Abstract
Covid-19 is associated with endothelial activation in the setting of a potent inflammatory reaction, immunological activation, and a hypercoagulable state. The result of this thrombo-inflammatory and immune-thrombosis state is an excess in thrombotic events, in particular venous thromboembolism. In this scenario, pulmonary embolism (PE) is associated with respiratory deterioration, increased risk of intensive care unit admission, prolonged hospital stays, and death. COVID-19-associated PE clearly differs from the conventional non–COVID-19-associated PE. This has led to define the preponderant role of "in situ" thrombosis in the pulmonary vessels, frequently peripheral, silent, and therefore difficult to detect. The diagnostic value of D-dimers has been confusing, and the normal values are not known for certain. The profitability of the predictive scales to assess pre-test probability, so widely used in conventional thromboembolism, have not been validated in thromboembolism associated with Covid-19 and the extent of anticoagulant treatment in proven pulmonary thromboembolism is still a matter of controversy. Prophylactic anticoagulation in patients with Covid-19 hospitalized in general wards and Intensive Care Units does not necessarily prevent thromboembolic events. Therefore, there are a series of fallacies in diagnosis, prevention, and treatment that have been derived from extrapolating data and concepts from conventional pulmonary thromboembolism to that associated with Covid-19. Furthermore, we briefly summarize the results from randomized controlled trials of the pathophysiology, epidemiology, diagnosis, prevention, and treatment of Covid-19 associated PE.
Keywords
pulmonary thromboembolism, immune-thrombosis, thrombo-inflammatory state, D-dimers, Covid-19
Introduction
In December 2019, a novel pneumonia with a high potential of transmissibility between humans was first reported [1,2]. The Chinese Center for Disease Control and Prevention, along with other related institutions, quickly identified the pathogen as a new type of coronavirus. To ensure that the information is shared quickly across the world, the first viral sequence was deposited into GenBank and made public on 26 December 2019 (LR757995, LR757998). On 11 February 2020, the International Committee of Taxonomy of Viruses named this virus severe acute respiratory syndrome (SARS)-CoV-2 based on the phylogenetic relationship of the coronavirus that caused the SARS outbreak in 2003. On the same day, WHO announced COVID-19 as the name of this novel disease caused by this virus following the guidelines of the World Organization for Animal Health and the Food and Agricultural Organization of the United Nations. At the time of writing this article (mid-March/2023) there are >676 million subjects globally with confirmed SARS-CoV-2 infection based on the molecular assay (from more than 210 countries), with 6,88 million of death with a lethality of 1.02% (Johns Hopkins University, USA) [3]. The previously reported lethality of around 3.4 has been reduced due to decreased mortality (prior vaccination and less lethal variants and sub-lineages) and increased infections (more contagious variants and sub-lineages). An unfavorable clinical evolution in patients with COVID-19 has been associated with risk factors known to the medical community and the public. There are basically three respiratory causes of death: severe pneumonia, acute respiratory distress syndrome (ARDS), and pulmonary thromboembolism (PE). This last entity is the one addressed in this review. Although COVID-19 is mainly associated with respiratory morbidity, patients with COVID-19 have higher D-dimer levels and an increased risk of thromboembolic events, especially venous thromboembolism (VTE), particularly in critically ill patients [4,5].
There are several potential hypotheses for the excess of thromboembolic events in COVID-19. The cytokine storm may be responsible for an imbalance in coagulation. These cytokines favor the increased production of fibrinogen in the liver, which serves as a substrate to produce fibrin, responsible for clot generation. There may also be increased production of fibrinogen at the endothelial level induced by IL-1 and TNF-α. This would favor “in situ” formation of thrombi. But there is also a reduction in fibrinolysis (which exerts control over thrombosis by undoing the thrombi formed). Plasminogen is a substrate that generates plasmin, the enzyme that breaks or fractures fibrin to break up clots. Plasminogen is activated by a plasminogen activating factor, which can be inhibited by PAI-1 (fibrinogen activator inhibitor 1) generated in the liver by cytokines, reducing fibrinolysis. The final balance is a polarization of the coagulation/fibrinolysis system towards thrombosis [6]. Direct invasion by the vascular endothelial virus (rich in angiotensin convertase 2 [ACE-2]) generates intravascular membrane phospholipids and this initiates the intrinsic coagulation pathway by activating factor XII (the origin of this loop). But the severe alveolar inflammatory process produced by the parenchymal invasion also generates inflammatory mediators that favor the expression of tissue coagulation factor which, by activating factor VII (the origin of this other loop), initiates the extrinsic pathway [7]. The antiphospholipid antibodies that can be generated in Covid-19 will damage the platelet phospholipids, producing thrombocytopenia but activating factor XII and therefore coagulation via the intrinsic pathway. Besides the production of neutrophil extracellular traps (NETs) may lead to heightened platelet activation and aggregation, and stimulate coagulation, which clinically manifests as thromboembolic events [8]. Angiotensin II stimulates the production of PAI-1, reducing fibrinolysis. Normally this effect is controlled by ACE-2, which degrades angiotensin II to angiotensin 1-7, which does not have this effect, but as in Covid-19, ACE-2 is glycosylated by the S protein of the SARS-CoV2 being internalized, its expression is reduced. This decreases the control over angiotensin II which can generate PAI-1 [9]. Deep hypoxia induces a prothrombotic state as it activates HIF (Hypoxia-Inducible Factor), which underregulates natural anticoagulants. Immobility, inflammation, and baseline comorbidities that increase the risk of thrombosis are among other potential reasons for the excess risk of thrombosis in COVID-19 [10]. Added to this is the clinical and pathophysiological associations of thrombosis with some vaccines used for preventing COVID-19.
