Bronchial asthma (BA) and chronic obstructive pulmonary disease (COPD) are two major health problems whose incidence is increasing. Both are recognized as major causes of morbidity and mortality worldwide. Never as now is better known the pathophysiology of both, as well as their inflammatory, cellular and immunological profiles. It has powerful diagnostic, image, gasometric, functional, cellular, and immunological tools, as well as abundant basic, clinical, genetic and therapeutic research. However, despite having international guidelines based on evidence, the responses to the treatments are sub-optimal in both. In BA the incidence of uncontrolled patients is high and in COPD, treatment with inhaled bronchodilators and glucocorticoids (IGCs) improves symptoms and quality of life but does not impact the deterioration in lung function or improve life expectancy. The problem is amplified by patients in which the two entities overlap and this creates problems of diagnosis and treatment. Therefore the research of new therapeutic tools (such as antioxidants, biological and those that impact the function of mitochondria) is vigorous. This monograph therefore aims to establish, as clearly as possible, the differences and similarities between BA and COPD to give the clinician a tool that brings him closer to a more precise diagnosis and a more rational treatment.
bronchial asthma, COPD, ACO
For a long time, it has been accepted that BA and COPD are distinct diseases that have different pathophysiological mechanisms as well as unique epidemiological features. BA is recognized as an allergic disease, usually starting in childhood and characterized by airway hyperreactivity leads to a variable airflow obstruction [1]. In contrast, COPD is usually caused by the consumption of tobacco or biomass / fuels, develops after the fourth decade of life and is characterized by a progressive and persistent airflow obstruction [2]. BA is characterized predominantly by type 2 inflammation with eosinophils and CD4 + T lymphocytes, whereas COPD is mainly characterized by neutrophilic inflammation and by CD8 + T lymphocytes and macrophages [3-5]. These features allow them to be recognized as distinct pathological entities (British hypothesis). However, in daily clinical practice, it is sometimes difficult to reach a final diagnosis, especially in older patients who have clinical features of asthma and COPD. Indeed, fixed obstruction develops over time due to airway remodeling in some patients with asthma, and some phenotypes in asthma are neutrophils or eosinophils and neutrophils combined; variable airway obstruction may occur in some patients with COPD, and respiratory viral infections in COPD may trigger eosinophilic inflammation or a type 2 immune response.
It has been recognized that the characteristics of asthma and COPD can coexist in patients with persistent airflow obstruction and asthma-like symptoms. This concept is old and new at the same time. In 1961, Orie, et al. stated that "bronchitis and asthma can be found in a patient of the same age, but as a rule there is a fluid development from bronchitis in young people to a more asthmatic picture in adults, which in turn becomes bronchitis in elderly patients "[6]. The hypothesis states that various forms of airway obstruction, such as BA, chronic bronchitis and emphysema, can be recognized as an entity called chronic nonspecific lung disease and different environmental factors drive it into different phenotypes. Many years later, Fletcher, et al. proposed the term "Dutch hypothesis" to describe the phenomenon of "asthma" becoming "COPD" [7]. After a while, there were many negative opinions about the "Dutch Hypothesis", because it was the main notion that asthma and COPD are unique conditions with different pathological mechanisms and separate treatments. However, in the 1990s, it was shown that the increase in the responsiveness of the respiratory tract is positively associated with the development of chronic respiratory symptoms, and the "Dutch hypothesis" has been recently re-evaluated [8]. In clinical practice, Gibson, et al. described "the syndrome of BA and COPD overlap" in 2009 [9]. After that, many manuscripts on the overlap of asthma-COPD were published.
In 2014, a joint project of the Global Initiative for Asthma (GINA) and Global Initiative for Chronic Obstructive Pulmonary Disease (GOLD) has coined the term "asthma-COPD overlap syndrome" (ACOS), defined as a persistent airflow limitation with several characteristics generally associated with BA and several characteristics usually associated with COPD [10]. Later, GINA / GOLD recommended the modified term “asthma-COPD overlap (ACO)", because ACOS has often been used as if it were a separate disease and because it is not at all clear that it is a separate and distinct entity. The statement provides a list of the common characteristics of BA and COPD, and a diagnosis of ACO is suggested if there are a similar number of characteristics for both BA and COPD. However, he mentioned that "a specific definition for ACO can’t be developed until more evidence is available about its clinical phenotypes and underlying mechanisms" [11]. The "Dutch hypothesis" is genetically unlikely because there is little genetic "background" between BA and COPD [12] (Figure 1).

Figure 1. BA, COPD, and ACO BA and COPD have distinct features, with different cells, mediators and consequences of inflammation as well as different response to treatment with corticosteroids. Approximately 15% of patients have features of BA and COPD (ACO)
Definitions, like the classifications of biological phenomena, are never perfect nor can they encompass the totality of the variables that make up these phenomena. This is valid for obstructive airway diseases. For example, the definition of GINA (2018) of asthma describes it as heterogeneous [13]. COPD is also heterogeneous with different phenotypes however the definition of COPD by GOLD (2018) does not consider this fact [14]. EPOC is described as a common entity. Asthma is also and is not mentioned in the definition. The definition of asthma states that it is inflammatory event. COPD is also and the definition does not mention it. Neither definitions clearly cites both have cyclic activations and critical inflammatory process ("flare-up" in asthma and AECOPD in COPD), and yet both events are part of the evolution of both diseases and objectives of treatment. In the definition of asthma, the four respiratory symptoms are mentioned, but not in COPD. Therefore, being such similar diseases, the definitions do not clearly establish the similarities and differences between the two.
Bronchial asthma is, therefore, a chronic and common inflammatory respiratory disease that affects 10% of the adult population in different countries [15). An estimated 315 million affected people worldwide and 346,000 deaths every year. It is more difficult to assess the prevalence in children under 5 years old for several reasons. It is difficult to make a reliable diagnosis of asthma in this age group because respiratory findings such as cough and bronchospasm are frequent in children without asthma, particularly in those 0-2 years of age. Some viral infections (respiratory syncytial and rhinovirus) are associated with recurrent wheezing throughout childhood. Therefore not all wheezing in this age means asthma. In addition, and routinely, it is not possible to investigate airflow limitation in this age group [13]. What is a fact is that it is the most common disease in childhood and causes a leading morbidity evidenced by school absenteeism, visits to emergency departments and hospitalizations [16]. Based on epidemiological studies, the global prevalence of COPD is estimated at 11.7%, with 384 million affected and 3 million deaths per year (6% of all deaths), becoming a leading cause of morbidity and mortality [17,18]. With the increase in smoking in developing countries and the increase in life expectancy in high-income countries, it is expected that the prevalence will increase in the next 30 years and it is likely that in the year 2030 there will be a total of annual deaths higher than 4.5 million [19]. In these evaluations there is a sub-registry, since most of the studies are carried out in smoking patients and nowadays the prevalence of COPD in non-smokers is increasing, associated with the use of biomass, other fuels and environmental pollution [20].
Allergic asthma usually starts in early childhood and at least 50% of asthmatic patients started their asthma at this stage and it starts earlier in men than in women [13]. This phenotype is usually the most recognized, however, the other asthmatic phenotypes such as non-allergic asthma, late-onset asthma, asthma with fixed airflow obstruction and asthma and obesity can begin in adult life [21,22]. In COPD, however, the prevalence is higher in smokers and ex-smokers than in non-smokers, generally in people over 40 years of age and predominantly in men. This is valid for countries in Europe, United States and Latin American countries, as evidenced by the PLATINO study carried out in 5 countries in this region [23]. Collectively these are the two non-neoplastic respiratory chronic inflammatory diseases that most affect the human species and it is likely that their real prevalence is greater than the data reported by GINA and GOLD and that in the coming years there is an increase in it, despite the fact that existence of guides that define their diagnoses and treatments.
The pattern of respiratory symptoms is one of the clinical elements that have most valuable for diagnosis. In asthma, dyspnea, chest tightness, coughing and wheezing are very common (occasionally a patient with asthma may only have cough). They worsen during the night or early morning, vary in time and intensity, can resolve spontaneously or with medication, may be absent for weeks or months and are triggered basically by exercise, allergens, irritating odors, laughter, climate changes, viral infections and stress. The key datum is variability [13]. In contrast, in COPD, dyspnea is progressive and chronic and the cough can be productive or non-productive, initially intermittent, then almost daily and mainly during the day. If the patient does not have an exacerbation, it usually does not get worse at night. There are "better days and worse days" but the symptoms are invariably present to a greater or lesser degree, and by far the most frequent triggers are weather changes and viral or bacterial infections. The key fact is persistence [14].
The demonstration of airflow variability in asthma and its persistence in COPD through pulmonary function tests are considered basic in the diagnostic work of both. However, there are practical realities and technical aspects that are not easy to solve that prevent a generalized application of this tool. The first is the availability of equipment. Doctors who work in developing countries know that few patients have access to them, particularly in primary care where they originally consulted. Neither GINA nor GOLD describe in their guidelines how many patients diagnosed worldwide of both diseases have access to functional documentation, much less patients with COPD not associated with smoking. And when the test is available there are technical aspects to be solved; for example, the principle of clinical decision should be applied. This means estimating the post-test probability after taking into account the pre-test probability, evaluating the quality of the results, assessing the inconvenience of false positives or false negatives. However, the main drawback is that the interpretation of the results is usually done without clinical information. Like any other medical exam, the results should be interpreted in the clinical context and not the other way around.
