg glycopyrronium, glycopyrronium 50 µg alone and tiotropium 18 µg alone (open label). The study showed that dual bronchodilation is superior in preventing moderate to severe AECOPD compared to each of the LAMAs alone. These results indicate the potential of dual bronchodilation in the treatment of COPD from severe to very severe [82].
A combination of inhaled dry powder containing umeclidinium bromide (62.5 µg) and vilanterol trifenatate (25µg) (Ellipta®) has recently been approved in the US and EU for maintenance treatment of COPD. This combination has been shown to improve lung function, dyspnea, health-related quality of life, and reduce AECOPD, without adding serious adverse effects [83]. TOVITO®, a large global phase III study, has been conducted to investigate the efficacy and safety of the fixed dose combination of tiotropium and olodaterol delivered via the Respimat® inhaler [43]. In May 2015, the FDA approved Stiolto® Respimat® inhaler (olodaterol 5µg plus tiotropium 5 µg) once daily for maintenance treatment of COPD.
GOLD recommends the use of the LABA / LAMA combination from group B of the COPD classification under certain circumstances. For example, if the patient persists with dyspnea despite monotherapy, the use of two bronchodilators is advisable. If dyspnea is severe from the start of diagnosis, combined treatment is suggested, and de-escalation to one if symptoms do not improve with the combination. Group C with AECOPD qualifies to use the combination as an alternative to the LABA / IGCs combination, especially since IGCs increase the risk of pneumonia. The same happens with group D where the combination is clearly superior to monotherapy, and even superior to prevent AECOPD with respect to LABA / IGCs. In addition, this group is particularly at high risk for developing pneumonia with IGCs [66].
LABA/LAMA in bronchial asthma
Even with the most recent therapeutic recommendations, there is a population of asthmatic patients who are not well controlled [84]. Patients with severe asthma make up 3-10% of the population of adult with asthma, but their care is estimated the account for more than 60% of the costs associate with asthma, which are primarily for medications [85,86]. Health care costs per patient for severe asthma are higher than those for type 2 diabetes, stroke and COPD [87]. Severe asthma is a subset of difficult-to-control asthma; the term is used to describe patients with asthma that remains uncontrolled despite treatment with high-dose of IGCs combined with LABA, a leukotriene modifier or theophylline for the previous year or treatment with systemic GCs for at least half the previous year. Criteria for uncontrolled asthma include exacerbations, poor symptom control, impaired lung function or a combination [85,86]. It excludes patients in whom asthma is vastly improved with optimization of adherence, inhalation technique, and treatment of coexisting conditions. These patients may have basically two immunological profiles. The eosinophilic profile, characteristic of airway inflammation type 2, characterized by the presence of cytokines originally recognized as being produced by type 2 helper T (Th2) cells. Th2 cytokines are Interleukin (IL) -4, IL-5 and IL-13. These cytokines are also produced by innate lymphoid cells (which do not express B or T cell receptors) in response to infectious agents, pollutants and other "non-allergic" stimuli. These patients persistently maintain this inflammatory profile despite high doses of IGCs [88-91]. The other profile is neutrophilic inflammation (defined as exceeding 40 to 60% neutrophils) in induced sputum samples. It appears that these two profiles have different patho-biological pathways and some patients overlap both profiles with more severe disease, greater airflow limitation and greater consumption of health resources [92,93].
Many of the recent approved biological interventions target type 2-inflammation. IL-5 plays a central role in promoting eosinophilic inflammation. Anti-IL-5 monoclonal antibodies are now available for the treatment of patients with severe eosinophilic asthma and recurrent exacerbations. Mepolizumab and reslizumab, both of which bind to IL-5, have been approved by several regulatory agencies in the US and EU. Benralizumab, which binds to the IL-5 receptor, producing eosinophilic apoptosis, is nearing Food and Drug Administration (FDA) approval [94]. Blockade of IL-13 has the potential to alter airway inflammation and smooth-muscle reactivity, but one of the two anti-IL-13 monoclonal antibodies, lebrikizumab, failed to provide consistent improvement in patients with type 2-inflammation. The other, tralokinumab, continues in development [95]. Dupilumab is a fully human monoclonal antibody to the alpha subunit of the IL-4 receptor that blocks both interleukins (IL-4 and IL-13) signaling [96]. One of the fundamental problems with adding biological therapies to these patients is the high price. And, if this is a limiting argument in high-income countries, much more in third world countries with meager health budgets. Two alternatives in this context exist. The first is the development of clinics specialized in severe asthma, within the organization chart of the pulmonology services, as well as the cessation of smoking, tuberculosis, cystic fibrosis, respiratory therapy, etc. Systematic evaluation and multidisciplinary treatment of these patients in these clinics increases the identification of coexisting conditions and improves outcomes [97,98]. The second is to add tiotropium.