For the purposes of this monograph, we will use the term COVID-19-associated PE to refer to said association and establish similarities, but above all differences, with conventionalPE. For example, conventional PE is classically associated with deep vein thrombosis (DVT), whereas in COVID-19-associated PE, a significant percentage of cases have thrombosis “insitu” in the lungs, which begins in the periphery, in segmental and/or subsegmental vessels, silently, progressively, and difficult to diagnose. Between 66-90% of peripheral locations of thrombi are reported in different series (occult thrombosis), and between 56-77% of patients do not have deep vein thrombosis and there are series that only document it between 3-12 %. This means two facts. The globally actual incidence of COVID-19 associated PE is unknown and PE may occur as a silent, hemodynamically stable disease, at least initially [11-14].
Pathophysiology
More than a century ago, a German physician and scientist Rudolf Virchow proposed that blood clots in the leg could travel to the lung and cause pulmonary embolism (PE), because he discovered at autopsy that the emboli in the lung and the leg usually coexisted. He also performed an experiment showing that foreign bodies in deep veins can be found in pulmonary arteries (Figure 1) [15]. Therefore, he coined the terms “PE” and “DVT” [16]. From then on, embolus from peripheral venous system, such as lower extremities and pelvis, has been deemed the predominant cause of pulmonary artery obstruction. However, from the contemporary perspective, Dr. Virchow’s proposal might be biased and excluded the possibility of “de novo” thrombosis in pulmonary vessels. Based on Virchow’s proposal, PE and DVT are disparate manifestations of the same disease, both belonging to VTE [17] (Figure 2). In recent years, multiple lines of evidence have indicated the possibility of generating “de novo” thrombus in pulmonary arteries without DVT in lower extremities [18]. Therefore, to distinguish from the PE associated with DVT, It has been proposed the term “in situ pulmonary artery thrombosis” (in situ PAT) to describe the pathology of “de novo “ thrombosis in proximal (main, lobar, and segmental arteries), distal (segmental, mid-segmental, and sub-segmental arteries, down to small pulmonary arteries of 2–5 mm in diameter), and micro (microvasculature of 0.1–0.5 mm in diameter) pulmonary arteries [19]. Other terms are PIC (pulmonary intravascular coagulation) and TIRAC (thrombotic immune response associated with Covid-19).

Figure 1. Rudolf Virchow MD
Famous German pathologist (1821-1902). Classic pulmonary thromboembolism (PTE) has been associated with many diseases. Clot formation is a consequence of hypercoagulability, blood stasis, and damage to the endothelium of blood vessels, a set of changes known as the Virchow triad.