An obstructive defect is a disproportionate reduction of the maximum air flow in relation to the maximum volume (vital capacity = VC), which can be displaced from the lung (the VC is slower for exhalation than the FVC [forced vital capacity] that obviously is to be forced is faster). This implies an obstruction of the airway during exhalation. The concept is clear, but the limits of normality are not. GOLD recommends FEV1 / FVC <0.70 post-bronchodilator (PB) as the spirometric criterion of COPD, which could lead to functional over-diagnosis of COPD in patients older than 60 years and without exposure to noxious particles or gases, and also the opposite; sub-diagnosis in younger patients [14)] Pellegrino insists that VC should be used instead of FVC and, if FEV1 / FVC PB is used, do not use the lower level of 0.70 but the reduction below the 5th percentile of the predicted value. This criterion does not lead to overestimation of the obstructive defect in elderly patients [24]. In a recent review of GOLD 2018, the authors suggest that the LLN (lower limit of normal) of FEV1 should be used to define obstruction as recommended by ATS and ERS and not by FEV1 / FVC PB [25]. Another controversy is that this last edition defines that for the diagnosis of COPD, it is necessary to document the limitation to airflow, but it excludes the functional severity of the obstruction of the classification of the groups (ABCD) and of the therapeutic guide [25].
The bronchial response to bronchodilator medication is an integrated physiological response that involves the epithelium of the airway, nerves, mediators and smooth muscle of the pathway and the relationship between bronchoconstriction and bronchodilator response is imperfect, which means that it can´t be infer one from the other. The response is usually reported as the percentage increase from the baseline spirometric value or the absolute improvement in milliliters (cc). An 8% or <150 cc of improvement are values that are within the normal variability and therefore are not statistically significant. It is accepted that an improvement >12% or >200 is a positive bronchodilator response [24]. Classically, it is accepted that the asthmatic patient will have this type of response, but not in COPD, that is, in asthma there is this bronchial hyperreactivity while in COPD the obstruction is not reversible in the test. However, facing a given patient the response to bronchodilator, by itself, is not able to discriminate the two types of obstruction. Again, the result must be interpreted in the clinical context. An asthmatic already remodeled and with fixed obstruction will not have hyperreactivity as a severely obstructed patient in an asthma attack and this does not invalidate the diagnosis of asthma and also a patient with the eosinophilic phenotype of COPD may have bronchial hyperreactivity [13,14]. In addition, these tests are performed in a very short time but do not predict an antegrade clinical response. The absence of response in the pulmonary function laboratory does not exclude the patient from a trial of 1-8 weeks with bronchodilators to see clinical results. Small-airways tests (<2mm in diameter) have very little value, since they are very nonspecific and variable because they depend on the behavior of FVC [24]. The diffusion capacity of CO (DLCO) is decreased in emphysema and increased in asthma. Of course, it is not necessary to measure volumes and capacities to identify an obstructive defect.
BA and COPD are characterized by chronic inflammation of the respiratory tract although the nature of the inflammation and localization are different [26]. In both there are two groups of cells that are activated. One group is inflammatory cells recruited from peripheral blood to the lungs by chemotactic factors released locally, and the other group is airway and lung structural cells such as epithelial cells, endothelial cells, fibroblasts and smooth muscle cells, which also release inflammatory mediators and actively participate in the inflammatory process. In both, BA and COPD, the inflammatory response involves innate immunity (eosinophils, neutrophils, macrophages, mast cells, killer natural cells, γð-T cells, innate lymphoid cells [ILCs] and dendritic cells [DC]) and adaptive immunity (B and T lymphocytes).
In BA the inflammation is located from trachea to peripheral airways. In COPD the inflammatory process is located in peripheral pathways and parenchyma and is associated with systemic inflammation. Bronchial obstruction in BA occurs due to smooth muscle contraction, vascular congestion (edema), remodeling of the airway (structural changes and fibrosis) and impaction of mucus (fatal asthma). This inflammation also leads to bronchial hyperresponsiveness, a physiological abnormality of asthma that is characterized by variable symptoms including nocturnal worsening. In COPD, the predominantly peripheral obstruction, due to fibrosis and collapse due to loss of pulmonary elasticity (emphysema), leads to gas trapping, which is an irreversible mechanism. However, there is an added cholinergic contraction ("cholinergic tone") that is reversible [26].
The predominant inflammatory pattern in asthma is the type 2 pattern. It is a type of immunity that provides protection against parasitic infections but is also active in allergic diseases such as asthma. It is orchestrated by the CD4 + T helper 2 (Th2) cells and the innate lymphoid cells group 2 (ILC2s) through the secretion of IL-4. IL-5, IL-9 and IL-13, which are regulated by the transcription factor GATA3. These interleukins will lead to airway infiltration with eosinophils, mast cells, B cells (which produce IgE in response to allergens) and alternatively activated macrophages, which will produce obstruction and symptoms. In this type of immunity the epithelial cells, in response to allergens, will release cytokines such as alarmins (IL-25, IL-33 and TSLP [thymic stromal lymphopoietin]) which program the immature DCs of the respiratory tract to process the allergens and migrate to the local lymph nodes, present the peptides resulting from the processing to the naive or uncommitted T lymphocytes and generate Th2. (Figure 2). Patients with BA who do not have this pattern qualify have non-T2 asthma (more severe BA). Molecular mechanisms (endotypes) do not necessarily correlate with clinical types (phenotypes) [27]. DCs are specialized cells, similar to macrophages, that are in the epithelium of the airway and are the major line of antigen presenting cells and mature when presenting the antigens to the T system [28,29].
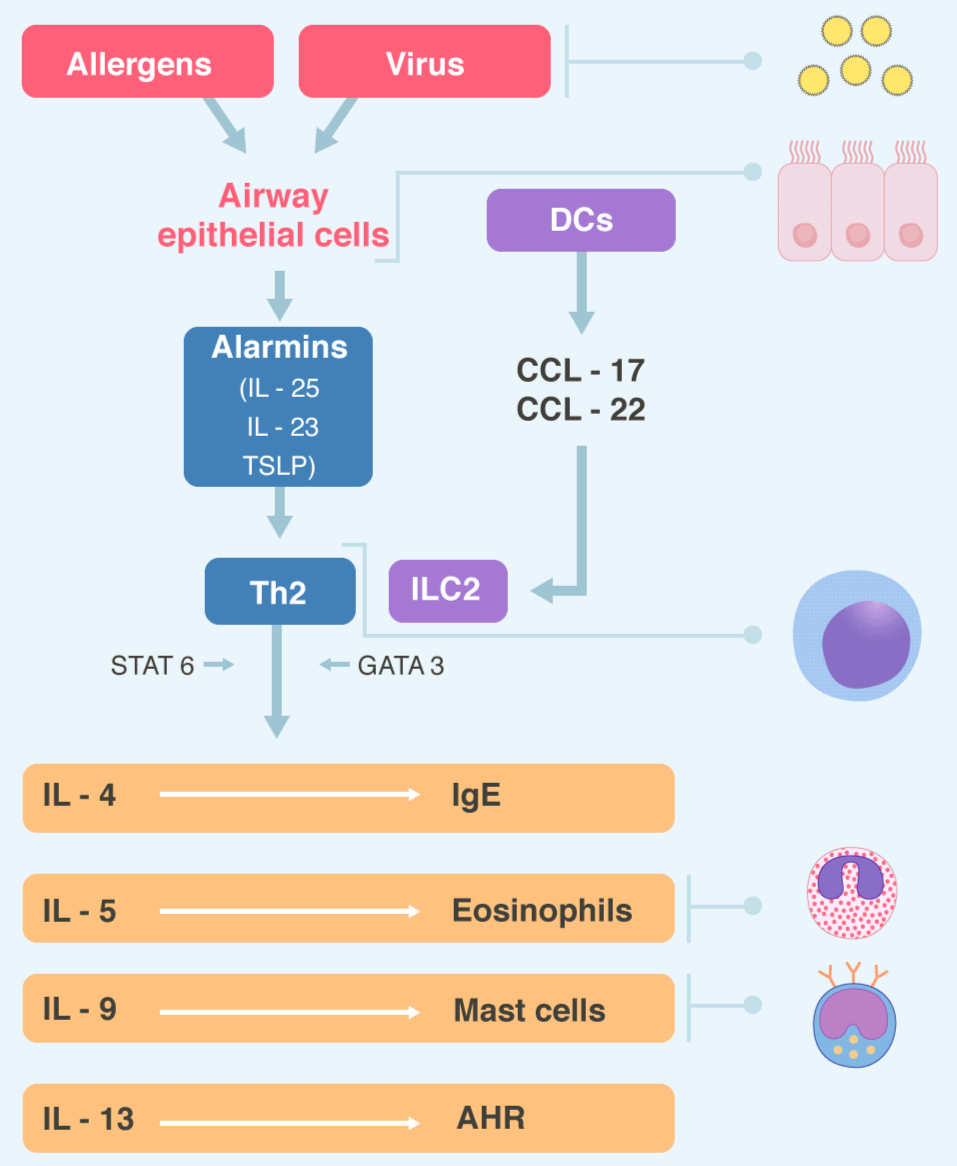
Figure 2. Type 2 immunity Virus and inhaled allergens activated lung dendritic cells (DCs) to release the CC-chemokines ligand 17 (CCL17) and CCL22 which recruit T helper 2 (Th2) cells and group innate lymphoid cells (ILC2s). Activation of the transcription factor GATA3 and STAT6 in Th2 cells and ILC2s leads to secret of the cytokines IL-4, IL-5, IL-9, IL-13. These cytokines regulated IgE synthesis, eosinophilic proliferation, mast cells proliferation, and airway hyperreactivity (AHR) respectively. Epithelial cells also producing alarmins, including thymic stromal lymphopoietin (TSLP), Il-25 and IL-33, which are “upstream” cytokines in the eosinophilic response. STAT= Signal transduction and activator of transcription 6
The mast cells are key effector cells in BA. Not so in classic COPD. They are considered, being near the smooth muscle in the airway and releasing multiple bronchoconstrictive mediators, the probable responsible for bronchial hyperreactivity. They do not seem to play a significant role in COPD, which may explain the absence of variable airway obstruction [30]. Eosinophilic inflammation is the hallmark of asthma [31]. They come from blood and bone marrow attracted by CCL11 (eotaxin), CCL5 (RANTES), and IL-5 (secreted by Th2 and ILC2s). Role in COPD is less clear. When present in COPD they predict a better response to bronchodilators and steroids and may indicate co-occurrence of asthma, ACO or a particular eosinophilic phenotype of COPD. This phenotype is characterized by peripheral eosinophilia greater than 2%, which is equivalent to approximately 150-200 eosinophils per cubic millimeter. It seems that this level of eosinophilia is associated with an increased risk of exacerbations [32].