In a recent and comprehensive review of the tiotropium literature on bronchial asthma from 149 papers published over 67 years, 5 randomized clinical trials were found that demonstrated that the use of tiotropium in combination IGCs alone or IGCs plus LABAs in uncontrolled asthma moderate to severe, improved lung function. This improvement was sustained even when the IGS were reduced and even when the LABAs were discontinued. It was considered beneficial in this type of complex asthma without adding safety problems [99]. The TALC study showed that tiotropium HandiHaler® (18 µg daily) plus low-doses of IGCs was more effective than doubling IGCs dose in improving morning and evening peak expiratory flow (p ≤ 0.001) and FEV1 (p ≤ 0.004) [100]. In the work of Kerstjens and collaborators, using tiotropium (5 µg per day Respimat®) increased by 56 days the time to first severe exacerbation, with a 21% reduction in the risk of severe exacerbations versus placebo (p = 0.03) [101]. It should also be taken into account that more than 80% of refractory asthma patients have poor adherence to controller medications, and the reasons that promote this behavior need to be solved before simply adding another drug [102].
The consistent improvement in lung function and the lack of significant side effects when tiotropium is added to the LABA / IGCs scheme, probably led the Medicines and Healthcare Products Regulatory Agency (MHRA) in the UK, to the approval of Spiriva Respimat® inhalation spray for asthma on September 13, 2014. In our country, Costa Rica, the Ministry of Health approved Respimat® inhaler for adult asthmatic patients with bronchial asthma who remain symptomatic despite the use of IGCs in April 2015. GINA 2015 guidelines recommended adding tiotropium Respimat® to the base therapy of maintenance in steps 4 and 5, considering the benefit of improvement in lung function and increased the time to the first severe exacerbation of asthma. [103] The latest GINA guidelines recommend it for adults (5 µg per day) and adolescents older than 12 years of age up to the age of 17 (2.5 µg per day). Not recommended for children under 12 years.
LABA/LAMA in ACOS
Recently, in a joint effort of GINA and GOLD the term ACOS was developed as a syndrome of overlap between asthma and COPD. ACOS definition is purely descriptive; it refers to a subgroup of patients with persistent airflow limitation that concomitantly shows several features, usually associated with asthma, and several features associated with COPD. The fundamental point of identifying and properly treating these patients is that they seem to have a poorer prognosis [104]. ACOS patients have frequent AECOPD, poor quality of life, fast decline in lung function, increased mortality and they also consume more health resources than patients with asthma or COPD alone. In the sputum of these patients there are neutrophils and eosinophils and there is an urgent need to investigate to what extent this syndrome is the result of a change in the pathogenesis of bronchial asthma towards COPD over time, or the entity has a different molecular biology and immunopathogenesis [105]. ACOS pharmacotherapy includes low to moderate doses of IGCs. The LABA / LAMA combination is recommended in this entity. The asthmatic component contraindicates the use of LABA without IGCs, given the debate about the safety of LABAs in patients with asthma.
