Molecular oxygen is vital for energy which is essential for life. The fact that we all need oxygen to live might make us think that the use of supplemental oxygen in every hypoxemic patient seems obvious. This is a fallacy. The survival of all metazoan organisms is dependent on the regulation of the delivery and use of oxygen to maintain a balance between the generation of energy and the production of oxygen radicals, potentially toxic. HIF (hypoxia inducible factor) is a transcription factor or nuclear messenger that should be conceptualized as a master regulator of oxygen homeostasis. Therefore, it is proceeded to discuss how its action in hypoxia conditions the activation of diverse genes that codify the synthesis of proteins that explain some of the clinical manifestations and the events of hypoxia. We also discuss how inflammatory diseases of the respiratory system can perpetuate, in the presence of oxygen at inappropriately high doses, the oxidative stress that in turn amplifies the inflammatory phenomenon. Subsequently, the doses of ambulatory oxygen are defined in conventional indications and in some unconventional indications such as air travel and high altitude. Finally, some research avenues are mentioned in this topic.
Inflammation, hypoxia, hyperoxia, oxidative stress, domiciliary oxygen
The atmosphere as we know it today as about 4.5 billion years old. At first probably it consisted of volcanic emanations almost no oxygen. Living organisms appeared on Earth were essentially anaerobic. However, their growth is inhibited and usually die from exposure to concentrations of 21% oxygen, the usual atmospheric level [1]. This level was reached because the primitive marine organisms acquired chlorophyll, a pigment able of convert light energy into chemistry, by photosynthetic combination of water with CO2 generating oxygen. Almost all of the free oxygen in today's air was formed by this combination in seawater. Approximately 570 million years ago, that the content of dissolved oxygen in the oceans allowed marine life to breathe. It was about 400 million years ago that the atmosphere acquired enough oxygen in the gas phase for the animals that emerged from the sea to breathe air [2].
The discovery of oxygen would have to await the appearance in the European scientific firm of Antoine-Laurent Lavoisier (1743-1794) and Joseph Priestly (1773-1804). Lavoisier with his precise and methodical approach, directed chemistry by the same way that physics had taken a century before with the monumental work of Isaac Newton and separated it from alchemy. Studying combustion with "mercury lime" (mercury oxide) found that an acid formed every time a substance bound with this active gas (air) and baptized it with the Greek roots meaning "gas producer", OXY -GEN. The idea that oxygen is the "acid-generating principle" is erroneous, but the element was already discovered and had a name. He also coined the name of HYDRO-GEN, which means water producer (had shown that water is actually hydrogen oxide). Who produced oxygen was Priestley in 1774, also working with mercury oxide; and predicted that it could be healthy for the lungs in certain morbid conditions. Lavoisier came from a wealthy parisian family and became a tax collector, which was an aristocratic privilege in prerevolutionary France. This was a corrupt and unpopular system, and used it partly to finance his experiments. In 1794 a Revolutionary Court condemned him for having participated in this collection system. Despite the pleas of his friends and the great services rendered to science and France, he was condemned to death and the judge commented: "The republic does not need sages." The guillotine claimed one of its greatest victims [3].
Later, West would teach us that there are 5 mechanisms of alveolar hypoxia and hypoxemia: diffusion disorders, hypoventilation, right-left intrapulmonary shunt, imbalance ventilation/perfusion, and decreased oxygen inspired fraction [4].
Despite the ancient acquisition of oxygen from the atmosphere and its discovery by Priestley and Lavoisier 250 years ago, the cellular and nuclear mechanism of the coordinated response to hypoxia has been unraveled in the last 25 years. The key to this knowledge is the family of HIFs, which are transcription regulators that respond primarily to oxygen levels and bind to specific DNA sequences, which control the rate of gene transcription.
All nucleated cells sense and respond to hypoxia and not just the glomus cells of the carotid body, as was originally believed. One of the first key elements was the discovery of the role of HIFs as proteins that bind to the hypoxia response element (HRE) of the gene encoding erythropoietin (EPO) synthesis under conditions of hypoxia [5]. It was then defined that HIF-1 is a heterodimer formed by two subunits: HIF-1α (oxygen regulatory unit) and HIF-1β (constitutional unit). HIF-1β is ubiquitous and is transcribed continuously, but HIF-1α is restricted, and under normoxic conditions, is present at very low levels in the cytoplasm [6]. Under normoxic conditions, prolyl-hydroxylases (PHDs) in the presence of oxygen, iron and 2-oxoglutarate, hydroxylates the HIF-1α proline, creating a binding site with the von Hippel-Lindau protein, which recruits the ubiquitin E3 ligase. Polyubiquitination signals protein for degradation by proteasome 26S [7] (Figure 1). HIF inhibitory factor (FIH) is an oxygen-dependent asparaginyl-hydroxylase (hydroxylates asparagine residue) that reduces HIF activity [8]. Therefore, both metabolic pathways reduce HIF activity and both are oxygen dependent, which explains the reduced expression of HIF in the presence of normal oxygen concentrations. HIF-2 consists of two subunits: HIF-1β and HIF-2α which is a paralogue of HIF-1α and expressed mainly in normal tissue macrophages and plays an important role in erythropoiesis and vascularization. Its expression is also regulated by oxygen-dependent hydroxylation [9].
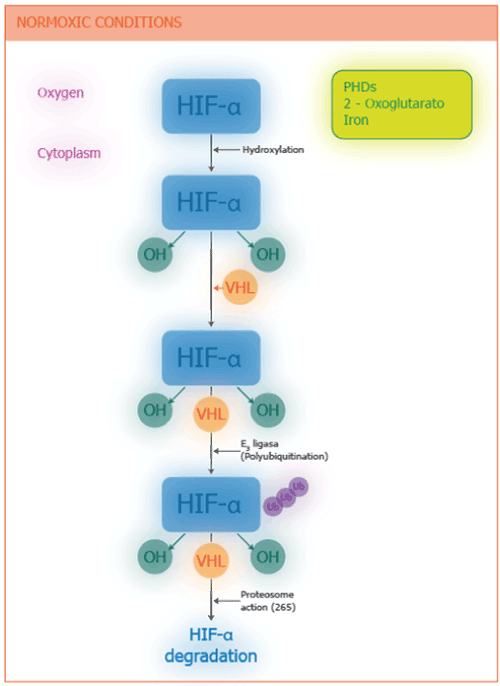
Figure 1. Role of Hypoxia-Inducible Factor α (HIF-α) under Normoxic Condition. Under normoxic conditions, HIF-α is hydroxylated by prolyl-hydroxylase domain 2 protein (PHD2). It then interacts with von Hippel-Landau protein (VHL), and recruits ubiquitin E3 ligase. The polyubiquitinitation of HIF-1α flags the protein for degradation by the 26S proteasome.