ILCs are immune cells that do not express B or T cell receptors for specific antigens. Three main subtypes are recognized depending on the expressed transcription factors and the pattern of cytokines released. GATA3 is a transcription factor expressed predominantly by Th2 cells and ILCs group 2 which is important for its differentiation and secretion of T2 cytokines (IL-4, IL-5, IL-9, IL-13). These cytokines regulate IgE synthesis, eosinophil proliferation, mast cell proliferation and bronchial hyperreactivity respectively. Th2 is a cellular subset of CD4 + T helper with an important role in humoral immunity and allergic responses [27].
The predominant inflammatory pattern in COPD is that of type 1 immunity and that of type 3 immunity, with predominance of macrophages and neutrophils and an increased number of cytotoxic T cells (Tc1) CD8 +, CD4 + Th1, Th17, ILC3s and B cells, which are organized with T cells in the lymphoid follicles in peripheral airways [33,34]. Type 1 immunity provides protection against microbial infection and is orchestrated by Th1 cells, cytotoxic T cells, ILCs group 1 and the transcription factor T-bet, which regulates the secretion of IFNγ. Type 1 immunity is associated with increased activation of pro-inflammatory macrophages (Figure 3). Type 3 immunity is a type of immunity directed against microorganisms, in particular fungi, and orchestrated by Th17 and ILCs group 3, which express RORγt and secrete IL-17 and IL-22 leading to neutrophilic inflammation (Figure 4). This inflammation is an amplification of the mucosal inflammatory response that occurs in response to inhaled irritants (cigarette smoke or biomass), but which persists even when exposure ceases. Patients with non-T2 asthma may have this neutrophilic pattern of inflammation, with less reversibility, a progressive decline in lung function and a poor response to corticosteroids [35].
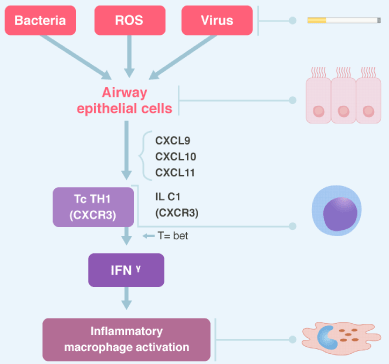
Figure 3. Type 1 immunity Bacteria, virus and reactive oxygen species (ROS) activate airway epithelial cells to release CXC-chemokine ligands (CXCL9, CXCL10, CXCL11) and ILC1s through CXCR3. Th1, Tc, ILC1 and transcription factor T bet regulate the secretion of INFγ. Type 1 immunity is associated with increased pro-inflammatory macrophage activation. CXCR3= CXC chemokine receptor 3.
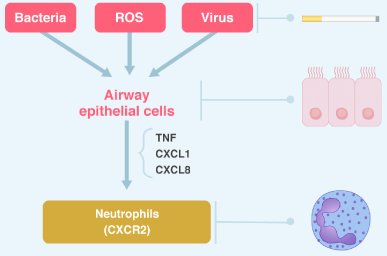
Figure 4. Type 3 immunity Bacteria, virus and ROS activate airway epithelial cells to release pro-inflammatory mediators such as tumor necrosis factor /TNF) and the chemokines CXC-chemokine ligand 1 (CXCL1) and CXCL8, which attract neutrophils by binding to CXC-chemokine receptor 2 (CXCR2). Th17 cells and ILC3s (it is not shown in the figure) through IL-17 also well recruit neutrophils. See text for discussion.
In the sputum of patients with ACO there are neutrophils and eosinophils, and there is an urgent need to investigate to what extent it is the result of a change in the pathogenesis of bronchial asthma towards COPD over the time, or the entity has a different molecular biology and immunopathogenesis. This may have diagnostic and therapeutic implications [35]. For example, anti-eosinophilic treatment may be of therapeutic value in eosinophilic asthma and COPD associated with eosinophils.
The severity of BA is evaluated retrospectively from the level of treatment required to control the symptoms and exacerbations (36). The severity is not static and could vary in months or years. This allows the step down treatment with the idea of finding the minimum effective level of treatment. So, mild asthma is a well-controlled asthma with step 1 or 2 of treatment. This is with a PRN reliever alone, or with low intensity controller treatment (low doses of IGCs, chromones or leukotriene receptor antagonists). Moderate asthma is asthma that is well controlled with step 3 of treatment, this is low doses of IGCs / LABA and severe asthma which requires step 4 or 5 of treatment (high doses of IGCs / LABA) to prevent it from becoming "out of control" or that remains "uncontrolled" despite treatment. The term severe asthma should be reserved for patients with asthma refractory to treatment and for those patients in whom the response to the treatment of co-morbidities is incomplete [36]. Note that the classification is retrospective and does not take into account the severity of functional obstruction as measured by pulmonary function tests.
In COPD, the classification and therapeutic guidance groups (ABCD) are based on dyspnea and exacerbations. The concept of severity is used for the spirometric classification of airflow obstruction (the term "stage" is abandoned), which is not taken into account in the construction of the groups as well as in asthma, despite recognizing the need for spirometric diagnosis in its definition. Contrary to asthma, the group is defined by dyspnea and AECOPD and on that basis the treatment is decided. The coding of a group with its airflow limitation is impractical for most readers who do not handle the GOLD language. As noted by Mirza, et al. It is easier to describe an individual patient as "severe obstruction with severe symptoms and frequent exacerbations" than to code it as "GOLD 3D stadium". There is little benefit of encryption and it only serves to confuse. Although the current review (2017-2018) offers a more centralized approach in the patient to guide pharmacotherapy, the pictorial representation is somewhat overwhelming (25). Recent data show that repeatedly changing the classification does not predict better respiratory mortality and all causes than the previous classifications from 2007 to 2011 [37-39].
Several comorbidities are commonly present in BA, particularly in patients with severe or difficult to treat asthma. The active management of them is recommended because they contribute to the symptomatic burden, alters the quality of life, lead to drug interaction and poor control of asthma [40]. Allergic rhinitis affects up to 40% of the world population [41]. 40% of patients with allergic rhinitis have BA and 80-90% of asthmatics have allergic rhinitis. Patients with allergic rhinitis are 3 times more likely to develop BA than those who do not have it and 40-60% of patients with BA have radiological data of chronic rhino-sinusitis [42]. Chronic rhino-sinusitis can occur with or without nasal polyposis. If it occurs with nasal polyposis, it is associated with more severe BA [43]. Up to 30% of children and 2-10% of adults are impacted by allergic dermatitis and this entity is associated with BA, rhinitis, conjunctivitis, eosinophilic esophagitis, systemic immune activation and food allergy [44]. Rarely food allergy is a trigger for asthma symptoms, but in patients with food-induced anaphylaxis, the co-existence of BA is a strong risk factor for more severe and even fatal reactions. Peanuts and nuts are the most commonly responsible foods [45]. Obviously these associations have a strong allergic and / immunological basis, reflecting type 2 immune inflammation. Quantification of peripheral eosinophils, sputum and nasal exudate and specific and non-specific IgE attempts to document this diathesis, but markers of systemic inflammatory disease are not used because in BA there does not seem to be such activation.
Obesity (there is a phenotype of asthma and obesity), gastroesophageal reflux, anxiety and depression are non-allergic comorbidities associated with asthma. All, to a greater or lesser degree, threaten an adequate control of BA [13].