Even before the description of ACOS, the potential benefit of tiotropium in patients with COPD and bronchial hyper-reactivity had been pointed early [106,107]. Magnussen and colleagues conducted a 12-week, prospective, randomized, double-blind, placebo-controlled, multicenter clinical trial involving 472 patients. Patients receiving tiotropium achieved significant improvement in FEV1 compared to those in the placebo group (p ≤ 0.001, AUC0-6 h). There was also a significant reduction in the use of rescue medication in the tiotropium group (108). The recommendation of the LABAs / LAMAs guides in ACOS as well as in COPD does not suggest any particular molecules of either LABAs or LAMAs. The indication must be adapted to each country, its resources and the availability of the association. If the association is not available in a single device obviously each can be used separately. Given the more central distribution of cholinergic receptors and the more peripheral adrenergic receptors it is recommended to first use the LAMA and 30-60 minutes later the LABA. In asthma the only LAMA approved is tiotropium.
The tone and patency of the airways depend on the activity of the autonomic nervous system.
From the therapeutic point of view, the muscarinic and adrenergic receptors can be manipulated with drugs to obtain an adequate bronchodilation in obstructive diseases of the airways.
Anti-muscarinic and adrenergic bronchodilators, used together, have complementary effects that can optimize bronchodilation of the airway.
This association can be used in exacerbations of obstructive diseases and as rescue therapy with short acting bronchodilators (SABA / SAMA).
Long-acting bronchodilators (LABA, ULTRA-LABA and LAMA) used in separate devices or in the same device are effective and is an accepted therapy as maintenance therapy in COPD and ACOS.
In patients with difficult to control asthma, tiotropium bromide, added to the LABA / IGC association, is an alternative to try to obtain control.
The use of triple therapy in the same device is foreseeable in the near future and for 24 hours, facilitating the delivery of drugs and improving adherence, as well as high costs are expected to limit their use, particularly in low income countries
No.
No.
This work was carried out only by author. Author AA contributed in the planning, data collection, data analysis, writing and critical review. The author read and approved the final manuscript.
- Rau JL (2002) Respiratory Care Pharmacology. (6th Edn) Georgia.
- Hansel TT, Barnes PJ (2004) An Atlas of Chronic Obstructive Pulmonary Disease. (1st Edn) The Parthenon Publishing Inc, New York.
- Alvarado-Gonzalez A, Arce I (2015) Tiotropium Bromide in Chronic Obstructive Pulmonary Disease and Bronchial Asthma. J Clin Med Res 7: 831-839. [Crossref]
- Marwick J, Adcock IM, Chun KF (2010) Overcoming reduced glucocorticoid sensitivity in airway disease: molecular mechanisms and therapeutic approaches. Drugs 70: 929-948.
- Combivent Inhalation Aerosol Study Group (1994) In chronic obstructive pulmonary disease, a combination of ipratropium and albuterol is more effective than agent alone. An 85-day multicentric trial. Chest 105: 1411-1419.
- Wilson JD, Seby CW, Menjoge SS, Witek TJ (1996) The efficacy and safety of combination bronchodilator therapy. Eur Respir Rev 6: 286-289.
- Combivent Inhalation Solution Study Group (1997) Routine nebulized ipratropium and albuterol together are better than either alone in COPD. Chest 112: 1514-1521.
- Campbell S (1999) For COPD a combination of ipratropium bromide and albuterol sulfate is more effective than albuterol base. Arch Intern Med 159: 156-160. [Crossref]
- Gross N, Tashkin D, Miller R, Oren J, Coleman W, et al. (2002) Inhalation by nebulization of albuterol-ipatropium combination (Dey combination) is superior to agent alone in the treatment of chronic obstructive pulmonary disease. Respiration 65: 354-362.
- Sakula A (1988) A history of asthma. The FitzPatrick lecture 1987. J R Coll Physicians Lond 22: 36-44. [Crossref]
- Bielory L, Lupoli K (1999) Herbal interventions in asthma and allergy. J Asthma 36: 1-65. [Crossref]
- Cazzola M, Matera MG (2009) Emerging inhaled bronchodilators: an update. Eur Respir J 34: 757-769. [Crossref]
- Ahlquist RP (1948) A study of the adrenotropic receptors. Am J Physiol 153: 586-600. [Crossref]
- Lands AM, Arnold A, McAuliff JP, Luduena FP,2021 Copyright OAT. All rights reserv of receptors systems activated by sympathomimetic amines. Nature 214: 597-598.