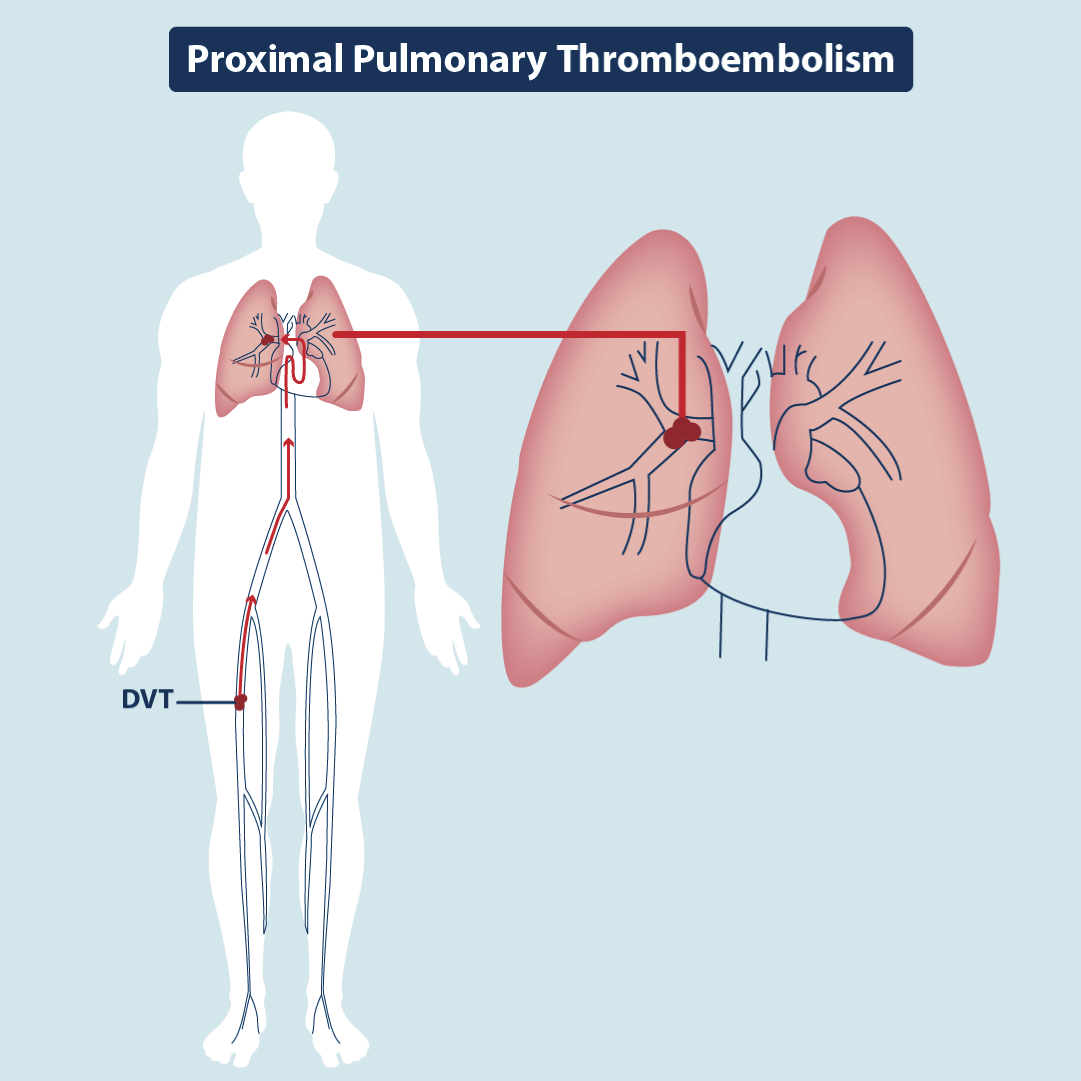
Figure 2.Classic Pulmonary Thromboembolism
Illustration demonstrating large-vessel occlusion and it´s anterograde consequence: pulmonary embolism.
VTE is known to be a common respiratory and/or cardiovascular complication in hospitalized patients with viral infections. Hospitalized patients with HIV, hepatitis C, Chikungunya, Zika, Varicella-Zoster, H1N1 and Covid-19 have an associated prothrombotic condition. Ebola-Marburg infection has been associated with PE and disseminated intravascular coagulation (DIC) [20]. Inflammation and immune response go hand in hand in human physiology and pathology. Vast to review the profound interrelationship of both processes in the innate immune response and the adaptive immune response [21].
The pathogenesis of COVID-19-associated PE is still not completely understood but encompasses the effects of thrombo-inflammation and immune-thrombosis at the alveolus, pulmonary interstitium, and pulmonary microvasculature level [22] (Figure 3). But it is obvious that under the mechanisms described in the introduction to produce thrombosis "in situ" underlie three highly interrelated phenomena, namely, the direct invasion of the virus in the microvascular environment, the cytokine storm, and the inflammatory process. Severe inflammation leads to a hypercoagulable state, platelet activation, and endothelial dysfunction, already initiated by direct virus invasion. It should be remembered that ACE-2 is found not only in type II pneumocytes but also that the endothelium is abundant in the expression of said receptor [23]. CoV-2 interacts with the type II pneumocytes through several membrane proteins, including the membrane-bound ACE-2. This interaction can lead to a pneumocyte activation, production of thrombo-inflammatory cytokines which have cross-communication with immune cells, so the immune-thrombi will be formed, in addition to fibrin, by immune cells such as monocytes, neutrophils, T and B lymphocytes, and platelets. At the same time, invasion by the endothelial virus will produce endotheliosis not only in the lung, but also in the heart, liver, and kidneys, amplifying the production of micro-thrombosis [24]. These inflammatory cytokines also activate platelets. Having no nucleus, the health of platelets depends on their mitochondria. Being dysfunctional, these mitochondria in Covid-19 undergo apoptosis, inducing clot formation 50 times faster than normal platelets, increasing thrombus formation [25]. Cardiolipin is as mitochondrial phospholipid that participates in maintaining the integrity of the membranes of these organelles. In patients with coagulopathy and thrombocytopenia in Covid-19, IgA anticardiolipin antibodies are detected in the serum [26]. Cytokines also favor the formation of NETs that are extracellular traps for neutrophils that also favor thrombosis "in situ". The hypercoagulable state of patients with Covid-19 is characterized by increased D-dimers, thrombocytopenia (usually not severe), mild elevation of PT and TPT, increased fibrinogen, thrombin and Leyden factor V and factor VIII of the coagulation. This profile is different from that of DIC and sepsis-induced coagulopathy, suggesting different mechanisms of coagulopathy genesis [27]. Although “in situ” thrombosis can be a specific mechanism in patients with COVID-19, both “in situ” thrombosis and embolic events can contribute to the overall COVID-19-associated PE pathogenesis This is why VTE is fundamentally an acute complication of Covid-19. The risk of VTE in non-hospitalized COVID‐19 patients is not known.