Under hypoxic conditions, hydroxylation of proline and asparagine is not activated, and HIF-1α accumulates rapidly, translocated to the nucleus, dimers with HIF-1β, recruit p300 (coactivating protein), binds to HRE and activates RNA polymerase II from hundreds of target genes. For example, EPO, which is the hormone that stimulates the production of red blood cells, VEGF encoding vascular endothelial growth factor (which is the angiogenic factor that stimulates the formation of blood vessels) and glycolytic enzymes, which adapt cellular metabolism to conditions of hypoxia [10] (Figure 2). Other genes that are activated by HIFs are those encoding the synthesis of nuclear factor Kβ (NF-kβ) (a pro-inflammatory factor) and Toll-like receptors (TLRs). Normally NF-kβ is inactive in the cytosol because there is a molecule that inhibits its activity, IKβα, but when hypoxia conditions (e.g., EPOC) are present, HIFs on translocation to the nucleus (in alveolar macrophages) activate the gene of the IKKβ, a kinase that phosphorylates IKβα and the inactive, releasing the control that was on NF-Kβ. NF-Kβ translocated to the nucleus and in turn, activates genes encoding the synthesis of pro-inflammatory proteins such as tumor necrosis factor-alpha (TNF-α) and interleukin-8 (IL-8), the largest chemotactic mediator and activator of neutrophils that infiltrate the airway, capitalizing the inflammation (Figure 3). Hypoxia therefore produces inflammation and this in turn tissue hypoxia generating a vicious circle [11]. In general, hypoxia amplifies the cell activity of innate immune response /while suppressing the adaptive immune system response [12]. The TLRs are a family of protein receptors that play a key role in recognizing molecules in the immune system [13].
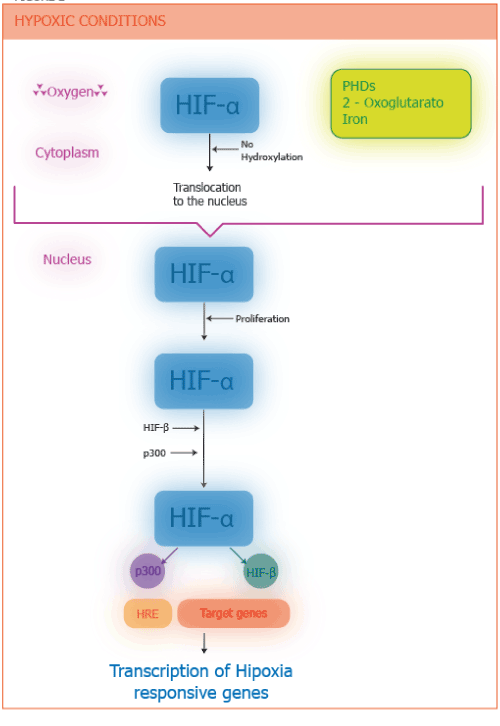
Figure 2. Role of the HIF-1α under hypoxic conditions. Under hypoxic conditions, HIF-α does not undergo degradation and rapidly accumulates, but instead translocated to the nucleus where dimerizes with HIF-β and the recruits coactivators (p300), binds to hypoxia response elements (HRE), and activates the transcription by RNA polymerase II of hundreds of target genes.
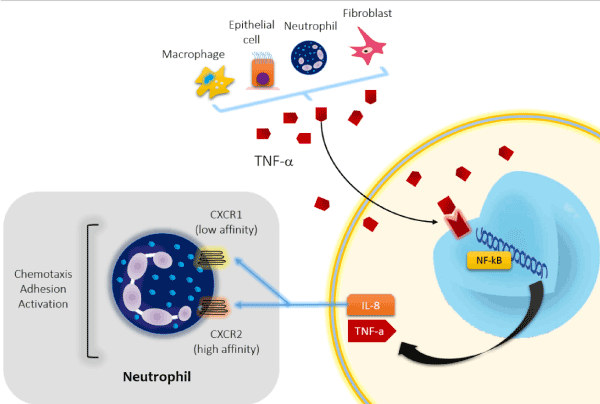
Figure 3. Activation of the Nuclear Factor-kβ (NF-kβ). NF-kβ has a central role in orchestrating the inflammatory response in COPD. NF-kβ is activated by oxidants and inflammatory mediators such as TNF-α produced by macrophages, epithelial cells, neutrophils and fibroblasts. Most of the inflammatory proteins that are upregulated in COPD macrophages are regulated by NF-kβ. Interleukin-8(IL-8) is ubiquitous inflammatory chemokine that mediates several inflammatory events in the lungs. It is a major chemotactic and activating mediator of neutrophils. TNF-α = tumor necrosis factor-α. NF-kβ = Nuclear Factor-kβ. CXCR1= chemokine C-X-C motif receptor 1. CXCR2 = chemokine C-X-C motif receptor 2.
Solid tumors contain increased levels of HIF-1α and HIF-2α, and these elevated levels correlate with cancer-related death. Inflammatory cells also contribute to the abnormal growth and activity of blood vessels in tumors by the release of VEGF [14,15].
One way in which HIF-1 promotes cell survival under hypoxic conditions is through the switch of oxidative to glycolytic metabolism (Figure 4). Glycolytic enzymes convert glucose into pyruvate, which can be converted, in well-oxygenated cells, into acetyl coenzyme A (CoA) (by the enzyme pyruvate dehydrogenase (PDH). Acetyl CoA is oxidized in mitochondria to tricarboxylic acid (TCA), generating electrons that are transferred through a series of protein complexes, electron transport chain (ETC), and are eventually transferred to oxygen to form water. The proton gradient established by the ETC is used to synthesize ATP. Under hypoxic conditions, pyruvate dehydrogenase kinase 1 (PDK1) inactivates PDH, and lactate dehydrogenase A (LDHA) converts pyruvate to lactate. HIF-1 activates the expression of LDHA and PDK1 genes, thus tipping the balance from oxidative to glycolytic metabolism [16]. Although glycolysis is a relatively inefficient path for the production of ATP (oxidative metabolism generates 18 times more ATP per mole of glucose than glycolytic metabolism), it can keep the cell alive as it reduces the oxygen consumption. Figure 5 summarizes the wide variety of effects of HIFs.
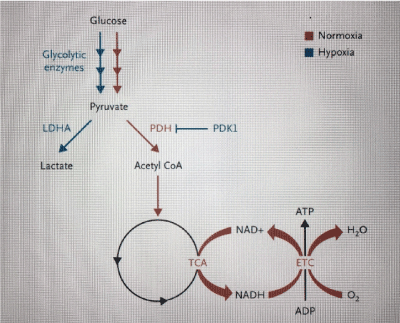
Figure 4. Regulation of Glucose metabolism in response to Changes in Cellular Oxygen Levels. Glucose is converted to pyruvate by the action of the glycolytic enzymes. In well-oxygenated cells (red pathway), PDH converts pyruvate to acetyl coenzyme A (CoA), which is oxidized in the mitochondrial tricarboxylic acid (TCA) cycle, generating electrons that are transported through a series of protein complexes (ETC) and are eventually transferred to oxygen to form water. The proton gradient established by the ETC is used to synthesize ATP. Under hypoxic conditions (blue pathway) PDK1 inactive PDH, and LDH converts pyruvate to lactate. See text for description [11].
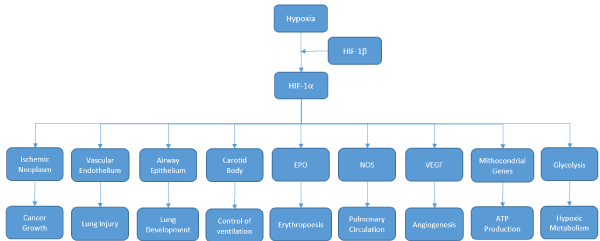
Figure 5. The spectrum of Responses to HIFs. Some targets genes activated by HIFs and the physiological responses are shown. EPO denotes erythropoietin, NOS nitric oxide synthase, and VEGF vascular endothelial growth factor.