- Johnson M (2001) Beta2-adrenoceptors: mechanisms of action of beta2-agonists. Paediatr Respir Rev 2: 57-62. [Crossref]
- Liggett SB (2002) Update on current concepts of the molecular basis of beta2-adrenergic receptor signaling. J Allergy Clin Immunol 110: S223-227. [Crossref]
- Johnson EN, Druey KM (2002) Heterotrimeric G protein signaling: role in asthma and allergic inflammation. J Allergy Clin Immunol 109: 592-602. [Crossref]
- Barnes PJ (2011) Biochemical basis of asthma therapy. J Biol Chem 286: 32899-32905. [Crossref]
- Zhou Y, Sondek J, Harden TK (2008) Activation of human phospholipase C-eta2 by Gbetagamma. Biochemistry 47: 4410-4417. [Crossref]
- McGraw DW, Ligget SB (1997) Heterogeneity in ß-adrenergic receptor kinase expression in the lung account for cell-specific desensitization of the ß2-adrenergic receptor. J Biol Chem 272: 7338-7344.
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ (2001) Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 294: 1307-1313. [Crossref]
- Hamid Q, Tulic M (2009) Immunobiology of asthma. Annu Rev Physiol 71: 489-507. [Crossref]
- Howarth PH, Beckett P, Dahl R (2000) The effect of long-acting beta2-agonists on airway inflammation in asthmatic patients. Respir Med 94 Suppl F: S22-25. [Crossref]
- Mak JC, Chuang TT, Harris CA, Barnes PJ (2002) Increased expression of G protein-coupled receptor kinase in cystic fibrosis lung. Eur J Pharmacol 436: 165-172.
- McGraw DW, Almoosa KF, Paul RJ, Kobilka BK, Liggett SB (2003) Antihetic regulation by ß-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway ß-agonist paradox. J Clin Invest 112: 619-626.
- Schmid CL, Bohn LM (2009) Physiological and pharmacological implications of beta-arrestin regulation. Pharmacol Ther 121: 285-293. [Crossref]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK (2007) Beta-arrestins and cell signaling. Annu Rev Physiol 69: 483-510. [Crossref]
- Violoin JD, Lefkowitz RJ (2007) ß-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci 28 :416-422.
- Liggett SB (2000) The pharmacogenetics of beta2-adrenergic receptors: relevance to asthma. J Allergy Clin Immunol 105: S487-492. [Crossref]
- [Crossref] Hawkins GA1, Weiss ST, Bleecker ER (2008) Clinical consequences of ADRbeta2 polymorphisms. Pharmacogenomics 9: 349-358.
- Israel E, Chinchilli VM, Ford JG, Boushey HA, Cherniak R, et al. (2004) Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomized, placebo-controlled cross-over trial. Lancet 364: 1505-1512.
- Bleeckeer ER, Postma DS, Lawrence RM, Meyers DA, Ambrose HJ, et al. (2007) Effect of ADRB2 polymorphisms on response to long acting ß2-agonist therapy: a pharmacogenetics analysis of two randomized studies. Lancet 370: 2118-2125.
- Anderson GP, Lindén A, Rabe KF (1994) Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J 7: 569-578. [Crossref]
- Gross NJ1 (1988) Ipratropium bromide. N Engl J Med 319: 486-494. [Crossref]
- Chapman KR (1993) History of anticholinergic treatment in airways diseases. In Gross NJ (Ed) Anticholinergic Therapy in Obstructive Airways Diseases. Franklin Scientific Publications, London pp: 9-17.
- Herxheimer H (1959) Atropine cigarettes in asthma and emphysema. Br Med J 2: 167-171. [Crossref]
- Gandevia B (1975) Historical review of the use of parasympatholytic agents in the treatment of respiratory disorders. Postgrad Med J 51: 13-20. [Crossref]
- Siefkin AD (1996) Optimal pharmacologic treatment of the critically ill patients with obstructive airways diseases. Am J Med 100(1A): 54S-61S.