In contrast, in COPD, many of the comorbidities have an inflammatory base. Several are linked by the same risk factors: smoking, age and inactivity. There are two different ways to visualize the relationship. EPOC generates a "spill-over" of inflammatory and reparative events that occur in the respiratory system generating comorbidities, but with the lung as the center of the process, or COPD is another form of expression of a systemic inflammatory state that involves multiple organs [46)] (Figure 5). These comorbidities can have an impact on the course of COPD. In general, the presence of comorbidities should not alter the treatment of COPD and comorbidities should be treated in a standard manner, regardless of the presence of COPD. Cancer is frequently found in COPD and is the main cause of death [14]. Inflammation and increased oxidative stress in COPD may enhance the growth and metastases of lung cancer. In addition, increased expression of epidermal growth factor receptors (EGFR) may accelerate cancer growth [46]. Five separate entities can be found within the cardiovascular diseases associated with COPD: heart failure, ischemic heart disease, arrhythmias, peripheral vascular disease and arterial hypertension. Osteoporosis, anxiety and depression, gastroesophageal reflux, bronchiectasis and obstructive sleep apnea also impact COPD.
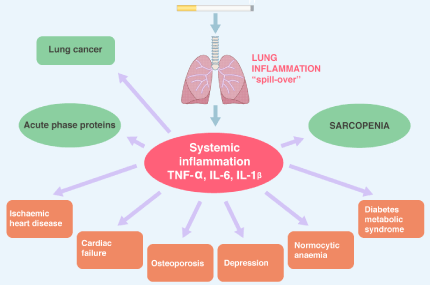
Figure 5. COPD morbidities Lung inflammation causes an “spill-over” of cytokines into the systemic circulation, which may increase acute phase proteins. Systemic inflammation may then lead to skeletal muscle atrophy and cachexia and may initiate and worsen comorbidities conditions. Systemic inflammation may also accelerate lung cancer.
Less well-known are diabetes mellitus and metabolic syndrome. While asthmatic patients do not have an increased risk of diabetes, patients with COPD, even minimal, have an increased risk (relative risk of 1.5-1.8) [47]. It does not seem to be related to the use of inhaled steroids, since patients with COPD who do not use them also develop diabetes and if it seems to be related to the systemic inflammatory process since IL-6 and TNF-α induce insulin resistance by blocking signals in the insulin receptor, increasing the risk of diabetes mellitus type 2. The same profile of systemic inflammatory cytokines is found in metabolic syndrome. Normocytic-normochromic anemia is associated with COPD and is due to the fact that systemic inflammation produces resistance to erythropoietin. Anemia does not respond to oral iron or exogenous erythropoietin. It is treated with blood transfusions. In severe disease it appears between 15-30% while polycythemia is rare (6%) [48]. IL-6, IL-1β, IL-8 and TNF-α are chronically elevated in the systemic circulation in all comorbidities and can increase acute phase reactive proteins (CRP, serum amyloid A, surfactant protein D and fibrinogen). Systemic inflammation leads to slow but substantial sarcopenia (loss of muscle mass), with skeletal muscle atrophy and cachexia [46].
A lot has been written about ACO. Some concepts are basic. First of all, it should be clear that it is not a single entity, but that, like asthma and COPD, it includes several different forms of disease (phenotypes) caused by different underlying mechanisms. The evidence of treatment is very limited due to the absence of pharmacotherapy studies in this population. The term is coined basically for two reasons: the difficulty of distinguishing asthma from COPD, particularly in older adults and smokers, especially for the primary care physician where these patients initially consulted, and due to the fact that it is clear that patients who have concomitant asthma and COPD changes have more frequent exacerbations, poor quality of life, a more rapid decline in lung function and higher mortality, in addition to consuming greater health resources than asthma and COPD alone [49-52]. The wide range in the prevalence of the epidemiological reports of ACO (15-55%) exposes the different criteria for diagnosis, and this is due to the absence of a clear definition.
Unlike asthma and COPD, ACO does not have a definition but a description: "persistent airflow obstruction with several features usually associated with asthma and several features usually associated with COPD." The patient must have a persistent airflow limitation. In addition to documenting the persistence of the obstruction, the diagnosis involves making a very good clinical history based on which 80% of the diagnosis of asthma, COPD or ACO rests. It is vital to take into account especially the findings that easily distinguish COPD asthma in clinical practice [53]. If several changes (three or more) strongly suggest COPD or if several changes (three or more) suggest asthma, in the absence of an alternative diagnosis; the diagnosis of classic COPD or classical asthma can be made. When the patient has a similar number of changes compatible with both asthma and COPD, the diagnosis of ACO should be considered and it is therefore essential to document the persistent airflow obstruction. GINA has in its document well-structured characteristics to be contrasted (Box 5-2a and Box 5-2b) [53].
Regarding the functional study, it is important to insist that it is a complement to the clinical diagnosis and not the opposite. In addition, as in any respiratory pathology, spirometric evolution has more value than a single assessment. Some data are practical help. A FEV1 / FVC pre or PB normal is not compatible with ACO, while a FEV1 / FVC <0.7 is usually present. An FEV1 >80% PB could be present in mild ACO. An FEV1 <80% is an indicator of the severity of airflow limitation and is a predictor of mortality and exacerbations. An improvement in FEV1 PB >12% or > 200 ml (a reversible obstruction) could occur in ACO if the initial FEV1 is low. An improvement in FEV1 PB >12% and >400 ml (exquisite reversibility) is compatible with ACO (if BA can be ruled out consistently) since it is really unusual in COPD. Among the symptomatic periods, patients with ACO will show a persistent limitation (non-variability) to airflow (FEV1/FVC <0.7), and when they are symptomatic, there may be reversibility in the response to bronchodilators (improvement in FEV1). If the primary care physician suspects ACO, BA or COPD, should treat to document the functional pattern.
Another criterion that has been recently described to define ACO is the use of biomarkers. Shirai and colleagues have proposed that the combined evaluation of serum periostin and YKL-40 may have value in identifying ACO [54]. Periostin is a matricellular protein downstream of IL-4 and IL-13 signals. It is highly expressed in several allergic diseases including bronchial asthma, particularly type 2 [55]. YKL-40 is a member of the chitinase family, although it lacks enzymatic activity [56]. YKL-40 is upregulated in patients with COPD, rather than in patients with asthma [57]. Shirai and colleagues have shown that patients with ACO have both biomarkers elevated, which is consistent with the concept that the inflammation type 2 (classic asthma) and neutrophilic inflammation (classic COPD) are involved in the pathogenesis of ACO). Although the finding is very interesting, and throws information on the pathogenesis of ACO, ACO is a heterogeneous phenomenon, as well as asthma and COPD, and there will be patients with "atypical" ACO who will not elevate the 2 biomarkers concomitantly, so the evaluation combined is not enough for an accurate diagnosis of ACO. It is obvious that future investigations of other biomarkers are required for diagnosis, classification, and therapeutic decisions more appropriate in these patients and also to evaluate the practical value in the clinic of their use [11].
Leonard Bernstein (1928-1990) started his asthma and allergic rhinitis at the age of 2 years. The books of famous asthmatics (eg., Wilhelm Grimm and Juan Calvino) note that he could not join the army and that when he conducted the concerts, his gasps and sneezes were evident to the public. A heavy smoker, he never gave up smoking, developing emphysema, a lung tumor and heart failure. It is likely that Leonard would have had ACO.
Exacerbations in asthma represent an acute and sub-acute worsening in the symptomatology and pulmonary function of the patient's usual state. GINA recommends the term "flare-up" instead of crisis, attacks, episodes or severe acute asthma for unclear reasons. FEV1 or PEF (peak expiratory flow) is not always available to document deterioration, but in exacerbations, the increase in the frequency of symptoms is a more sensitive measure for the onset of exacerbation than PEF [13]. AECOPD is defined as a worsening of respiratory symptoms that requires additional therapy. Note that the definition does not incorporate, ex officio, a functional assessment [14].
In asthma the crisis is classified as mild to moderate if the patient can speak in sentences, does not shake sitting or lying down, has increased respiratory rate without the use of accessory muscles, the heart rate is between 100-120 per minute, has a saturation of oxygen between 90-96% and PEF >50% of the predicted. These patients can be treated in primary care. If the crisis is severe he will speak with words but not with phrases, he will be sitting, agitated, with respiratory frequency more than 30 per minute, use of accessory muscles, desaturated (<90%) and with a PEF <50% or can’t perform it . Asthma that compromises life involves patient who is drowsy, confused or has a silent chest. The last two groups need treatment in an acute care unit and eventually an intensive care unit. Note that the classification depends on the initial clinical presentation, oxygenation and pulmonary function and not necessarily the response to treatment. In contrast, the classification of the AECOPDs does not incorporate oxygenation or lung function at the beginning, and if, the response to treatment. Thus, it will be mild if it is resolved with installing or increasing bronchodilators, moderate if it is resolved with bronchodilators plus antibiotics and / or oral steroids and it will be severe if it requires attention in an emergency service, hospitalization or is associated with acute respiratory failure.