Hypoxic pulmonary hypertension is a progressive and often fatal complication of chronic lung disease. Unlike systemic arterioles, which dilate in response to hypoxemia in order to increase tissue perfusion, pulmonary arterioles constrict in order to shunt blood away from regions of the lung that are not ventilated. This is an adequate adaptive response in patients with a localized pathology (lobar pneumonia), but not in those with chronic lung disease, in whom alveolar hypoxia occurs throughout the lungs, producing a generalized contraction of the pulmonary arterioles and subsequent pulmonary hypertension, leading to right heart failure and progression of hypoxemia. HIFs regulate target genes that play key roles in the pathogenesis of hypoxic pulmonary hypertension (World Health Organization-WHO-group III) and idiopathic pulmonary hypertension (WHO group I) [17,18]. HIF-1 induced by alveolar hypoxia (chronic lung disease or residence at high altitudes) activates vascular smooth-muscle cells (SMCs), leading to decreased expression of voltage-gated potassium channels (Kv1.5 and Kv2.1), increased expression of transient receptor-potential calcium channels (TRPC1 and TRPC6), and increased expression of sodium-hydrogen exchanger 1 (NHE1). The resulting alterations in the intracellular concentrations of potassium, calcium and hydrogen ions trigger SMCs depolarization, contraction, hypertrophy, and hyperplasia, which lead to increased pulmonary vascular resistance. Endothelin 1 (EDN1), which is produced by hypoxic vascular cells in an HIF-dependent manner, also contributes to reduced expression of the Kv channel genes. Altered redox homeostasis also plays an important role in pulmonary hypertension [19].
The physiological effects and clinical manifestations of hypoxemia are logical consequences of the above-mentioned alterations. There is no significant increase in ventilation with the decrease in PaO2 while levels remain above 8 Kp (> 60 mmHg), but if PaO2 falls below 7.5 Kp (< 55 mmHg) a marked increase in minute ventilation occurs, with a subsequent fall in PaCO2 and respiratory alkali. Hypoxemia results in peripheral vasodilation, which induces compensatory tachycardia and a subsequent increase in cardiac output to improve oxygen delivery. Regional pulmonary vasoconstriction (in response to alveolar hypoxia) occurs in an effort to match ventilation and perfusion. EPO secretion increases causing erythrocytosis, thereby increasing oxygen carrying transport. These compensatory mechanisms can cause detrimental long-term effects, such as polycythemia, pulmonary hypertension, and right ventricular failure. On a cellular level, mitochondrial function declines, anaerobic glycolysis occurs, and lactate / pyruvate ratio is increased with metabolic acidosis. Patients may have impaired judgment with mild to moderate levels of hypoxemia, with progressive loss of cognitive and motor functions, and eventually loss of consciousness as hypoxemia worsens. Other non-specific symptoms include headache, breathlessness, palpitations, angina, fatigue, and tremor [20].
Anaerobes today are presumably descendants of those primitive organisms, "adapting" to increased atmospheric levels of oxygen for restricted themselves to environments where oxygen does not penetrate. Other organisms have developed defense systems to protect themselves from oxygen toxicity using the same to metabolic transformations (hydroxylase, oxidase and oxygenase enzymes are examples) and for efficient energy production by using ETC with oxygen as the terminal electron acceptor. For example, those present in mitochondria, where 80% of our cellular ATP needs are produced [21].
It is remarkable that we have developed antioxidant defenses against concentrations of 21% oxygen, but not more. All aerobic species suffer injury when exposed to concentrations above 21%. For example, if humans breathe pure oxygen, before six hours they develop chest tightness, cough and sore throat [22]. Exposure periods greater than 48 hours with concentrations greater than 50% oxygen would damage the pulmonary alveoli and premature infants exposed to high doses of oxygen develop lenticular fibroplasia (which can occur with blindness) [23].
In 1954, Gerschman and colleagues in the United States (US) proposed that the harmful effect of oxygen was due to the formation of oxygen radicals, the superoxide oxygen theory, followed in 1968 the discovery of the enzyme superoxide dismutase (SOD), specific for the removal of a free radical substrate. In its simplest form, this theory states that oxygen toxicity is due to the excess formation of the superoxide radical (O2•¯) and that SODs are important antioxidant enzymes [24]. Under aerobic conditions, the electrons are transferred through ETC to oxygen to form water. Under hypoxia conditions, the electron release is increased before being transferred to the mitochondrial complex, resulting in the production of the superoxide anion, which is then converted to hydrogen peroxide (H2O2) and other reactive oxygen species (ROS). The cost is the loss of redox homeostasis [25]. The human body generates, therefore, superoxide anion by adding a single electron to oxygen.
O2 + e¯ ------------------- O2•¯
ROS are highly toxic to cells producing various types of stress, generating intracellular, structural and functional damage, perpetuating and amplifying inflammatory processes such as COPD, which become autonomous and continuous, although the initial stimulus (smoking and/or exposure to fuels of biomass) has been discontinued [26]. Therefore, humans have evolved complex circulatory, respiratory and neuroendocrine systems to ensure that the oxygen level is exquisitely and precisely maintained, since a deficiency or excess can result in the death of cells, tissue or the organism.
In addition to alleviating arterial hypoxemia and alleviating the consequences listed previously, supplemental oxygen has several therapeutic benefits. Oxygen improves respiratory distress in normal subjects and in COPD patients during exercise [27]. Effort tolerance is improved in patients who develop significant hypoxemia [28]. In EPOC, improvement in both events may be due to decreased minute-ventilation, decreased dynamic hyperinflation, relief of hypoxic pulmonary vasoconstriction, and improvement in hemodynamics (e.g., decreased pulmonary vascular resistance and increased cardiac output), or increased oxygen delivery [29]. Proposed alternative mechanisms include improvement in the function of ventilatory muscles and altered ventilatory muscle recruitment [20]. Oxygen flow can stimulate upper airway receptors and facial receptors of the trigeminal nerve and reflexively inhibit central ventilatory drive. Finally, there also appears to be a direct effect of oxygen administration on the perception of dyspnea independent of any change in minute ventilation [27]. However, the current evidence does not support the routine use of oxygen in normoxemic patients with COPD during exercise [30]. These effects of oxygen on ventilation and work of breathing may help prevent respiratory muscle fatigue during acute respiratory failure, particularly in patients with chronic respiratory insufficiency who have a higher baseline ventilatory demand.
Hypoxia plays a critical role in pathobiology of heart disease, cancer, stroke and chronic lung disease, which are responsible for 60% of deaths in the US. Although the majority of evidence comes from the use of oxygen in patients with COPD, the scope of the guidance includes patients with a variety of long-term respiratory illness, and other groups in whom oxygen is currently ordered, such as those with heart failure, cancer, end-stage cardiorespiratory disease, terminal illness or cluster headache [31]. Medicare reimbursements for oxygen-related costs for patients with COPD exceeded $ 2 billion in 2011, in the US [32].
For more than 30 years the use of long-term oxygen therapy (LTOT) was based on studies conducted in very select cohort of patients. Two landmark studies non-blind, randomized trials that were conducted in 1970s: the Nocturnal Oxygen Therapy Trial (NOTT) [33] and the British Medical Research Council (MRC) Long-Term Domiciliary Oxygen Therapy Trial [34] examined the effects of LTOT on survival and physiological function in patients with severe chronic bronchitis and emphysema. Together, both studies involved 290 patients. LTOT given for 15 hours or more per day prolonged survival, as compared with only nocturnal use or not such therapy. Patients had COPD and chronic severe hypoxemia (a partial pressure of arterial oxygen [PaO2] of < 55 mm Hg or an oxyhemoglobin saturation level as measured by pulse oximetry [SpO2] of approximately 88%) or moderate hypoxemia (PaO2 of 56-59 mm Hg or SpO2 between 88% and 90%) with signs of right heart failure or polycythemia. Of note, patients in both studies were younger and had fewer comorbidities than patients starting LTOT in current practice [35]. A survival benefit was not seen in two small trials in the 1990s of nocturnal oxygen (in 76 patients) or LTOT (in 135 patients) among patients with mild-to-moderate hypoxemia [36,37].