- Rodrigo GJ, Rodrigo C (2002) The role of anticholinergics in acute asthma treatment: an evidence-based evaluation. Chest 121: 1977-1987. [Crossref]
- Gross NJ, Skorodin MS (1984) Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 129: 856-870. [Crossref]
- Wessler I, Kirkpatrick CJ (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 154: 1558-1571. [Crossref]
- Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, et al. (2008) A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 359: 1543-1554. [Crossref]
- Wise RA, Anzueto A, Cotton D, Dahl R, Devins T, et al. (2013) Tiotropium Respimat inhaler and the risk of death in COPD. N Engl J Med 369: 1491-1501. [Crossref]
- Kerwin E, Hébert J, Gallagher N, Martin C, Overend T, et al. (2012) Efficacy and safety of NVA237 versus placebo and tiotropium in patients with COPD: the GLOW2 study. Eur Respir J 40: 1106-1114. [Crossref]
- Jones PW (2015) Clinical potential of aclidinium bromide in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 10: 677-687.
- Buhling F, Lieder N, Reisenauer A, Welte T (2004) Anti-inflammatory function of tiotropium mediated by suppression of acetycholine-induced release of chemotactic activity. Eur Respir J 24(Suppl 48): 318s.
- Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, et al. (2007) Effect of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J 30: 472-478. [Crossref]
- Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, et al. (2007) Inhibition of allergen-induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J 30: 653-661. [Crossref]
- Shen LL, Liu YN, Shen HJ, Wen C, Jia YL, et al. (2014) Inhalation of glycopyrronium inhibits cigarette smoke-induced acute lung inflammation in a murine model of COPD. Int Immunopharmacol 18: 358-364.
- Milara J, Serrano A, Peiró T, Gavaldà A, Miralpeix M, et al. (2012) Aclidinium inhibits human lung fibroblast to myofibroblast transition. Thorax 67: 229-237. [Crossref]
- Milara J, Serrano A, Peiró T, Artigues E, Gavaldá A, et al. (2013). Aclidinium inhibits cigarette-smoke induced lung fibroblast to myofibroblast transition. Eur Respir J 41: 1264-1274.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliot WM, et al. (2004). The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350: 2645-2653.
- Price D, Fromer L, Kaplan A, van der Molen T, Román-Rodríguez M (2014) Is there a rationale and role for long-acting anticholinergic bronchodilators in asthma? Prim Care Respir Med 24: 1-9.
- Zhou XB, Wulfsen I, Lutz S, Utku E, Sausbier U, et al. (2008) M2 Muscarinic receptors induced airway smooth muscle activation via dual, Gß? -mediated inhibition of large conductance Ca2+-activated K+ channel activity. J Biol Chem 283: 21036-21044.
- Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ (1989) A muscarinic agonist inhibits reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol 67: 2461-2465.
- GINA (2016) Global strategy for asthma management and prevention. Global Initiative for Asthma.
- Newman KB, Milne S, Hamilton C, HALL K (2002) A comparison of albuterol administered by metered-dose inhaler and spacer with albuterol by nebulizer in adults presenting to an urban emergency department with acute asthma. Chest 121: 1036-1041.
- Cates CJ, Welsh EJ, Rowe BH (2013) Holding chambers (spacers) versus nebulizers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 9: CD000052.
- Selroos O (2014) Dry-powder inhalers in acute asthma. Ther Deliv 5: 69-81. [Crossref]
- Pierat F, Wildhaber JH, Vrancken I, Devadason SG, Le Souef PN, et al. (1999). Washing plastic spacers in household detergent reduce electrostatic charge and improves delivery. Eur Respir J 13: 673-678.
- Camargo CA Jr, Spooner CH, Rowe BH (2003) Continuous versus intermittent beta-agonists in the treatment of acute asthma. Cochrane Database Syst Rev: CD001115. [Crossref]
- Rodrigo GJ, Castro-Rodríguez JA (2005) Anticholinergics in the treatment of children and adults with acute asthma following emergency department discharge. Thorax 60: 740-746.
- Griffiths B, Ducharme FM (2013) Combined inhaled anticholinergics and short-acting beta2-agonists for initial treatment of acute asthma in children. Cochrane Database Syst Rev: CD000060. [Crossref]
- National Institute for Health and Care Excellence (2010) Chronic obstructive pulmonary disease in over 16s: diagnosis and management.