The therapeutic options of both are similar, and what varies is the posology pause, the use or not of antibiotics, the use of magnesium sulfate and mechanical ventilation. GINA states that the drug of choice in cases of mild to moderate crisis is salbutamol (SABA). The dose is 4-10 puffs every 20 minutes the first hour and then 4-10 puffs every 3-4 hours to a maximum of 6-10 every 1-2 hours. (the puffs has 100 micrograms = 100μg) (13). Most cost-effective delivery route is pMDI with spacer [58]. If the crisis is severe or life-threatening, the evidence of cost-effectiveness of pMDI is less robust and conflicting with respect to nebulizations. Recent work found reduced hospitalizations, and better pulmonary function with continuous nebulizations, compared with intermittent, in patients with poor lung function, which is what patients have in this degree of crisis [59]. A reasonable approximation, in this scenario, is after the first hour of continuous nebulizations, continue with intermittent nebulizations every 4-6 hours with salbutamol solution to nebulize. 2.5-5 milligrams (0.5-1 cc) are used in each nebulization diluted with 3.5-3 cc of saline. If the patient is older than 50 years, 0.5 cc and if less than 50 years old, 1.0 cc of SABA. Beta-2 intravenous agonists are not used. Adrenaline (epinephrine in USA) I.M. (0.5-1 milligram) can be added to standard therapy for acute asthma associated with anaphylaxis and angioedema. It is not indicated for other asthmatic exacerbations. It should not be used in AECOPD.
For the patient who gets worse in primary care and transfers to a hospital, and for adults and children who report to an emergency department with a severe or life-threatening crisis, a cycle of ipatropium bromide (SAMA) can be added. This tool is associated with great improvement in FEV1 and PEF and with a reduction in hospitalizations compared with SABA alone [60,61]. GINA states that ipatropium should be used basically in severe crises (13). If used in an inhaler, 4 puffs (80 μg) should be given every 20 minutes, during the first hour (together with the inhaled SABA) and then 2 puffs (40 μg) every 4 hours (together with the inhaled SABA). The more severe the crisis, the nebulization may be more effective and it is recommended to mix salbutamol (solution for nebulization 0.5-1 cc) with 2 cc of nebulization solution ipatropium (500 μg) with the same dosage regimen: three continuous nebulizations the first hour and then one nebulization every 4 hours. From 1-17 years the dose of ipatropium is reduced by half (1cc = 250 μg). The solution prepared to nebulize the mixture of salbutamol with ipatropium (Combiventr in which the vial to nebulize has 2.5 cc of volume with 2.5 milligrams of salbutamol and 0.5 milligrams of ipatropium) is available in the market, making the scheme very easy to use as each ampoule is diluted with 1.5 cc of saline and a vial is nebulized every 20 minutes the first hour (3 ampoules) and then one every 4 hours. One drop of the mixture has 10 μg of ipatropium and 50 μg of salbutamol. Since the dose per kilogram of weight in children is 30 μg of ipatropium and 150 μg of salbutamol, it is very easy to dose the mixture: 3 drops per kg of weight diluted with saline to a total volume of 3-4 cc, until the 17 kg. The whole vial is used with a higher weight [62].
In AECOPD the scheme is very similar, varying only doses. As in asthma, the use of SAMA is elective. The use of SABA is the initial bronchodilator treatment [63]. There are no significant differences between using pMDI (with or without a spacer) or nebulization to deliver the drug. However, in patients with moderate to severe AECOPD and adults, nebulization may be the most appropriate method for delivery. If pMDI is used you should receive 2-3 puffs every 20 minutes in the first hour (and not 4-10 as in asthma) and then one every 3-4 hours (and not 4-10 as in asthma). If the nebulized route is used, 0.5 cc of SABA (2.5 milligrams of salbutamol) is used with or without 1cc (250 μg) of ipatropium, plus 2.5 cc of saline solution number three in the first hour and then one every 4 hours, according to the answer. If the mixture is available, one ampoule is diluted with 1.5 c of saline and one ampoule is used by nebulization [64]. In patients with hypertrophic cardiomyopathy, tachyarrhythmias, myocardial ischemia, and / or recent infarction, SAMA is the choice [14].
Oxygen is a vital part of the treatment. In asthma, oxygen saturation should preferably be monitored by pulse oximetry. Arterial gas is not routinely required. Its use should be considered if the initial PEF or FEV1 is <50%, when there is no satisfactory response to the initial treatment, or if the patient deteriorates. A saturation <90% indicates the need for aggressive therapy. Regardless of the severity of the crisis, sufficient oxygen must be given to maintain saturation between 93-95% in adults and 94-98% in children, with nasal cannula or oxygen mask. Weaning of oxygen should be done once the crisis is resolved and guided by pulse oximetry [65]. In COPD, 80% of AECOPD is managed externally, with bronchodilators, corticosteroids and antibiotics [66]. In the patient in emergencies or hospitalized a basal arterial gas should be made. Oxygen should be given by Venturi mask with FiO2 of 28-35% if PaCO2 is not increased, 25-30% of FIO2 with Venturi mask is indicated if PCO2 is between 50-60 mm Hg (acute respiratory insufficiency that does not compromise life) and FIO2 will be prescribed equal to o or >40% with severe hypoxemia and associated hypercapnia (>60 mm Hg and / or acidosis [pH <7.25]) in a patient with tachypnea >30 per minute, altered mental status and use of accessory muscles (acute respiratory failure that compromises life). The target saturation in AECOPD is lower than in asthmatic crisis; 88-92% [67]. Patients in asthma attacks or AECOPD should receive oxygen therapy regardless of the baseline level because when using bronchodilators (which are also vasodilators) they will release hypoxic vasoconstriction of the pulmonary arterioles, displacing blood to poorly ventilated areas, deteriorating the ventilation / perfusion relationship and deepening hypoxemia until the crisis stabilizes [65].
In asthmatic crisis, if mechanical ventilation is used, it must be invasive. The evidence of non-invasive ventilation indication (NIV) is weak. In contrast, in AECOPD, the NIV is preferable to invasive ventilation and is considered the method of choice in AECOPD with respiratory insufficiency since it has no absolute contraindications, improves gas exchange, reduces the work of breathing, the need for intubation, decreases the hospital stay and improves survival [68].
The use of corticosteroids (GCs) in asthmatic crisis accelerates recovery, reduces exacerbations and should be used in all exacerbations, except the milder ones [69]. The oral route is as effective as the intravenous route and is preferred since it is easier to deliver the drug, less invasive and less expensive. For children the liquid formulation is preferred. The systemic route will be implemented if the patient is too dyspneic to swallow, is vomiting, or intubated. In children, the standard dose of oral prednisolone is 1-2 mg / kg / day for 3-5 days. 0-2 years maximum 20 mg per day, 2-5 years maximum 30 mg per day, 6-11 years maximum 40 mg per day and in adults 1 mg / kg / day, maximum 50 mg for 5-7 days [13]. The I.V use should be hydrocortisone (or equivalents), 200 mg / day divided into three or four doses. It has recently been shown that GCs have biological effects that are independent of the gene transcription process [70]. The local use of GCs occurs in the smooth muscle of the blood vessels of the mucosa of the airways, producing vasoconstriction and reducing mucosal edema [71,72]. When used in the first hour, they reduce the need for hospitalization in patients who do not receive systemic corticosteroids, although cost is a significant factor [73]. Nebulized budesonide can be used at a dose of 1 mg / 12 hours for 7-10 days. In severe cases and exquisitely selected 1.5 mg / 6 hours can be used. On the fifth day, overlap to an inhaled steroid should be attempted, stay together for 3-5 days and then suspend the nebulization [74]. In some works the efficiency is comparable and the safety clearly superior with the nebulized route. More pharmacoeconomic studies are needed but the cost-benefit ratio could favor nebulized suspensions, as well as the advantage of using IGCs in the chronic treatment of the entity [75]. In AECOPD, GCs shorten the recovery, improve lung function, oxygenation, the risk of early exacerbation and hospital stay. That is, the impact on the evolution of the crisis is very similar to that of asthma. So are the doses: 40 mg / day / 5 days, and the oral route is as effective as the intravenous one [76,77]. Nebulized budesonide is an alternative to oral therapy, although more expensive. Recent work suggests that GCs may be less effective in treating AECOPD in patients with reduced levels of peripheral eosinophilia [78].
Antibiotics are not recommended routinely in asthma attacks unless there is a strong pulmonary infection (fever, purulent sputum or radiological evidence of pneumonia). Crass error is to delay the use of steroids by giving time to a cycle of antibiotics. In contrast, in AECOPD, it is indicated whether there is sputum purulence (which suggests bacterial infection), increased sputum volume, frank increase in dyspnea and in moderate to severe exacerbations that require mechanical ventilation [79]. The choice is empirical since sputum culture takes at least 48 hours and often does not give reliable results for technical reasons. CRP does not discriminate between bacterial and viral infection and procalcitonin which is more specific for bacterial infection, is more expensive and not widely available. The duration goes from 5-7 days. Aminopenicillins with clavulanic acid, macrolides, and tetracyclines are of choice. If the patient has frequent exacerbations, severe airflow limitation or is under mechanical ventilation, sputum or other lung material cultures are clearly indicated to identify bacteria resistant to the aforementioned antibiotics. The oral route is the preferred one [14]. The use of intravenous aminophylline or oral theophylline is not indicated in exacerbations, neither in asthma nor in COPD.
Magnesium sulfate is not indicated ex officio in asthmatic crises. GINA recommends it in severe crises, with severe obstruction to airflow, with failure to the initial therapy and with persistent hypoxemia. It can indicate 2 grams in infusion in 20 minutes, or 10 grams in 500 c of physiological saline to pass to 100 cc / hour for maintenance or dilute the salbutamol to nebulize, not in physiological serum, but in isotonic magnesium sulfate [13]. It has not indication in AECOPD.