The recent Long-Term Oxygen Treatment (LTOT) randomly assigned 738 patients with COPD (73% of whom were men), and mild-to- to moderate hypoxemia at rest or during 6-minute walk test to receive LTOT or not long-term supplemental oxygen [38]. The supplemental oxygen was prescribed at 2 liters of oxygen per minute continuously in participants with resting hypoxemia (57%) and as adjusted oxygen dose during exercise and 2 liters of oxygen per minute during sleep in patients with exertional hypoxemia only (43%). There were 368 participants with oxygen and 370 without oxygen. During a median follow-up of 18.4 months, there was no significant between-group difference in the rate of death or first hospitalization in the time-to-event analysis primary (primary outcomes) or in the mortality and the rate of hospitalizations separately, COPD exacerbations, quality of life, anxiety, depression or functional status. From available information LTOT should be prescribed to patients with chronic hypoxemia (> 3 weeks) and severe, at rest (PaO2 <55 mm Hg [< 7.5 Kp] or SpO2 <88%) while breathing in ambient air. Therefore, LTOT should not be prescribed to patients with moderate hypoxemia at rest or during exercise [39]. However, there are authors who preach that a lack of evidence of effect is not evidence of a lack of any clinical effectiveness, suggesting that a trial of oxygen use might still be appropriate in selected patients with moderate exertional hypoxemia and intractable breathlessness, despite appropriate evidence-based treatment. One million citizens (US) use oxygen for moderate hypoxemia. Should the prescription be discontinued? If hypoxemia is severe during exercise, oxygen should be prescribed [20]. Moderate hypoxemia qualifies for LTOT if there is at rest, polycythemia (Hto > 55%), congestive heart failure or pulmonary hypertension [40]. There also appears to be no evidence that giving 24-hour oxygen is greater than 15 hours a day, so the latter duration is adequate for LTOT [41] (Figure 6). In addition, there is a risk of an increase in CO2 levels in patients receiving oxygen 24 h/day. The possible reasons for patients with severe hypoxemia to benefit from supplemental oxygen are the effects of the nonlinear oxygen saturation threshold on pulmonary vasoconstriction, mediator release, and ventilatory control, which occurs with SpO2 of 88% or less and probably more important, in patients with chronic hypoxemia [42]. Adding oxygen to patients with mild to moderate saturation levels will not benefit tissue oxygen delivery and may increase CO2 levels and oxidative injury in COPD patients.

Figure 6. Prescription of supplemental oxygen. Partial pressure of oxygen and carbon dioxide are measured using kilopascals (kPa) and millimeters of mercury (mm Hg). 1 kPa = 7.5 mm Hg and 1 mm Hg = 0.133 kPa.
Sleep disorders are common in COPD and COPD patients tend to develop nocturnal oxygen desaturation (NOD). The mechanisms responsible are: hypoventilation, mismatch ventilation / perfusion (difficult to quantify during sleep), increased upper airway resistance, and decreased respiratory muscle drive [43]. Night Oxygen Therapy (NOT) refers to oxygen prescribing only at night for patients who are normoxemic at day or have minimal hypoxemia and do not qualify for LTOT, but have NOD. Theoretically (NOT) could prevent NOD and the development of pulmonary hypertension, however, NOT has not been shown to improve sleep quality, pulmonary hemodynamics and survival in patients with COPD. For these reasons it is not recommended ex officio in the international guidelines and rather if NOD is present, other causes of NOD such as hypoventilation-obesity syndrome (HOS), neuromuscular weakness and obstructive sleep apnea (OSA) should be investigated [31]. Screening for NOD in normoxemic COPD patients should probably be reserved for those with hypercapnia, polycythemia, or evidence of pulmonary hypertension (20). Using NOT of trade is a fallacy just as it is the prescription of oxygen in mild to moderate chronic hypoxemia and during the exercise, except for very qualified exceptions.
For other pathologies, indications for home oxygen use are generally the same as for COPD. However, some aspects of oxygen therapy in these indications should be emphasized. For example, the use of LTOT in pulmonary hypertension (PAH), if not associated with COPD, does not impact survival and only improves tissue oxygenation and prevents some complications. It should be started when there is moderate hypoxemia (PaO2 < 8 Kp) and do not wait until severe hypoxemia, and some guides recommend it 24 hours a day [31]. Outpatient oxygen therapy (AOT) is defined as the use of supplemental oxygen during exercise and / or activities of daily living outside the home. It is not routinely recommended if the patient is not in the LTOT program. It may be offered during a formal or supervised exercise or pulmonary rehabilitation program and demonstrates improved endurance with exercise. It could be offered to patients with cystic fibrosis and active lifestyles or for some patients with interstitial lung disease (ILD) who do not qualify for LTOT and who desaturate during exercise and for patients with LTOT who can’t leave the home without supplemental oxygen. For example, to go to a medical appointment [44]. Patients with neuromuscular diseases that compromise ventilation with HOS and OSA when using NOT should be associated with noninvasive ventilation (NIV) and only if NOD does not correct with the latter tool. NOT could be used in patients with heart failure (CHF) with NOD, although they do not meet LTOT criteria and have evidence of sleep disordered breathing without other causes of NOD (OSA, HOS) and in whom CHF treatment has been optimized [45]. Short burst oxygen therapy (SBOT) refers to the use of oxygen for the relief of breathlessness not relieved by any other treatments. It should not be used before or after exercise, hypoxemic patients at rest, nor in normoxemic patients with COPD, nor when the patient leaves hospital for an acute exacerbation of COPD. Cluster headache is the most severe of primary headache syndromes. It is characterized by periodic attacks of strict unilateral pain associated with autonomic unilateral cranial symptoms. Most patients have episodic cluster headache (ECH), with periods of cluster that typically occur in a circannual rhythm, while 10% have the chronic form (CCH), with no remissions between periods of cluster. Patients are provided with a static oxygen cylinder for the home. Generally, the headache sends with high doses of 12 liters per minute for 10-20 minutes with a non-rebreather mask [46].
The term palliative oxygen therapy (POT) refers to the use of oxygen to alleviate persistent and refractory respiratory distress in advanced or life-limiting disease, regardless of background pathology where all reversible causes have been or are being treated optimally. Dyspnea is common in patients with advanced disease that limits life, of any type [47]. There is no objective evidence of improvement in these patients in dyspnea or general well-being, and should not receive POT if they are not hypoxemic or hypoxemia is minimal (SpO2 > 92%). Opioids are significantly better than POTs in reducing the intensity of dyspnea in hypoxemic and non-hypoxic patients. Very exceptionally (more than often) POT can be considered for patients with dyspnea that does not respond to other treatment modalities. In these cases, a formal evaluation of the palliative effect of oxygen should be done to reduce respiratory distress and improve quality of life [48].
Household oxygen is an additional economic burden for health systems. It is not only expensive but also a psychological and social burden. The oxygen-dependent patient is depressed, socially isolated, and prolonged oxygen therapy is associated with physiological and structural deterioration of the lung. Therefore, oxygen under these conditions should be very carefully assessed, using scientific criteria rather than as a placebo or ritualistic use [49]. From another perspective, there are recent studies that do not rule out the potential benefit for patients with certain comorbidities that report symptomatic improvement. It will be very interesting in the future as the guides will incorporate these results [50]. It is obvious that in this area there is no black or white color but a spectrum of grays.