Epidemiology
The reported incidence of COVID-19-associated PE varies significantly among the studies and meta-analyses [28,29]. This variability is due to multiple reasons. First, the sample size and methodology of the included individual studies differ significantly. Second, the modality for diagnosis is highly varied. Third, the clinical status of the patients has been strongly correlated with the PE incidence. ICU patients had a higher risk of PE than non-ICU hospitalized patients; whereas non-hospitalized patients have a lower risk compared with hospitalized patients with COVID-19. Fourth, the concomitant anti-inflammatory and prophylactic antithrombotic therapy may affect the incidence [30]. Fifth, the incidence of PE with the new variants such as delta (B.1.617.2) and omicron (B.1.1.529) and their sub-lineages and the impact that vaccination has had on Covid-19 associated PE are not known. Some studies suggest that the incidence is declining with new variants and vaccination [31-33]. The incidence of COVID-19-associated PE was reported in 13.6 to 16.7% of critically ill patients and 2.2 to 8.3% of hospitalized patients who were not admitted directly to the intensive care unit (ICU), but the numbers vary greatly with the various reports. There are works that report 24.7% in patients in the ICU and 10% in hospitalized in rooms [34-37]. There is greater uncertainty regarding the PE rates in outpatients and patients post-discharge with COVID-19 because of limited data. A meta-analysis assessing post-discharge rates of PE estimated an incidence of 1.5% (95% CI: 0.5–4.0%) at a mean of 68 days of follow-up [38]. However, data from placebo arms in randomized controlled trials (RCTs) of outpatients with COVID-19 have found rates of PE at approximately 1% up to 30 days of follow-up [39,40]. But the risk results in this group of outpatients are highly controversial. A study carried out with the National Registries in Sweden suggests that the risk extends up to 70 days for DVT and 110 days for PE, but in male patients, with severe Covid-19, admitted to the hospital and/or ICU and between 50-70 years of age of age [22]. Other reports suggest that the "persistent hypercoagulable state" can last from 2 weeks to 12 weeks [41,42]. An epidemiological report using data from the US Department of Veteran Affairs provides some evidence that the one-year risk and burden of CVD (coagulation vascular disorders) in survivors of acute Covid-19 is substantial [43]. But there are also studies that conclude that in post-discharge patients from a hospital due to Covid-19, the risk of PE is not increased, compared to the post-discharge risk for other diseases [44]. This risk appears to be related to the severity of Covid. In patients with mild Covid-19, reports suggest that there is no clear relationship between viral infection and VTE. In a large network of studies from 5 European countries, the incidence at 90 days-term is 0.2-0.8%. It is also not clear that late-onset cases of PE are necessarily related to the virus. There may be undetermined factors such as occult cancer in older patients, prolonged bed rest due to chronic fatigue (risk factor for conventional PE), or obesity and undetected thrombophilia in young people [45]. What is clear from this varied information is that the actual incidence of Covid-19 associated PE is not known and that the risk is greater the greater the severity of Covid. In a patient with mild, ambulatory Covid-19, or in a post-discharge patient who did not have PE during his hospital admission, the persistence of a chronic hypercoagulable state that predisposes to VTE is not convincing or is at most, debatable. A very recent publication supports this statement [46]. Results from other recently completed trials will provide further clarity on this topic.