BA, COPD, and ACO are mainly treated with inhaled therapy (ie, pressurized metered dose [PMDI] or dry powder [DPI] inhalers). The pillar anti-inflammatory treatment in bronchial asthma is IGCs in all patients with persistent symptoms. SABA of rescue, without IGCs, those patients with symptoms less than 2 times a month and of short duration, who are not awakened by asthma in the last month, without risk factors for exacerbation, including absence of them in the last year (Step 1). Except for these patients, the rest of the patients with asthma should use maintenance IGCs as a controlled drug, LABA will be added to patients with moderate to severe asthma, which are nowadays dispensed in fixed doses (together with the steroid in the same device) 1-2 times a day (Step 2). 5% have severe asthma that requires additional treatment, which includes LAMA, leukotriene receptor antagonist or low dose oral theophylline (Steps 3-4). 1% may require maintenance doses of oral steroids, and anti-IgE therapy with omalizumab in patients with high levels of plasma IgE; or with anti-IL-5 therapy (subcutaneous mepolizumab, or intravenous reslizumab) or anti-IL-5 receptor such as subcutaneous benralizumab (Step 5). But the basic anti-inflammatory maintenance drug is IGCs. The rest of the therapeutic tool is added according to the severity [13,27]. Most patients with COPD have minimal disease that only requires stopping smoking, immunizations and rescue with SABA and/or SAMA. It is the minority that requires more treatment. In these patients, the most effective treatment is not IGCs but long-acting bronchodilators (LABA and LAMA) that reduce hyperinflation, symptoms and AECOPDs. LABA and LAMA have additive effects and can be used in combination in the same inhaler at fixed doses. IGCs are added in cases of frequent exacerbations. The presence of peripheral eosinophilia can predict a beneficial response. Additional therapy for patients with very severe COPD includes the phosphodiesterase 4 inhibitor roflumilast, mucolytics or macrolide antibiotics, which can reduce the AECOPDs. Antioxidants in COPD have been revised recently [80]. The "step-wise algorithm" based on the usual clinical practice proposed by Mirza, et al. is more friendly than GOLD's ABCD tool, or at least complements it. Basically, it proposes using SABA and/or SAMA as rescue and adding LAMA and/or LABA according to clinical evolution (without such numbering or ABCD groups) and adding IGCs if there are frequent exacerbations. The other tools will be added if the symptoms or AECOPDs are not controlled, with the advantage that it can be staggered or de-staggered in any step that agrees with the severity of symptoms and clinical evolution [25]. ACO has changes in asthma and COPD, with more aggressive evolution, which requires maximum bronchodilator therapy with LABA and LAMA, and IGCS, which could conveniently be dispensed in a single inhaler.
Biological
Inactivate type 2 immunity is a strategy for the treatment of refractory eosinophilic asthma. Block IL-5, or it receptor (IL-5Rα), and even the GATA3 transcription factor has been implemented. Mepolizumab and reslizumab are monoclonal antibodies that block IL-5 and benralizumab blocks the IL-5 Rα receptor and inhaled DNAzyme ST010 inactivates the transcription factor GATA3, blocking the GATA3 mRNA. In severe refractory eosinophilic asthma, peripheral and sputum eosinophilia is reduced, and asthma exacerbations by a 50-75% and also the need for oral steroids, but the improvement in quality of life and pulmonary function is low [81]. Mepolizumab is also effective in patients with eosinophilic granulomatosis with polyangiitis (also known as Churg-Strauss syndrome) that is associated with severe asthma and in which it induces prolonged remissions. There is no clear established effect in allergic rhinitis, rhinosinusitis, nasal polyposis or atopic dermatitis. Blocking IL-13 has the potential to alter airway inflammation and smooth muscle reactivity, but the two monoclonal antibodies, lebrikizumab and tralokinumab have yielded poor results. This may be partly due to the fact that the goal of this treatment is preferably airway remodeling and mucus hypersecretion rather than eosinophilic exacerbations and that the results are difficult to measure in short-term clinical studies. In contrast, dupilumab (which blocks the alpha subunit of IL-4Rα) is more effective in moderate to severe asthma with eosinophilia [82,83] and in severe eczema, nasal polyposis, and rhinosinusitis [84]. Anti-IL-4 and anti-IL-13 antibodies have not been studied in COPD or ACO.
Anti-IL-5 therapy has been used in COPD (benralizumab and mepolizumab) and two large studies that have shown small reductions in AECOPDs in patients with this eosinophilic phenotype [32,85]. Blocking the alarmines (TSLP, IL-25 and IL-33) or their receptors (for example, TSLPR) is a strategy that seems more effective than blocking the Th2 cytokines, since the blockage would occur earlier in the cascade of the immune reaction type 2. Tezepelumab, which blocks TSLP, has been used in severe asthma, in dermatitis and rhinosinusitis [86,87]. Antibodies that block IL-25 and IL-33 are under clinical development. Long-term studies are required because of the possible reduction in the immune response. Antibodies against GM-CSF (granulocyte-macrophage series stimulating factor) (KB003) improve lung function in patients with blood eosinophilia. COPD could be objective in the future since macrophages maintain neutrophilic inflammation [88].
Neutrophilic inflammation has also been targeted by biologics. Immunity type 1 and 3 are involved in most patients with COPD and in some patients with asthma. This type of inflammation is insensitive to corticosteroids, so there are unresolved therapeutic needs. The cytokines TNFα, IL-17 and IL-23 are involved but the markers of their activity in plasma and sputum are highly variable [27]. Anti-TNF therapy (etanercept, infliximab and golimumab) has unacceptable side effects (infection and cancer). The anti-IL-17 strategy has scarcely been explored in airway diseases but blocking the IL-17Rα receptor (brodalumab) has not improved lung function, symptomatology or quality of life, nor blocked IL-17 (CNTO-6785) has impacted FEV1 in COPD. Antibodies to block IL-23 are under scrutiny. Blocking other proinflammatory cytokines such as IL-1 (canakinumab), IL-1R (anakinra), IL-1β, IL-18 and the NLRP3 inflammasome has not been successful [89,90]. Targeting neutrophilic inflammation in airways disease has not shown clear clinical benefit, although these same therapies have been effective in other chronic non-eosinophilic inflammatory diseases such as rheumatoid arthritis, inflammatory bowel disease and psoriasis.
Mitochondrial dysfunction and mitochondria as therapeutic objective
Mitochondria are complex cellular organelles assembled from proteins encoded by two distinct genomes: nuclear chromosomal DNA and the mitochondrial DNA (mtDNA). Despite the small size of the mtDNA (16.5 kb in humans), it encodes 13 potential oxidative phosphorylation subunits (OXPHOS) which interact with more than 70 units encoded by nuclear DNA; they concerted action is necessary to produce ATP, which is required for all active cellular processes [91].
Historically, the major role of mitochondria has been to catalyze the oxidation of metabolites for the production of ATP, via OXPHOS, involving the transference of high energy electrons derived from NADH and FADH2 to the four mitochondrial complexes, where they react with oxygen to form water. During this process, the protons are pumped through the inner mitochondrial membrane they are used by the FOF ATP synthase to generate ATP [91]. Additional critical functions are: regulation, proliferation, differentiation, cell death, redox and calcium homeostasis and recently, research has pointed to mitochondria as controlling the immune responses and determinants of immune cell phenotypes and their functions, including CD4+ T cells differentiation and CD8+ memory T cell formation [92]. Within the mitochondrial proteome there are key proteins such as mitochondrial antiviral signaling protein (MAVS), which is the first protein located in the mitochondria involved in the innate immune response and inflammatory response [93]. NOD-like receptor (NLRX1) is a member of the NLR family of PRRs that has a unique N-terminal domain and is the first, and only so far, of the PRRs located in the mitochondria and by therefore, it establishes a connection between mitochondrial function and innate immunity [94]. Apparently, the inhibition of inflammation and remodeling in COPD is performed by NLRX1 regulating MAVS [95].
In COPD, exposure to smoke reduces mitochondrial OXPHOS in airway smooth muscle cells, quadriceps, and external intercostal muscles, compromising oxidative function. Alteration of mitochondrial biogenesis may be associated with a significant reduction in body mass index and lower extremities muscle mass, a common phenomenon in COPD [96]. Loss of mtDNA has been associated with COPD [60]. Several independent studies have shown that NLRX1 protein expression and activity are suppressed by cigarette smoke in patients with COPD and this correlates with airflow obstruction. Basically, the idea is that chronic smoking alters NLRX1, inducing a mitochondrial dysfunction mediated by the NLRX1 / MAVS interaction. Chronic smoking, on inactivation of NLRX1, increases inflammatory activity (IL-18) favoring emphysematous destruction, mitochondrial ROS and proteases matrix metalloproteinase [97,98].
Mitochondria offer research objectives not only for diagnosis but for treatment of lung diseases. Transfer of mitochondria from bone marrow-derived mesenchymal stem cells to injured alveolar cells could be beneficial in COPD [99]. Antioxidants directed against mtROS may be more effective than extracellular antioxidants like NAC. Attempts to block mtROS should be careful since low doses of mtROS are cytoprotective (mithormesis). OXPHOS could be an objective to intervene as well as to create strategies to reduce mitophagy [91,100]. Several proteins have been implicated in the communication between the ER and the mitochondria and that contact occurs prior to the phenomenon of fission. Contact is important for the transmission of ER calcium signals to mitochondria. Therefore, designing strategies that refine and perpetuate transmission may improve the function and survival of mitochondria [101]. The attempt to modulate mitochondrial function as a therapeutic target is only initiated.