LTOT results in patients who continue to smoke at home may be of very limited benefit. Continuing smoking predisposes to secondary polycythemia, accelerates the decline of lung function and increases morbidity in COPD. The beneficial effect of LTOT may be counterbalanced by increased levels of carboxyhemoglobin produced by persistent smoking. However, the evidence is insufficient to determine the adverse effects of smoking persistence in LTOT compared to non-smokers, but if LTOT is to be prescribed to patients who continue to smoke, it should be discussed the possible more limited clinical outcomes than if they did not smoke [51]. The risk of serious burns, injury by inhalation or death from the use of oxygen while smoking or the use of oxygen near pilot light, stoves, gas or candles should be clearly explained and smoking cessation should be strongly encouraged.
Family members who smoke are an additional risk. Safety must be a crucial factor and educational and written information must be provided to the patient and family members with a written and signed contract that ensures that the patient and the family understand and assume the risks. The risk of fire spreading to children and neighbors and heavily overcrowded housing should be a criterion that can be taken into account for non-prescription. The use of carbon monoxide and / or urinary cotinine measurements can help identify patients who continue to smoke. The testimony of non-smoked is not reliable [52]. Oil-based emollients and petroleum jelly enable and perpetuate combustion in the presence of oxygen, so that only aqueous-based products should be used on hands, face and nose while using oxygen. Patient and family should be instructed not to remove the fire sensor (which blocks the delivery of oxygen from the equipment in the presence of smoke), nor to change the flow or connections recommended by the company or the technicians who visit the home. Telephone numbers and location of the home should be reported to the nearest fire station. Regular visits every 3-6 months should be implemented to monitor compliance with the rules. If there is a reasonable doubt regarding risks vs benefits, therapy should not be prescribed, even if the patient qualifies for gasometric criteria for LTOT [53].
Pulse oximetry should not be used as a sole criterion to indicate LTOT, but to justify an arterial gas (ABG). If SpO2 <92%, or SpO2 < 94% with peripheral edema, Hto > 55% or PAH data, ABG should be indicated (8 weeks after stabilization of an acute exacerbation), and should be repeated at 3 weeks. If both samples define described criteria, LTOT is considered. 1 liter per minute should be initiated and SpO2 can be used to titrate doses. The intervals should be at least 20 minutes (minimum time for PaO2 to equilibrate with the PAO2 [oxygen alveolar pressure]) [54]. It is increased by 1 liter per minute each time until a SpO2 > 90 or a PaO2 > 60 mmHg (> 8 Kp) (ABG) is obtained. It is advisable to increase 1liter above the daytime level during the night to reduce NOD. If the patient is active, requirements during exercise should be investigated. If there is cognitive deficit, visual or coordination problems, patient should have a fixed flow and not adjustable by himself. The selection of equipment is based on costs, efficiency, resources and safety. Oxygen concentrators are the most cost-effective for home use but the efficiency becomes variable up to 4 liters per minute. They are the choice if the required flow is less than or equal to that level. Portable equipment allows greater daily use and better quality of life than static equipment. Nasal cannulas, by mixing oxygen from the device with atmospheric air, increase oxygen concentration by 3-4% (up to 21% of atmospheric air) per liter, so nasal cannulas allow delivery between a 24- 35% oxygen and are the choice before the Venturi mask, which become necessary if higher flows are required. If very high flows are required the oxygen-conserving (devices deliver oxygen during inspiration only) is the choice because they increase the time the cylinder lasts. There is no evidence that patients benefit from continuous humidification of home oxygen unless they are tracheostomized [31]. In third-world countries electricity costs can be a limiting factor for the use of concentrators.
There is always concern of worsening CO2 retention when the oxygen is given to patients with chronic hypercapnia. The mechanism that is usually believed to underlie is the reduction of the ventilatory response to hypoxia and subsequent hypoventilation. Already since the 1980s, Aubier and colleagues demonstrated, administering 100% oxygen to COPD patients, that this mechanism contributes only 22% to hypercapnia, and 30% is due to the reduced availability of hemoglobin to bind and transport CO2, also known as Haldane effect (oxyhemoglobin displaces the hemoglobin dissociation curve with CO2 to the right by increasing the amount of CO2 dissolved in blood, which determines PaCO2). The largest responsible for the increase of carbon dioxide is an increase in dead-space ventilation (48%) by release of hypoxic vasoconstriction in sub-ventilated areas, causing imbalance ventilation / perfusion and not hypoventilation [55].
However, with controlled oxygen administration, respiratory distress can be alleviated and hypoxemia treated without significant increases in PaCO2. If the patient has basal hypercapnia, when the oxygen is titrated, a basal ABG should be done each time it is increased by one liter, and when the titration is completed. If the patient develops respiratory acidosis and / or an increase of PaCO2 of >1 Kp (> 7.5 mm Hg) during LTOT, he should be evaluated for clinically unstable disease, optimized medication and re-evaluated in 4 weeks. Patients who develop 2 repeated episodes of increase of PaCO2 >1Kp (>7.5 mm Hg) during evaluation of LTOT, being clinically unstable, may receive home oxygen but with nocturnal NIV [56].
Oxygen and air travel: Ascent to high altitude via air travel exposes patients to hypobaric conditions, with an increased risk of hypoxemia. Pressurizing the aircraft cabin limits the fall in atmospheric pressure to an altitude of 8000 feet (2438 meters), allowing it ascent to much higher altitudes. If SpO2 > 95% or PaO2 > 72 mm Hg, breathing in ambient air and on land will not require supplemental oxygen during the flight. For those who do not meet these criteria, an equation has been described that allows predicting PaO2 at an altitude starting at 8000 feet [57].
PaO2 at 8000 ft = [0.238 X (PaO2 at sea level)] + [(20,098 X (FEV1 / FVC) + 22.258.
If the patient, on land and breathing ambient air, has PaO2 < 50 mm Hg, predictable at 8000 feet, will require supplemental oxygen during flight.
Determining the flow of oxygen in liters per minute that will be required during the flight is more difficult. The hypoxic challenge test (HCT) on land allows an acceptable estimate [58]. The inspired oxygen pressure at sea level is 150 mm Hg (21% O2), but at 8000 feet it falls to 108 mm Hg. This equals as if the patient, on land, breathed 15% O2. Therefore, on the ground, the HCT is made by breathing the patient not 21% (atmospheric level of O2) but 15% of O2. The test can be implemented in several ways. Breathing in a plethysmograph a mixture of gases containing only 15% oxygen and the rest N2 (nitrogen), or breathing from a cylinder of gases prepared with 15% O2 and the rest of N2 through a non-rebreather mask or a Douglas bag. Another strategy is to breathe from a cylinder of N2 to 100% with a Venturi mask at 40%. This will reduce the inspired fraction of O2 to 15%. This method is probably less than the plethysmograph but it is easier to implement, cheaper and better tolerated. When performing the arterial gas under these conditions we will know the predictable PaO2 and be titrated the oxygen flow that will require until obtaining an acceptable PaO2. That will be the flow that will require in the flight. In one series, the use of 2 liters per minute of supplemental oxygen during flight was sufficient for those who did not require oxygen ashore [59].