Diagnosis
Generalities
In Covid-19, most PEs occur in patients admitted to the ICU or in wards, and usually in the first 15-30 days and, it can occur aggressively despite prophylactic (still widespread) and therapeutic anticoagulation [47]. If the diagnosis in conventional PE is not easy, it is even less so in Covid-19. The diagnostic suspicion is strictly clinical and there is no systemic screening test for diagnosis. The clinical findings of PE are not specific. Dyspnea, cough, chest pain, tachycardia, hemoptysis (rare), and hypoxemia can be caused by pneumonia, which is itself much more common than PE in Covid-19 (is what the doctor usually thinks first). Acute dried cough in the inflammatory process (66-70% of patients) occurs due to invasion of the sensory vagal pathways, which conduct stimuli to the brainstem (nucleus of the solitary tract and spinal trigeminal nucleus). In addition, the virus infects the glial cells of the vagus and other cranial nerves (dysgeusia and anosmia). The released neuropeptides initiate the reflex. Virus interaction with dorsal root ganglion neurons explains pain and dyspnea in inflammation (nociceptive receptors). The virus has been found in CSF orchestrating brain inflammation and providing a central basis for hypersensitivity. That is, there is central and peripheral inflammatory hypersensitivity without the need to invoke PE from the outset to explain the symptoms [48].
Therefore, the clinical findings of PE overlap with those of pulmonary inflammation. There are several clinical scenarios that can suggest the diagnosis. The most obvious would be a patient with the described symptoms and hypoxemia with a normal chest X-ray. If there is no picture of pneumonia that explains the findings, the suspicion of PE is mandatory [49]. Another scenario would be that of a patient with symptoms of covid-19 pneumonia and "disproportionate" hypoxemia, not attributable only to lung inflammation. Again, PE should be thought of as a mechanism superimposed on pneumonia [50]. The same happens in “lack of clinical improvement in ventilated patients” or in the patient with covid-19 pneumonia who develops right heart failure [51]. Another clinical scenario that suggests pulmonary thromboembolism in a patient with Covid-19 is a rapid deterioration in lung function, heart function, neurological function, and loss of perfusion (blood supply) to an extremity [52].
The profile of a patient with Covid-1-associated PE is generally a male patient, between 60-70 years of age, with obesity and immobilization, with cardiovascular and/or metabolic risk factors Covid-19-associated. A history of deep vein thrombosis and other risk factors associated with conventional PE is rare. Poor outcomes are associated with ICU, need for mechanical ventilation, long hospital stays, and right ventricular dysfunction [53]. On the other hand, conventional PE happens more in women aged 60 to 70 years, with a history of VTE and lower extremity disease or with a history of malignancy, postoperative status, late in pregnancy or the puerperium, or with a history of contraceptive use. The higher frequency in men has been associated with the protective effect that estrogens have on the integrity and immunity of the vascular endothelium (higher T cells) in women. It appears for this reason that the cytokine storm is more dramatic in the male gender (low T cells and more expression of ACE-2). In addition, men accumulate more risk factors and comorbidities for PE than women due to their lifestyle [54]. Another significant difference between both types of PE is the fact that in contrast to conventional PE, more patients with COVID-19-associated PE developed PE despite receiving standard-intensity thromboprophylaxis. In a Spanish study that included patients treated in emergency departments before hospitalization, COVID-19-associated PE had a higher incidence when compared with conventional PE (310 vs. 35 per 100,000 person-years), representing an almost ninefold increase in risk[44]. Remarkably, these rates may reflect the studied sample incidence rather than the actual disease incidence in the overall population [55].At the intra-hospital level, Covid-19 associated PE is responsible for 16.0% of all causes of death and 18.2% of ICU admissions. This contrasts with conventional PE which is responsible for 6.5% of all causes of death and 11.4% of ICU admissions. Another aspect that establishes differences between both types of PEs is that in conventional PE between 2.5-4% can evolve to CTEPH (chronic thromboembolic pulmonary hypertension). In Covid-19-associated PE the relationship with CTEPH and long-Covid-19 is unknown [56]. For these reasons, some authors have proposed that COVID-19-associated PE exhibits a different disease phenotype than conventional PE [57]. There is no clear a rapid diagnostic strategy that allows differentiating between classic PE and immune thrombosis. The probabilities of the other two vascular complications of Covid-1 9 associated PE, which are chronic thromboembolic disease and pulmonary hypertension secondary to diffuse interstitial lung disease produced by SARS-CoV-2, are not known [58,59].