- Celli BR, MacNee W (2004) Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 23: 932-946. [Crossref]
- GOLD (2017) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Global Initiative for Chronic Obstructive Lung Disease.
- Zhou Y, Zhong N, Xiaochen L, Chen S, Zheng J, et al (2017). Tiotropium in early-stage chronic obstructive pulmonary disease. N Engl J Med 377: 923-935.
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, et al (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010. A systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2095-2128.
- Wheaton AG, Cunningham TJ, Ford ES, Croft JB (2015) Employment and activity limitations among adults with chronic obstructive pulmonary disease-United States. MMWR Morb Mort Wkly Rep 64: 289-295.
- GOLD (2015) Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. Global Initiative for Chronic Obstructive Lung Disease.
- D'Urzo A, Ferguson GT, van Noord JA, Hirata K, Martin C, et al. (2011) Efficacy and safety of once-daily NVA237 in patients with moderate-to-severe COPD: the GLOW1 trial. Respir Res 12: 156. [Crossref]
- Cazzola M, MacNee W, Martinez FJ, Rabe KF, Franciosi LG, et al. (2008) Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J 31: 416-469. [Crossref]
- Beeh KM, Singh D, Di Scala L, Drollmann A (2012) Once-daily NVA237 improves exercise tolerance from the first dose in patients with COPD: the GLOW 3 trial: Int J Chron Obstruct Pulmon Dis 7: 503-513.
- Melani AS (2015) Long-acting muscarinic antagonists. Expert Rev Clin Pharmacol 8: 479-501. [Crossref]
- Murphy L, Rennard S, Donohue J, Molimard M, Dahl R, et al. (2014) Turning a molecule into a medicine: the development of indacaterol as a novel once-daily bronchodilator treatment of patients with COPD. Drugs 74: 1635-1657.
- Dhal R, Chung KF, Buhl HR, Magnussen H, Nonikov V, et al. (2010) Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 65: 473-478.
- Koch A, Pizzichini E, Hamilton A, Hart L, Korducki L, et al. (2014) Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat® versus placebo and formoterol twice daily in patients with GOLD 2-4 COPD: results from two replicates 48-week studies. Int J Chron Obstruct Pulmon Dis 9: 697-714.
- Kempsford R, Norris V, Diederer S (2013) Vilanterol trifenatate, a novel inhaled long-acting beta2-adrenoreceptor agonist, is well tolerated in healthy subjects and demonstrates prolonged bronchodilation in subjects with asthma and COPD. Pulm Pharamacol Ther 26: 256-264.
- Mahler DA, D’Urzo A, Bateman ED, Ozkan SA, White T, et al. (2012) Concurrent use of indacaterol plus tiotropium in patients with COPD provides superior bronchodilation compared with tiotropium alone: a randomized, double-blind comparison. Thorax 67: 781-788.
- Vincken W, Aumman J, Chen H, Henley M, McBryan D, et al. (2014) Efficacy and safety of coadministration of once-daily indacaterol and glycopyrronium versus indacaterol alone in COPD patients: the GLOW6 study. Int J Chron Obstruc Pulmon Dis 9: 215-228.
- Bateman ED, Ferguson GT, Barnes N, Gallagher N, Green Y (2013) Dual bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE study. Eur Respir J 42: 1484-1494.
- Wedzicha JA, Decramer M, Ficker JH; Niewoehner DE, Sandstrom T, et al (2013) Analysis of chronic obstructive pulmonary disease exacerbations with the dual bronchodilator QVA149 compared with glycopyrronium and tiotropium (SPARK). A randomized, double-blind, parallel-group study. Lancet Respir Med 1: 199-209.
- Spyratos D, Sichletidis L (2015) Umeclidinium bromide/vilanterol combination in the treatment of chronic obstructive pulmonary disease: a review. Ther Clin Manag 11: 481-487.