Oxygen at great heights: 15 million people are diagnosed with COPD in the US, the second leading cause of death in that country today [60]. Tissue hypoxia in these cases is due to arterial hypoxemia which in turn is secondary to imbalance ventilation / perfusion. But at high altitudes, it is the environmental hypoxia (decreased inspired fraction of O2 [FIO2]) which leads to hypoxemia and consequently to tissue hypoxia. The WHO estimates that 140 million people live at 2,500 meters or more, and it is likely that in the future this amount will increase as the population seeks residence in the heights due to global warming. They live in states of chronic alveolar hypoxia and severe hypoxemia. For example, over 3000 meters people live in Leadville (Colorado), Bolivia, Peru and Tibet (Asia), with PaO2 ranging from 58 mm Hg in Leadville (3100 meters high) to 43 mm Hg in La Rinconada (Perú) (5100 meters above sea level), when the sea level PaO2 is 95 mm Hg. For this reason, West believes that given the large number of people living at great heights, one of the main mechanisms of hypoxemia and chronic tissue hypoxia is possibly the reduced FIO2 [61,62].
Exposure to high altitudes may be for short periods of time for recreational purposes, or people residing in low altitude locations and moved to higher elevations for months or years (mine employees, hospitals, schools, embassies, corporations, etc.) and permanent residents at great heights (the Andes in South America and the plateaus of Tibet in Asia). The latter is the largest group chronically exposed to environmental hypoxia. The first two groups experience exertional dyspnea, impaired exercise capacity, and mild neurophysiological alterations. It is assumed that the permanent residents are adapted to the maximum at high altitudes. For thousands of years humans have lived on the plateaus of Tibet at great heights, exceeding 3000 meters. For example, in Lhasa in Tibet, the inspired oxygen pressure is 94 mm Hg (150 mm Hg sea level), the PaO2 is 61 mm Hg and the PaO2 is 54 mm Hg. When normal lungs are exposed to hypoxia, pulmonary vascular tone increases, increasing resistance with PAH and heart failure. Also, the increase in mass of red blood cells occurs but both mechanisms are poor adaptive responses. In adapted Tibetans these responses to hypoxia are erased and many of them have normal pulmonary arterial pressure and normal hemoglobin levels. Moreover, many have normal tissue aerobic metabolism despite hypoxemia [63].
With the advent of powerful DNA sequencing methods, loci have been identified in Tibetans where genetic selection has occurred in those living at high altitudes relative to those living in the lowlands. The strongest evidence for selection was found at the locus encoding HIF-2α [64-66]. Selection of loci encoding PHD2, FIH-1 and target HIF genes has also been reported [67]. As the selected genetic variants alter the function of these proteins is not known. The variants may result in reduced PHD2 and HIF-2α activity, the net effect of which may be an altered balance between HIF-2α, which mediates the erythropoietic and vascular responses to hypoxia, and HIF-1α, which mediates metabolic and vascular responses. In any case, these findings support the central role of the PHD-HIF system in maintaining oxygen homeostasis [16].
However, a perfect adaptation to chronic hypoxia is a fallacy. The three groups of residents at great heights mentioned above improve their maximum oxygen consumption if they descend to low altitudes. This has been clearly demonstrated in careful and well-conducted studies in Bolivian patients (3600 meters) and Peruvian patients (4300 meters) who, when descending at sea level, increase the maximum oxygen consumption [68,69]. These results indicate that permanent residents are not maximally adapted since their physical capacity improves to low altitude oxygenation levels. Neuropsychological functions are also altered in high-altitude residents compared with low-level resident controls, and cognitive development of children is decreased at high altitudes, although additional studies are required [70-74]. The results suggest that permanent residents at great heights can improve their neuropsychological function by descending to low altitudes. When increasing FIO2, when it descends, it improves the oxygenation in the muscles and in the brain, this suggests that the same effect could be obtained by increasing the oxygen concentration in the air at great heights, a process known as oxygen enrichment [75].
Oxygen enrichment is now used in dormitories, mines, luxury hotels, ski hotels, rooms where telescope operators live in Chile (5000 meters above sea level), trains from China to Lhasa in Tibet (> 5000 meters) [76]. Oxygen is obtained from air with synthetic zeolite which preferably absorbs nitrogen. For each increase of 1% of O2 the "physiological" altitude is reduced by 300 meters [75]. A worker living at an altitude of 5000 meters breathing 28% of O2 would reduce his "physiological" altitude to 3200 meters (it is as if he lived at that altitude), an altitude physiologically more tolerable than 5000 meters. Concentrations greater than 30% are not used [77]. "Oxygen conditioning" (to emphasize its similarity with air-conditioning) can be implemented in buildings at great heights. Already the benefit of air conditioning has been more than proven in hot climates and oxygen-conditioning could have beneficial effects on human production at great heights. The costs require studies but the principle is the same of both processes: to compress the gas. In air conditioning is the refrigerant gas in chiller, in oxygen-conditioning is zeolite to absorb nitrogen from atmospheric air [77]. Possible more general applications may be schools because the learning is compromised at high altitudes [78], hospitals (since, for example, wound healing is compromised at great heights and neonatal mortality is higher [79-81], boardrooms, courts, banks and embassies [62].
LTOT has potential consequences. In addition to emphasizing hypercapnia, for which a strategy is already defined in the international guidelines [31], chronic oxygen therapy can also accelerate oxidative injury. Hyperoxia can produce cellular injury through the production of ROS, which results in inflammation and cell death [82]. Recent studies have shown that supplemental oxygen increases the exhaled biomarkers of oxidative stress and airway inflammation [83]. Interestingly, the group of researchers in the National Emphysema Treatment Trial (NEET) found that oxygen use was an independent risk factor for death compared to those who did not use oxygen (hazard ratio, 1.36, 95% confidence interval, 1.02-2.10) in those who were randomized to NETT's medical arm [84]. To what extent oxygen contributed to early mortality or was simply an epimarker of a sicker group of patients is difficult to discern from the data. In light of these findings, the progressive growth of literature involving oxidative injury in the pathogenesis of COPD makes the potential toxicity of LTOT difficult to ignore and underscores the importance of identifying the optimal candidates for this therapy.
On the other hand, the failure of current bronchodilators and steroid therapy to attenuate the natural evolution of the disease and the progressive deterioration of COPD patients indicate the need to develop new and powerful drugs with innovative effects [85]. In this context, research on different antioxidant strategies that try to block oxidative stress in this entity and that is responsible for the perpetuation and autonomy of the inflammatory process is also increasing [86-90].
Molecular oxygen is vital for energy that is essential for life. No other molecular factor has had a singular influence on the development and progress of animal life as oxygen. Humans have evolved complex circulatory, respiratory, and neuroendocrine systems to ensure that oxygen levels are precisely maintained, since excess or deficiency may result in death of cells, tissue, or the organism. For these reasons the oxygen candidates must be suitably selected.
Oxygen therapy offers significant short- and long term benefits. Immediate benefits include relief of hypoxemia and its sequelae, improvement in exercise capacity, reduction of dyspnea and possibly sleep consolidation. Long-term it has shown to improve survival in patients with severe hypoxemia. Patients with a resting PaO2 < 55 mm Hg (< 7.5 Kp, SaO2 < 88%) and those with PaO2 between 55-59 mm Hg (7.5-8 Kp) with signs of tissue hypoxemia (PAH, CHF, polycythemia) are eligible for LTOT. Other indications are very controversial or do not have frank evidence supporting them such as NOT, oxygen during exercise and oxygen at the end of life. Inadequate use or inadequate doses of oxygen have the risk, in addition to hypercapnia, of oxidative injury that would increase the oxidative stress prevalent in LTOT candidate diseases.
Research into chronic respiratory diseases seeks new and powerful drugs that impact sub-cellular compartments and metabolic pathways to provide more effective, early and safe therapy in these entities, since conventional therapy fails to reduce natural and progressive deterioration of many patients.
The knowledge of the molecular mechanisms of action of oxygen has allowed to define other non-conventional applications.