BA and COPD are the two most prevalent chronic, inflammatory and obstructive airways diseases, worldwide; however, there is a gap between biological knowledge and response to treatment.
It is not even clear enough whether they are completely different entities or have a common pathophysiological base, with different clinical expressions conditioned by environmental factors.
Both have heterogeneous endotypes and phenotypes that sometimes overlap, which has led to the coining of the ACO term, which until now seems more a term in which to group the patients that we can’t or do not know how to classify, than a different obstructive pathophysiological entity.
It is possible that future investigations in basic molecular biology of both are designed with the aim of finding inflammatory or immunological markers that allow to differentiate them and to make a more precise diagnosis.
Two other basic areas that should be priorities are: education and new treatments. Education is vital because the main reason for the failure of treatment is not to know how to use inhalers, the abandonment of it, the ignorance of chronic and progressive evolution and the risk of exposure to environmental noxas. Regarding treatment, new and powerful drugs must be sought that impact the subcellular compartments and block the various metabolic pathways of both entities, since the current treatment has not allowed to obtain satisfactory control of both or to impact the torpid evolution.
This work was only carried out by the author. Author AA contributed in the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
No
No.
- GINA (2017) Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma.
- GOLD (2017) Global Strategy for the Diagnosis, Management and Prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease.
- Barnes PJ (2008) Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183-192.
- Liesker JJ, Ten Hacken NH, Zeinstra-Smith M, Rutgers SR, Postma DS, et al. (2009) Reticular basement membrane in asthma and COPD: similar thickness, yet different composition. Int J Chron Obstruct Pulmon Dis 4: 127-135.
- Louie S, Zeki AA, Schivo M, Chan AL, Yoneda KY, et al (2013). The asthma-chronic obstructive pulmonary disease overlap syndrome: pharmacotherapeutic considerations. Expert Rev Clin Pharmacol 6: 197-219.
- Orie NG, Sluiter H, de Vries K, Tammeling G J, Wikop J (1961) The host factor in bronchitis. In: Orie N, Sluiter H. Editors. Bronchitis. The Netherlands: Royal Van Gorcum pp. 43-59.
- Kraft M (2006) Asthma and chronic obstructive pulmonary disease exhibit common origins in any country. Am J Respir Crit Care Med 174: 238-240.
- Xu X, Rijcken B, Schouten JP, Weiss ST (1997). Airway responsiveness and development and remission of chronic respiratory symptoms in adults. Lancet 350: 1431-1434.
- Gibson PG, Simpson JL (2009) The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax 64: 728-735. [Crossref]
- GINA (2015) Diagnosis of Diseases of Chronic Airflow Limitation: Asthma, COPD and Asthma-COPD Overlap Syndrome (ACOS). Global Initiative for Asthma.
- Izuhara K, Barnes PJ (2019) Can We Define Asthma-COPD Overlap (ACO) by Biomarkers? J Allergy Clin Immunol Pract 7: 146-147. [Crossref]
- Barnes PJ (2016) Asthma-COPD Overlap. Chest 49: 7-8.
- Bozek A, Jarzab J (2011) Improved activity and mental function related to proper antiasthmatic treatment in elderly patients with Alzheimer's disease. Allergy Asthma Proc 32: 341-345. [Crossref]
- GOLD (2018) Global Initiative for Chronic Obstructive Lung Disease, Global strategy for the diagnosis, management and prevention of COPD.
- To T, Stanojevic S, Moores G, Gerson AS, Bateman ED, et al. (2012). Global asthma prevalence in adults: finding from the cross-sectional World Health Survey. NMC Public Health 12: 204.
- Masoli M, Fabian D, Holt S, Beasley R (2004) The global burden of asthma: executive summary of the GINA Dissemination Committee Report. Allergy 59: 469-478.
- Adeloye D, Chua S, Lee C, Basquill C, Papana A, et al. (2015). Global and regional estimates of COPD prevalence: systematic review and meta-analysis. J Glob Health 5: 020415.
- Global Burden of Diseases Study Collaborators (2015) Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Diseases Study 2013. Lancet 385: 117-171.
- WHO (2015) Projection of mortality and causes of death, 2015 and 2030, World Health Organization.
- Alvarado A (2018) Chronic obstructive pulmonary disease in non-smokers: An update. Clin Res Trials 4: 1-8.
- Wenzel SE (2012) Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18: 716-725. [Crossref]
- Alvarado A (2018) Respiratory pathologies associated with obesity. Clin Res Trials 4: 1-10.
- Menezes AM, Perez-Padilla R, Jardim JR, Muiño A, Lopez MV, et al. (2005). Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study). Lancet 366: 1875-1881.
- Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, et al. (2005) Interpretative strategies for lung function tests. Eur Respir J 26: 948-968. [Crossref]
- Mirza S, Clay RD, Koslow MA, Scanlon PD (2018) COPD Guidelines: A Review of the 2018 GOLD Report. Mayo Clin Proc 93: 1488-1502. [Crossref]
- Barnes PJ (2017) Cellular and molecular mechanisms of asthma and COPD. Clin Sci (Lond) 131: 1541-1558. [Crossref]
- Barnes PJ (2018) Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 18: 454-466.
- Upham JW, Xi Y (2017) Dendritic Cells in Human Lung Disease: Recent Advances. Chest 151: 668-673. [Crossref]
- Hammad H, Lambrecht BN (2015) Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 43: 29-40. [Crossref]
- Ballarin A, Bazzan E, Zenteno RH, Turato G, Baraldo S, et al. (2012). Mast cell infiltration discriminates between histopathological phenotypes of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 186: 233-239.
- Rosenberg HF, Dyer KD, Foster PS (2013) Eosinophils: changing perspectives in health and disease. Nat Rev Immunol 13: 9-22. [Crossref]
- Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Kor S, et al. (2017) Mepolizumab for eosinophilic chronic obstructive pulmonary disease. Epub 377: 1613-1619.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, et al. (2004). The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350: 2645-2653.
- Brusselle GG, Joos GF, Bracke KR (2011) New insights into immunology of chronic obstructive pulmonary disease. Lancet 378: 1015-1026.
- Postma DS, Rabe KF (2015) The Asthma-COPD Overlap Syndrome. N Engl J Med 373: 1241-1249. [Crossref]
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, et al. (2014) International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 43: 343-373. [Crossref]
- Gedebjerg A, Szépligeti SK, Wackerhausen JH, Horvath-Puho E, Dahl R, et al. (2018). Prediction of mortality in patients with chronic obstructive pulmonary disease with the new Global Initiative for Chronic Obstructive Lung Disease 2017 classification: a cohort study. Lancet Respir Med 6: 204-212.
- Cabrera Lopez C, Casanova Macario C, Mann Trigo JM, de-Torres JP, Sicilia Torres R, et al. (2018). Comparison of the 2017 and 2018 Global Initiative for Chronic Obstructive Lung Disease reports: impact of grouping and outcomes. Am J Respir Crit Care Med 197: 463-469.
- Hwang HG (2018) Does the GOLD 2017 classification improve the ability to predict exacerbation and mortality? Tuberc Respir Dis (Seoul) 81: 256-257.
- Boulet LP (2009) Influence of comorbid conditions on asthma. Eur Respir J 33: 897-906. [Crossref]
- Tong MCF, Lin JSC (2015) Epidemiology of allergic rhinitis throughout the world. In: Akdis CA, Hellings PW, Agache I (Eds) Global Atlas of Allergic Rhinitis and Chronic Rhinosinusitis. Zurich, Switzerland: European Academy of Allergy & Clinical Immunology pp. 62-63.
- Schmitt J, Stadler E, Küster D, Wüstenberg EC (2016) Medical care and treatment of allergic rhinitis: a population-based cohort study based on routine health care utilization data. Allergy 71: 850-858.
- Hamilos DL (2011) Chronic rhinosinusitis: epidemiology and medical management. J Allergy Clin Immunol 128: 693-707.
- Lyons JJ (2015) Atopic dermatitis in children: clinical features, pathophysiology, and treatment. Immunol Allergy Clin Nort Am 35: 161-183.
- Bock SA, Munoz-Furlong A, Sampson HA (2007). Further fatalities caused by anaphylactic reactions to food, 2001-2006. J Allergy Clin Immunol 1191016-1018.
- Barnes PJ, Celli BR (2009) Systemic manifestations and comorbidities of COPD. Eur Respir J 33:1165-1185.
- Mannino DM, Thorn D, Swensen A, Holguin F (2008) Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in chronic obstructive pulmonary disease. Eur Respir J 32: 962-969.
- Shorr AF, Doyle J, Stern L, Dolgitser M, Zilberberg MD (2008). Anemia in chronic obstructive pulmonary disease: epidemiology and economic implications. Curr Med Res Opin 24: 1123-1130.
- Alshabanat A, Zafari Z, Albanyan O, Dairi M, FitzGerald JM (2015) Asthma and COPD Overlap Syndrome (ACOS): A Systematic Review and Meta Analysis. PLoS One 10: e0136065. [Crossref]
- Gibson PG, Simpson JL (2009) The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax 64: 728-735. [Crossref]
- Kauppi P, Kupiainen H, Lindqvist A, Tammilehto L, Kilpeläinen M, et al. (2011) Overlap syndrome of asthma and COPD predicts low quality of life. J Asthma 48: 279-285. [Crossref]
- Andersén H, Lampela P, Nevanlinna A, Säynäjäkangas O, Keistinen T (2013) High hospital burden in overlap syndrome of asthma and COPD. Clin Respir J 7: 342-346. [Crossref]
- GINA (2018) Diagnosis and initial treatment of asthma, COPD and asthma-COPD overlap (ACO). Global Initiative for Asthma.