- Braman SS (2012) Refractory asthma: lessons learned from the bronchoscope. Chest 141: 575-576. [Crossref]
- Hekking PP, Wener RR, Amelink M, Zwinderman AH, Bouvy ML, et al. (2015) The prevalence of severe refractory asthma. J Allergy Clin Immunol 135: 896-902. [Crossref]
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, et al. (2014) International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 43: 343-373. [Crossref]
- O’Neill S, Sweeney J, Patterson CC, Menzies-Gow A, Niven R, et al. (2015) The cost of treating severe refractory asthma in the UK: an economic analysis from the British Thoracic Society Difficult Asthma Registry. Thorax 70: 376-378.
- Wu W, Bleecker E, Moore W, Busse WW, Castro M, et al. (2014). Unsupervised phenotyping of Severe Asthma research Program using expanded lung data. J Allergy Clin Immunol 133: 1280-1288.
- Wenzel SE (2012) Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18: 716-725. [Crossref]
- Lambrecht BN, Hammad H (2015) The immunology of asthma. Nat Immunol 16: 45-56. [Crossref]
- Brusselle GG, Maes T, Bracke KR (2013) Eosinophils in the spotlight: Eosinophilic airway inflammation in nonallergic asthma. Nat Med 19: 977-979. [Crossref]
- Haldar P, Pavord ID (2007) Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol 119: 1043-1052. [Crossref]
- Fahy JV (2015) Type 2 inflammation in asthma--present in most, absent in many. Nat Rev Immunol 15: 57-65. [Crossref]
- Pavor ID, Korn S, Howarth P, Bleecker ER, Keene ON, et al. (2012) Mepolizumab for severe eosinopilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 380: 651-659.
- Hanania NA, Noonan M, Corren J, Korenblat P, Zhen Y, et al. (2015) Lebrikizumab in moderate-to-severe asthma: pooled data from two randomized placebo-controlled studies. Thorax 70: 748-756.
- Wenzel S, Castro M, Corren J, Maspero J, wang L, et al (2016) Dupiloumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus long-acting ß2 agonist: a randomized, double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 388: 31-44.
- Radhakrishna N, Tay TR, Hore-Lacy, Stirling R, Hoy R, et al. (2017) Validated questionnaires heighten detection of difficult asthma comorbidities. J Asthma 54: 294-299.
- van der Meer AN, Pasma H, Kempennar-Okkema W, Pelinck JA, Storm H, et al. (2016) A 1-day visit in a severe asthma centre: effect on asthma control, quality of life, and healthcare use. Eur Respir J 48: 726-733.
- Befekadu E, Onofrei C, Colice GL (2014) Tiotropium in asthma: a systematic review. J Asthma Allergy 7: 11-21. [Crossref]
- Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, et al. (2010) Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med 363: 1715-1726. [Crossref]
- Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, et al. (2012) Tiotropium in asthma poorly controlled with standard combination therapy. N Engl J Med 367: 1198-1207. [Crossref]
- Gamble J, Stevenson M, McClean E, Heaney LG (2009) The prevalence of nonadherence in difficult asthma. Am J Respir Crit Care Med 180: 817-822. [Crossref]
- GINA (2015) Global strategy for asthma management and prevention. Global Initiative for Asthma.
- Diagnosis of Asthma, COPD, and Asthma-COPD Overlap Syndrome (ACOS) (2015) A joint project of GINA and GOLD.
- Postma DS, Rabe KF (2015) The Asthma-COPD Overlap Syndrome. N Engl J Med 373: 1241-1249. [Crossref]
- Terzano C, Petroianni A, Ricci A, D’Antoni L, Allegra L. (2004) Early protective effects of tiotropium bromide in patients with airways hyperresponsiveness. Eur Rev Med Pharmacol Sci 8: 259-264.
- Yoshida M, Nakano T, Fukuyama S, Matsumoto T, Eguchi M, et al. (2013) Effects of tiotropium on lung function in severe asthmatic with or without emphysematous changes. Pulm Pharmacol Ther26: 159-166.
- Magnussen H, Bugnas B, van Noord J, Schmidt P, Gerken F, et al. (2008) Improvements with tiotropium in COPD patients with concomitant asthma. Respir Med 102: 50-56. [Crossref]