NO
NO
This work was carried out only by author. Author AA contributed in the planning, data collection, data analysis, writing and critical review. The author read and approved the final manuscript.
- Herman JR, Newman PA, McPeters R, Krueger AJ, Bhartia PK, et al. (1995) Meter 3/total ozone mapping spectrometer observation of the 1993 ozone hole. J Geophys Res 100: 2973-2983.
- Alvarado A (1998) El Enos, el efecto invernadero, la capa de ozono y los inhaladores. Una revision, Rev Med Costa Rica y Centroamérica 544: 125-133.
- Hamilton J (1995) They Made Our World. (1st Edn) BBC World Service and Broadside Books Ltd, London.
- West JB (2000) Respiratory Physiology: the essentials. (6th Edn) Lippincott Williams & Wilkins, USA.
- Semenza GL, Wang GL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12: 5447-5454.
- Semenza GL (2007) Life with oxygen. Science 318: 62-64. [Crossref]
- Kaelin WG (2007) Von Hippel-Lindau disease. Annu Rev Pathol 2: 145-173. [Crossref]
- Coleman ML, Ratcliffe PJ (2009) Signalling cross talk of the HIF system: involvement of the FIH protein. Curr Pharm Des 15: 3904-3907. [Crossref]
- Patel SA, Simon MC (2008) Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ 15: 628-634. [Crossref]
- Semenza GL (2010) Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med 2: 336-361. [Crossref]
- Taylor CT (2008) Interdependent roles for hypoxia inducible factor and nuclear factor-kappa B in hypoxic inflammation. J Physiol 586: 4055-4059.
2021 Copyright OAT. All rights reserv
- Eltzschig HK, Carmeliet P (2011) Hypoxia and inflammation. N Engl J Med 364: 656-665. [Crossref]
- Alvarado A, Arce I (2016) Immune recognition in lung diseases: basic research and clinical application. Clin Infect Immun 1: 31-40.
- Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721-732. [Crossref]
- De Bock K, De Smet F, Leite De Oliveira R, Anthonis K, Carmeliet P (2009) Endothelial oxygen sensor regulate tumor vessel abnormalization by instructing phalanx endothelial cells. J Mol Med 87: 561-569.
- Semenza GL (2011) Oxygen sensing, homeostasis, and disease. N Engl J Med 365: 537-547. [Crossref]
- Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, et al. (2006) An abnormal mitochondrial-hypoxia inducible factor-1a-Kv channel pathway disrupt oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630-2641.
- Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, et al. (2010) Hypoxia-inducible factor-1a regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130-1138.
- Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, et al. (2010) Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and new therapeutic target. Circulation 121: 2661-2671.
- Kim V, Benditt JO, Wise RA, Sharafkhaneh A (2008) Oxygen therapy in chronic obstructive pulmonary disease. Proc Am Thorac Soc 5: 513-518. [Crossref]
- Hallwell B (1996) The radical view: free radicals, health and disease. Odyssey 2: 10-15.
- Bryan CL, Jenkinson SG (1988) Oxygen toxicity. Clin Chest Med 9: 141-152. [Crossref]
- Tobin MJ (1986) Update on strategies in mechanical ventilation. Hosp Pract (Off Ed) 21: 69-84. [Crossref]
- GERSCHMAN R, GILBERT DL, NYE SW, DWYER P, FENN WO (1954) Oxygen poisoning and x-irradiation: a mechanism in common. Science 119: 623-626. [Crossref]
- Hamanska RB, Chandel NS (2009) Mitochondrial reactive oxygen species regulate hypoxic signaling. Current Opin Cell Biol 21: 894-899.
- Alvarado A, Arce I (2016) Antioxidants in respiratory disease: basic science research and therapeutic alternatives. Clin Res Trials 3: 1-11.
- Swinburn CR, Mould H, Stone TN, Corris PA, Gibson GJ (1991) Symptomatic benefit of supplemental oxygen in hypoxemic patients with chronic lung disease. Am Rev Respir Dis 143: 913-915.
- Somfay A, Porszasz J, Lee SM, Casaburri R (2001) Dose-response effect of oxygen on hyperinflation and exercise endurance in nonhypoxaemic COPD patients. Eur Respir J 18: 77-84.
- O’Donnell DE, D’Arsigny C, Webb KA (2001) Effects of hyperoxia on ventilator limitation during exercise in advance chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 892-898.
- Nonoyama HL, Brooks D, Lacasse Y, Guyatt GH, Goldstein RS (2007) Oxygen therapy during exercise training in chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2: CD005372.
- Hardinge M, Annandale J, Bourne S, Cooper B, Evans A, et al. (2015) British Thoracic Society guidelines for home oxygen use in adults. Thorax 70 Suppl 1: i1-43. [Crossref]
- CMS statistics (2013) CMS publication. U.S. Department of Health & Human Services, Washington, DC.
- Nocturnal Oxygen Therapy Trial Group (1980) Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Ann Intern Med 93: 391-398.
- Medical Research Council Working Party (1981) Long-term domiciliary oxygen therapy in chronic hypoxic cor pulmonale, complicating chronic bronchitis and emphysema. Lancet 1: 681-686.
- Ekström MP, Wagner P, Ström KE (2011) Trends in cause-specific mortality in oxygen-dependent chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183: 1032-1036.
- Chaouat A, Weitzenblum E, Kessler R, Charpentier C, Ehrhart M, et al. (1999) A randomized trial of nocturnal oxygen therapy in chronic obstructive pulmonary disease patients. Eur Respir J 14: 1002-1008.
- Górecka D, Gorzelak K, Sliwinski P, Tobiasz M, Zielinski J (1997) Effect of long-term oxygen therapy on survival in patients with chronic obstructive pulmonary disease with moderate hypoxaemia. Thorax 52: 674-679.
- Long-Term Oxygen Treatment Trial Research Group, Albert RK, Au DH, Blackford AL, Casaburi R, et al. (2016) A Randomized Trial of Long-Term Oxygen for COPD with Moderate Desaturation. N Engl J Med 375: 1617-1627. [Crossref]
- Ekström M (2016) Clinical Usefulness of Long-Term Oxygen Therapy in Adults. N Engl J Med 375: 1683-1684. [Crossref]
- (GOLD) (2017) Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. Global Initiative for Chronic Obstructive Lung Disease.
- Armadi Z, Sundh J, Bornefalk-Hermansson A, Ekström M (2016) Long-term oxygen therapy 24 hs vs 15 hs/day and mortality in chronic obstructive pulmonary disease. PLoS One 11: e0163293.
- Williamson W, Fuld J, Westgate K, Sylvester K, Ekelund U, et al. (2012) Validity of reporting oxygen uptake efficiency slope from submaximal exercise using respiratory exchange ratio as secondary criterion. Pulm Med 874020.
- Krachman S, Minai OA, Scharf SM (2008) Sleep abnormalities and treatment in emphysema. Proc Am Thorac Soc 5: 536-542. [Crossref]
- Arnold E, Bruton A, Donovan-Hall M, Fenwick A, Dibb B, et al. (2011). Ambulatory oxygen: why do COPD patients not use their portable systems as prescribed? A qualitative study. BMC Pulm Med 11: 9.
- Sasayama S, Izumi T, Matsuzaki M, Matsumori A, Asanoi H, et al. (2009) Improvement of quality of life with nocturnal oxygen therapy in heart failure patients with central sleep apnea. Circ J 73: 1255-1262.
- NICE (2012) Guideline for the of diagnosis and management of the headaches in young people and adults. NICE.