- Shirai T, Hirai K, Gon Y, Maruoka S, Hikichi M, et al. (2019). Combined assessment of serum periostin and YKL-40 may identify asthma-COPD overlap. J Allergy Clin Immunol Pract 7: 134-145.
- Izuhara K, Nunomura S, Nanri Y, Ogawa M, Ono J, et al. (2017) Periostin in inflammation and allergy. Cell Mol Life Sci 74: 4293-4303. [Crossref]
- Lee CG, Da Silva GA, Dela Cruz CS, Ahangari F, Ma B, et al. (2011). Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol 73: 479-501.
- Gon Y, Maruoka S, Ito R, Mizumura K, Kozu Y, et al. (2017) Utility of serum YKL-40 levels for identification of patients with asthma and COPD. Allergol Int 66: 624-626. [Crossref]
- Newman KB, Milne S, Hamilton C, Hall K (2002) A comparison of albuterol administered by metered-dose inhaler and spacer with albuterol by nebulizer in adults presenting to an urban emergency department with acute asthma. Chest 121: 1036-1041.
- Camargo CA Jr, Spooner CH, Rowe BH (2003) Continuous versus intermittent beta-agonists in the treatment of acute asthma. Cochrane Database Syst Rev CD001115.
- Rodrigo GJ, Castro-Rodríguez JA (2005) Anticholinergics in the treatment of children and adults with acute asthma following emergency department discharge. Thorax 60: 740-746.
- Griffiths B, Ducharme FM. (2013). Combined inhaled anticholinergics and short-acting beta2-agonists for initial treatment of acute asthma in children. Cochrane Database Syst Rev CD000060.
- Rodrigo GJ, Rodrigo C (2002) The role of anticholinergics in acute asthma treatment: an evidence-based evaluation. Chest 121: 1977-1987. [Crossref]
- NICE (2010) Chronic Obstructive pulmonary Disease in over 16s: diagnosis and management. National Institute of Health and Care Excellence.
- Alvarado A (2017) Dual bronchodilator therapy. Clin Res Trials 3: 7-12.
- Alvarado A (2017) Domiciliary oxygen: facts and fallacies. Clin Res Trials 3: 1-10.
- Jones PW (2009) Health status and the spiral of decline. COPD 6: 59-63. [Crossref]
- Austin MA, Wills KE, Blizzard L, Walters EH, Wood-Baker R (2010). Effects of high flow oxygen on mortality in chronic obstructive pulmonary diseases patients in prehospital setting: randomized controlled trial. BMJ 341: c5462.
- Chandra D, Stamm JA, Taylor B, Ramos RM, Satterwhite L, et al (2012). Outcomes of noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease in the United States, 1998-2008. Am J Respir Crit Care Med 185: 152-159.
- Rowe BH, Spooner CH, Ducharme FM, Bretzlaff JA, Bota GW (2007) Corticosteroid for preventing relapses following acute exacerbations of asthma. Cochrane Data Base Syst Rev: CD000195.
- Marwick J, Adcock IM, Chun FK (2010) Overcoming reduced glucocorticoid sensitivity in airway disease: molecular mechanisms and therapeutic approaches. Drugs 70: 929-948.
- Zhu Y, Rice CD, Pang Y, Pace M, Thomas P (2003) Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in mitotic maturation of fish oocystes. Proc Natl Acad Sci USA 100: 2231-2236.
- Hernández Alcalá-Pérez M, Cormack J, Mallison D, Mutungi G (2013) A membrane glucocorticoid receptor mediated the rapid/non-genomic actions of glucocorticoids in mammalian skeletal muscle fiber. J Physiol 591: 5171-5185.
- Edmonds LM, Milan SJ, Camargo CA Jr, Pollack CV, Rowe BH (2012) Early use of inhaled corticosteroids in the emergency department treatment of acute asthma. Cochrane Database Syst Rev 12: CD002308.
- Szefler SJ, Eigen H (2002) Budesonide inhalation suspension: a nebulized corticosteroid for persistent asthma. J Allergy Clin Immunol 109: 730-742.
- O’Connell EJ (2003) Review of the unique properties of budesonide. Clin Ther 25: C42-C60.
- Alia I, de la Cal MA, Esteban A, Abella A, Ferrer R, et al. (2011) Efficacy of corticosteroid therapy in patients with an acute exacerbation of chronic obstructive pulmonary disease receiving ventilator support. Arch Intern Med 171: 1939-1946.
- Leuppi JD, Schuetz P, Bingisser R, Bodmer M, Briel M, et al. (2013). Short-term versus conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 309: 2223-2231.
- Hurst JR, Vestbo J, Anzueto A, Locantore N, Müllerova H, et al. (2010) Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 363: 1128-1138. [Crossref]
- Wilson R, Anzueto A, Miravitlles M, Arvis P, Alder J, et al. (2012). Moxifloxacin versus amoxicillin/clavulanic acid in outpatient acute exacerbations of COPD: MAESTRAL results. Eur Respir J 40:17-27.
- Alvarado A, Arce I (2016) Antioxidants in respiratory diseases: basic science research and therapeutic alternatives. Clin Res Trials 3: 1-11.
- Cabon Y, Molinari M, Marin G, Gamez AS, Chanez P, et al. (2017) Comparison of anti-interleukin-5 therapies in patients with severe asthma: global and indirect meta-analysis of randomized placebo-controlled trials. Clin Exp Allergy 47: 129-138.
- Wenzel S, Ford L, Pearlman D, Spector S, Sher L, et al. (2013) Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med 368: 2455-2466. [Crossref]
- Wenzel S, Castro M, Corren J, Maspero J, Wang L. et al. (2016) Dupilumab efficacy and safety in adult with uncontrolled persistent asthma despite use of medium-to high-dose inhaled corticosteroids plus long acting ßeta 2-agonist: a randomized, double placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 388: 31-44.
- Bachert C, Mannent L, Naclerio RM, Mullol J, Ferguson BJ, et al. (2016). Effect of subcutaneous dupilumab on polyp burden in patients with chronic sinusitis and nasal polyposis: a randomized clinical trial. JAMA 315: 469-479.
- Dasgupta A, Kjarsgaard M, Capaldi D, Radford K, Aleman F, et al. (2017). A pilot randomized clinical trial of mepolizumab in COPD with eosinophilic bronchitis. Eur Respir J 49: 1602486.
- Corren J, Parnes JR, Wang L, Mo M, Roseti SL, et al. (2017) Tezepelumab in Adults with Uncontrolled Asthma. N Engl J Med 377: 936-946. [Crossref]
- Verstraete K, Peelman F, Braun H (2017) Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma. Nat Commun 8: 14937. [Crossref]
- Molfino NA, Kuna P, Leff JA, Oh CK, Singh D, et al. (2016). Phase 2 randomized placebo-controlled trial to evaluate the effectivity and safety of an anti-GM-CSF antibody (KB003) in patients with inadequately controlled asthma. BMJ Open 6: e007769.
- Rennard SI, Fogarty C, Kelsen S, Long W, Ramsdell J, et al (2007). The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 175: 926-934.
- Ricciardolo FLM, Sorbello V, Folino A, Gallo F, Massaglia GM, et al. (2017) Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol 140: 395-406. [Crossref]
- Mizumara K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, et al. (2014). Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987-4003.
- Lommatzsch M, Cicko S, Muller T, Lucattelli M, Bratke K, et al. (2010). Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 181: 928-984.
- van der Toorn, Slebos DJ, de Bruin HG, Leuvenink HG, Bakker SJ, et al. (2007). Cigarette-smoked induced blockage of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol 292: L1211-1218.
- Tashkin DP, Celli B, Seen S, Burkhart D, Kesten S, et al. (2008). A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 359: 1543-1554.
- Magnussen H, Disse B, Rodriguez-Roisin R, Kirsten A, Watz H, et al. (2014) Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med 371: 1285-1294. [Crossref]
- Dekhuijzen PN, van Beurden WJ (2006) The role of N acetyl-cysteine in the management of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 1: 99-106.
- Decramer M, Rutten-van Molken M, Dekhuijzen PN, Trooster T, van Herwaarden, et al. (2005). Effect of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (bronchitis randomized on NAC cost-utility study, BRONCUS): a randomized controlled trial. Lancet 365: 1552-1560.
- Tse HN, Raiteri L, Wong KY, Yee KS, Ng LY, et al. (2013). High-dose N-acetylcysteine in stable COPD 1 year double-blind, randomized placebo-controlled HIACE study. Chest 144: 196-118.
- Kang MJ, Shadel JS (2016) A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis (Seoul) 78: 207-213.
- Hutterman M, Lee I, Gao X, Pecina P, Pecinova A, et al. (2012). Cytochrome C-oxidase subunit 4 isoform 2-knockout mice show reduce enzyme activity airway hiperreactivity, and lung pathology. FASE BJ 25: 3916-3930.
- Mishra P, Chan DC (2014) Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15: 634-646. [Crossref]