D-dimers
In its purest conception, D-dimers are not a test of coagulation but of fibrinolysis (is a fragment of the fibrillar protein). When fibrin is degraded, D-dimers are released. Fibrin has molecules with D and E domains. The D are in the central part of the fibrin mesh (cross-linked by coagulation factor III) and the E are on the periphery. In addition, the E-dimer bond is single, but the D-dimer bond is double, in such a way that when plasmin fractures the fibrin, the single bonds are easily broken but the double bonds, which are the D-dimers, persist. The measurement is made by means of a monoclonal antibody directed towards a particular epitope of D-dimers. Its measurement is used in VTE, PE and DIC [60]. When elevated by clot formation that is later lysed, D-dimers rise 1 hour after formation and remain elevated for about 7 days if there is no recurrence of the clot [61]. Normally there is formation of small clots in the circulation and rupture by fibrinolysis so that they do not produce pathology. Therefore, there is a normal value of D-dimers in plasma (<500 ng/ml [nanograms/milliliters]). D-dimers are a marker of clot rupture and not necessarily of clot formation, but if they are elevated it is assumed that there is a hypercoagulable state [62]. In pulmonary thromboembolism not associated with Covid-19, values less than 500 ng/ml (nanograms/milliliter ) practically rule out PE if the pretest probability of PE is low according to predictive scales, but higher values do not confirm it because these dimers may be elevated due to the activation of the fibrinolytic system normal at a faster rate or fibrin mesh was formed in a compartment outside the blood, as in an inflammatory process and the fibrin is being broken down and the dimers (which are absorbed into the vascular compartment) will be elevated, but there is no PE [63]. Although it is true that they have a sensitivity of 95% (very low false negative rate) so they are helpful in ruling out classic PE (particularly if the pretest probability of PE is low), they have very low specificity (less than 50%) and therefore high rates of false positives [64]. There are many clinical scenarios and medical situations in which D-dimers can be elevated, without the necessary presence of PE. I cite a few examples. Old age, nicotine use, pregnancy, contraceptives, ethnicity (African American), exercise, recent trauma, or surgery (hematoma), atrial fibrillation, acute coronary syndrome, sickle cell crisis, malignancy, infection (severe pneumonia), renal failure, liver dysfunction, myocardial infarction, cerebral thrombosis and DIC. Therefore, its elevated levels are not specific for venous thrombosis and PE. An inflammation of the lung by Covid-19 will produce a sequestration to the lung, from the blood by means of inflammatory molecules (cytokines), of fibrin and its precursor (fibrinogen) with the aim of trapping inflammatory cells. When this fibrin (extravascular, in the lung and not in the blood), degrades due to fibrinolysis that also occurs in that compartment) D-dimers will be produced that will pass into the blood and give the positive test, without necessarily the patient have thrombosis and intravascular emboli (false positive). Elevated D-dimers in patients with pulmonary inflammation by Covid-19 do not discriminate whether the patient has pulmonary thromboembolism [65]. They can tell us if they are elevated about the extent of the inflammation, but they are not a reliable sign of thrombosis. The usefulness of this test in pulmonary thromboembolism in Covid-19 is a question without an answer yet. If a patient has a pulmonary thromboembolism (without pneumonia), they usually appear elevated at the time of the event, then remain elevated for 7 days afterward, and then return to normal. But if the patient has persistent inflammation in the lung, they will remain elevated whether the patient had thromboembolism [66].
The other problem with this marker in Covid-19 is that the normal values are not known. The 500 ng/ml are validated and accepted for conventional thromboembolism, but in those associated with Covid-19, that value is used by extrapolation, but in reality, no one knows the exact cut-off point from which they should be considered high. The PE D-dimer threshold not associated with Covid-19 should not be used. The statement “the diagnostic accuracy of d-dimer testing in patients with coronavirus disease 2019 (Covid-19) remains unchanged” could expose patients to unnecessary CTPA [67]. Published papers give “elevated” values as low as 350 ng/ml to as high as 10,000 ng/ml, and it is also not known whether these values should be adjusted for the patient's age, pregnancy, or other conditions that may impact the normal value [68-73]; values and settings if known for D-dimers in classic PE. It’s clear that elevation of D-dimers in covid-19 is a predictor of severity and mortality, but it must be understood that this predictive value does not necessarily depend on the presence of PE but rather on the extent of the inflammatory process. Of course, if PE is added to pneumonia the prognosis and mortality will increase. But a CTPA, a lung ventilation/perfusion scintigraphy or a venous ultrasound of the lower limbs should not be indicated only for an isolated elevated value of D-dimers. It is the clinical picture, the serial trend towards increasingly elevated values of D-dimers and the evolution that should guide decision-making. Nor should a patient be anticoagulated solely for an isolated elevated D-dimer value. The "cut-off point" for D-dimers in Covid-19 has not yet been established and future studies of this biomarker are required to define its role in diagnosis and anticoagulation in Covid-19 associated PE.