- Ripamonti C, Bruera E (1997) Dyspnea: pathophysiology and assessment. J Pain Symptom Manage 13: 220-232. [Crossref]
- Collier A, Breaden K, Phillips JL, Agar M, Lister C (2017) Caregivers perspectives on the use of long-term oxygen therapy for the treatment of refractory breathlessness: a qualitative study. J Pain Symptom Manage 53: 33-39.
- Jindal SK (2017) Long term oxygen therapy-it is still relevant? J Thorac Dis 9: E266-266E268. [Crossref]
- Balkissoon R (2017) Journal Club: COPD and Oxygen Therapy. Chronic Obstr Pulm Dis 4: 71-75. [Crossref]
- Hilberink SR, Jacobs JE, van Opstal, van der Weijden T, Keegstra J, et al. (2011) Validation of smoking cessation self-reported by patients with chronic obstructive pulmonary disease. Int J Gen Med 4: 85-90.
- Goldbart J, Yohannes AM, Woolrych R, Caton S (2013) “It is not going to change his life but it has picked him up”: a qualitative study of perspectives on long term oxygen therapy for people with chronic obstructive pulmonary disease. Health Qual Life Outcomes 11: 124.
- Amani H, Lozano DD, Blome-Eberwein S (2012) Brother, have you got a light? Assessing the need for intubation in patients sustaining burn injury secondary to home oxygen therapy. J Bur Care Res 33: e280-285.
- Muñoz X, Torres F, Sampol G, Rios J, Marti S, et al. (2008) Accuracy and reliability of pulse oximetry at different arterial carbon dioxide pressure levels. Eur Respir J 32: 1053-1059.
- Aubier M, Murciano D, Milic-Emili J, Toutay E, Daghfous J, et al. (1980) Effects of the administration of O2 on ventilation and blood gas in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis 122: 747-754.
- Chiang LL, Hung TC, Ho SC, Lin HC, Yu CT, et al. (2002) Respiratory response to carbon dioxide stimulation during low flow supplemental oxygen therapy in chronic obstructive pulmonary disease. J Formos Med Assoc 101: 607-615.
- Dillard TA, Moores LK, Billelo KL, Phillips YY (1995) The preflight evaluation: a comparison of the hypoxia inhalation test with hypobaric exposure. Chest 107: 352-357.
- Ahmedzai S, Balofou-Lynn IM, Berick T, Buchdanl P, Coker RK, et al. (2011) Managing passenger with stable respiratory disease planning air travel: British Thoracic Society recommendations. Thorax 66: i1-i30.
- Cramer D, Ward S, Geddes D (1996) Assessment of oxygen supplementation during air travel. Thorax 51: 202-203. [Crossref]
- Wheaton AG, Cunningham TJ, Ford ES, Croft JB. (2015) Employment and activity limitations among adults with chronic obstructive pulmonary disease-United States. 2013. MMWR Morb Mortal Wkly Rep 64: 289-295.
- Introduction to mountain regions. In: Human health impacts from climate variability and climate change in the Hindu Kush-Himalaya Region. Geneva: World Health Organization, report of an inter-regional workshop, Mukteshwar, India pp: 2-12.
- West JB (2017) Physiological Effects of Chronic Hypoxia. N Engl J Med 376: 1965-1971. [Crossref]
- Beall CM (2007) Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc Natl Acad Sci U S A 104 Suppl 1: 8655-8660. [Crossref]
- Simonson TS, Yang Y, Huff CD, Yun H, Qin G, et al. (2010) Genetic evidence for high-altitude adaptation in Tibet. Science 329: 72-75. [Crossref]
- Yi X1, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, et al. (2010) Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329: 75-78. [Crossref]
- Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, et al. (2010). Natural selection on EPAS1 (HIF-2a) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA 107: 11459-11464.
- Bighman A, Bauchet M, Pinto D, Mao X, Akey JM, et al (2010) Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet 6: e1001116.
- Elsner RW, Bolstad A, Forno C (1964) Maximum oxygen consumption of Peruvian Indians native to high altitude. In: Weihe WH (Ed) The Physiological effects of high altitude, New York: MacMillan.
- Favier R, Spielvogel H, Desplancher D, Ferretti G, Kayser R, et al. (1995) Maximal exercise performance in chronic hypoxia and acute normoxia in high-altitude natives. J Appl Physiol 78: 1868-1874.
- Yan X, Zhang J, Gong Q, Weng X (2011) Adaptive influence of long term high altitude residence on spatial working memory: an fMRI study. Brain Cogn 77: 53-59.
- Yan X, Zhang J, Gong Q, Weng X (2011) Prolonged high-altitude residence impacts verbal working memory: an fMRI study. Exp Brain Res 208: 437-445. [Crossref]
- Yan X (2014) Cognitive impairments at high altitudes and adaptation. High Alt Med Biol 15: 141-145. [Crossref]
- Hogan AM, Virues-Ortega J, Botti AB, Bucks R, Holloway JW, et al. (2010) Development of aptitude at altitude. Dev Sci 13: 533-544. [Crossref]
- Virués-Ortega J, Buela-Casal G, Garrido E, Alcázar B (2004) Neuropsychological functioning associated with high-altitude exposure. Neuropsychol Rev 14: 197-224.
- West JB (1995) Oxygen enrichment of room air to relieve the hypoxia of high altitude. Respir Physiol 99: 225-232. [Crossref]
- West JB (2008) A new approach to very-high-altitude land travel: the train to Lhasa, Tibet. Ann Intern Med 149: 898-900. [Crossref]
- West JB (2016) Oxygen Conditioning: A New Technique for Improving Living and Working at High Altitude. Physiology (Bethesda) 31: 216-222. [Crossref]
- Rimoldi SF, Rexhaj E, Duplain H, Urben S, Billieux J, et al. (2016) Acute and Chronic Altitude-Induced Cognitive Dysfunction in Children and Adolescents. J Pediatr 169: 238-243. [Crossref]
- Sen CK (2009) Wound healing essentials: let there be oxygen. Wound Repair Regen 17: 1-18. [Crossref]
- Mazess RB (1965) Neonatal mortality and altitude in Peru. Am J Phys Anthropol 23: 209-213. [Crossref]
- West JB (2015) A strategy for reducing neonatal mortality at high altitude using oxygen conditioning. J Perinatol 35: 900-902. [Crossref]
- Kirkham PA, Barnes PJ (2013) Oxidative stress in COPD. Chest 144: 266-273. [Crossref]
- Hansel TT, Barnes PJ (2003) An atlas of chronic obstructive pulmonary disease: COPD. A resource for, teaching and lecturing (1st edn) Taylor & Francis, London.
- Martinez FJ, Foster G, Curtis JL, Criner G, Weinmann G, et al. (2006) Predictors of mortality in patients with emphysema and severe airflow obstruction. Am J Respir Crit Care Med 173: 1326-1334. [Crossref]
- Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, et al. (2008) A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 359: 1543-1554. [Crossref]
- Alvarado A, Arce I (2015) Molecular biology of chronic obstructive pulmonary disease from the bases to the therapeutic decision: a review. Br J Med Medic Res 10: 1-14.
- Cloonan SM, Choi AM (2016) Mitochondria in lung disease. J Clin Invest 126: 809-820. [Crossref]
- Chinnery PF1, Zeviani M1 (2016) Mitochondrial Matchmaking. N Engl J Med 375: 1894-1896. [Crossref]
- Kang MJ, Shadel GS. (2016). A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis 79: 207-213.
- Falk MJ, Decherney A, Kahn JP (2016) Mitochondrial Replacement Techniques--Implications for the Clinical Community. N Engl J Med 374: 1103-1106. [Crossref]