Computed Tomography Pulmonary Angiography (CTPA)
In conventional PE noninvasive tests to rule out the diagnosis that are based on the assessed clinical probability of pulmonary embolism are extremely effective in safely reducing the use of computed tomography pulmonary angiography (CTPA) resulting in only 30 to 40% of patients with suspected pulmonary embolism subsequently undergoing diagnostic imaging [74,75]. These data are not so clear in Covid-19 associated PE. The anatomical location of Covid-19-associated PE has been related to different radiologic patterns compared with conventional PE. Covid-19-associated PE has been particularly observed in segmental and subsegmental pulmonary arteries, whereas a smaller proportion of patients, compared with conventional PE, have thrombi int the main pulmonary arteries (ie, central vasculature) [76,77]. To illustrate the concept, the pulmonary vascular tree consists of a main pulmonary artery, two pulmonary arteries, five lobar arteries, eighteen segmental arteries, and hundreds of subsegmental arteries where the precision of radiological studies is reduced. These findings align with the lower frequency of DVT observed in patients with Covid-19-associated PE and “in situ” thrombosis in the distal pulmonary vasculature on postmortem analyses [78,79]. The incidence of false positive results from CT screening varies among providers and may be as high as 5% [80]. Small artery defects visualized on CTPA may be technique artifacts, and inaccurate interpretation leads to reporting of indeterminate findings. Movement artifacts (lines), adjacent lymph nodes, peripheral mucus plugs that compress the arteries can be confused with peripheral embolism or "in situ" thrombosis. Also, the inability to take a deep breath can harm the quality of the image and the reliability of the report [81].
CTPA has the risk of exposing the breasts and lungs to radiation and can worsen kidney damage if the patient has previous risk factor nephropathy (DM/HTA) or kidney damage from the SARS-Cov-2 virus. In this sense, the risk of inducing kidney injury is 0% if eGFR (estimated glomerular filtration rate) is >45 milliliters/minute/1.73 m2 (ml/m). If eGFR is between 30-44 ml/m/1.73m2 the risk varies between 0-2% but it is uncertain if the eGFR is less than 30 ml/m/1.73 m2. Prophylaxis with intravenous saline solution is indicated in these cases [82]. CT pulmonary angiography is usually the most timely and accessible imaging technique; however, to minimize lung and breast-tissue irradiation in younger patients, ventilation–perfusion single-photon-emission CT (SPECT) is a low-radiation option. CTPA failure rates are higher in Covid-19-associated PE (compared with conventional PE) and there are no robust data to justify screening CTPA, regardless of coagulation profile. In other words, a CTPA is not requested because D-dimers or another coagulation marker are elevated, unless the patient has a clinical suspicion of PE. If PE is not suspected, routine CTPA can lead to overdiagnosis of Covid-19-associated PE (false positives) and unnecessarily expose patients to anticoagulation with the risk of complications. This leads us to another well-defined concept: the performance of the CTPA must be left to the physician's discretion [83].
PE Predictive Scales
In conventional PE, in which physicians have an implicit sense that their patient is very unlikely to have pulmonary embolism (estimated likelihood, <15%), large cohort studies have shown that the Pulmonary Embolism Rule-out Criteria (PERC) rule can safely rule out pulmonary embolism without further diagnostic imaging [84]. In practice, however, implicit estimation typically overestimates the probability of pulmonary embolism, which can limit the use of the PERC rule [85]. Among patients with a low structured clinical probability score — a Wells score of 4.0 or less (found in 80% of patients tested in North America) [60], a revised Geneva score of 10 or less (on a scale ranging from 0 to 22, with higher scores indicating a greater probability of pulmonary embolism), and a simplified Geneva score of 4 or less (on a scale ranging from 0 to 9, with higher scores indicating greater probability of pulmonary embolism) — pulmonary embolism can be safely ruled out on the basis of D-dimer levels when manufacturer-recommended cutoffs were used (sensitivity, 98 to 99%; specificity, 37 to 40%) [64]. The YEARS algorithm uses adjusted D-dimer threshold for ruling out pulmonary embolism (sensitivity, 96 to 98%; specificity, 54 to 61%) [86]. In Covid-19-associated PE, the sensitivity and specificity of these scales are not reliable and have not been validated, especially in critically ill patients, so it is often unavoidable to reach CTPA in this subgroup of patients [87-90]. Many variables that make up the predictive scales (they are present or absent) are compromised by the severe inflammatory component of Covid-19 without there necessarily being PE.
In the broad population of patients with COVID-19, several risk factors have been associated with increased short-term mortality. These factors include elevated D-dimer, C-reactive protein (CRP), cardiac troponin I, dementia, diabetes mellitus with insulin treatment, chronic obstructive pulmonary disease, peripheral arterial disease, active smoking, chronic liver disease, and rheumatologic diseases [91,92]. Over 300 prediction models have been described, but many are of limited utility [93]. Studies assessing risk factors for poor outcomes in patients with COVID-19-associated PE are limited. Patients with COVID-19-associated PE may have a higher risk of ICU admission, mechanical ventilation, and prolonged hospitalizations than patients without PE. Moreover, a higher simplified Pulmonary Embolism Severity Index score and the presence of right ventricle dysfunction have been related to an increase in in-hospital mortality [55].
Sudden death
Death that occurs unexpectedly (not anticipated) and that generates a catastrophic and challenging duel is defined as such. In terms of time, death appears suddenly 24-48 hours after the onset of symptoms, which may even be non-significant or non-specific. For example, 25% of acute myocardial infarctions (AMI) that have sudden death do not have coronary pain [94,95]. The 5 most common causes of sudden death are: AMI, cardiac arrhythmias, stroke, aortic catastrophe, and PE. Sudden death can be the first manifestation of PE in 25% of cases and in cases of fulminant PE, 90% of deaths occur within 1-2 hours of the onset of symptoms. Massive pulmonary thromboembolism may be a significant cause of death in COVID-19 infection [96,97]. In addition, SARS-CoV-2 can cause severe pneumonia, generate cardiac arrest due to myocardial ischemia or a poor response to myocardial hypoxia. Cardiomyopathy due to sepsis can also lead to sudden death and in asthmatic patients with infection if there is a cardiomyopathy of unknown origin [98]. Many pulmonary emboli invariably prevent diagnosis because of their occult nature or because they cause rapid death from cardiopulmonary arrest. PE remains an elusive diagnosis despite technological advances, extensive research, and high indexes of suspicion. We still do not know what is the real percentage of pulmonary embolisms that debut as a sudden death in Covid-19.
Chronic Covid-19
The World Health Organization (WHO) defines chronic Covid-19 (long-Covid-19) as the persistence or onset of symptoms in the first 3 months of a confirmed or probable diagnosis of SARS-CoV-2, lasting at least 2 months without an alternate explanation. The British define it as the presence of such symptoms for more than 12 weeks after diagnosis. More than 50 symptoms presumably attributable to this chronic complication have been described, but the most frequent are: fatigue (very similar to chronic fatigue syndrome caused by other viruses), dyspnea, joint pain, and chest pain. Another frequent finding is cognitive deficit ("brain fog"). Chronic Covid-19 occurs in 10-20% of patients who have had Covid-19, generally in women, with severe disease, and not vaccinated [99]. The definition is strictly descriptive and in terms of time, since neither its pathophysiology, diagnostic test, specific treatment nor why some patients develop it and others do not are known. It is debilitating and disabling and there are not enough data to show thyroid hormone compromise, although it does show adrenal insufficiency (due to chronic use and subsequent suspension of steroids), and diabetes mellitus (pancreatic invasion by the virus). POTS (posterior orthostatic tachycardia syndrome) is part of chronic Covid-19, CFS (chronic fatigue syndrome) and MEL (myalgia encephalomyelitis) [100]. Respiratory sequelae may appear as early as 2 weeks after hospital discharge, and pulmonary fibrosis, arrhythmias, and cardiomyopathies, 2 months into the post-discharge recovery period. For this reason, at 1-2 months, if there are persistent or new symptoms, do a respiratory function test, CAT, and cardiac studies to try to document chronic sequelae [101].
It is estimated that 10 million North Americans have chronic Covid-19. The reports of evolution towards pulmonary fibrosis give very dissimilar results (28-77%) possibly because the diagnostic criteria are not standardized, but 33% at 3 months and 15.7% at 6 months of patients who had Covid-19 they have altered respiratory function. Little is known regarding appropriate patient selection for and clinical outcomes with lung transplantation for respiratory failure due to Covid-19. In USA, approximately 7% of lung transplantations nationally were performed (August 2020 through September 2021) in patients with respiratory failure due to Covid-19. Because the 3-month survival among these patients approached that among patients who underwent lung transplantation for reasons other than Covid-19, we believe that lung transplantation may be an acceptable treatment for selected patients with irreversible respiratory failure due to Covid-19 [102]. The considerations that should be carefully weighed when assessing a patient with COVID-19-associated ARDS regarding potential candidacy for lung transplantation have been delineated as well as the ethical aspects of the tool [103-106]. In a placebo-controlled trial, treatment with either alone phosphodiesterase 4B Inhibitor or with background use of an antifibrotic agent, prevented a decrease in lung function in patients with idiopathic pulmonary fibrosis [107]. The impact that this strategy could have on pulmonary fibrosis associated with Covid-19 is unknown.
A truly controversial aspect of chronic Covid-19 is whether a chronic hypercoagulable state in various organs or systems of the human economy plays any role in its pathophysiology. Additionally, it is still unclear whether PE following COVID-19 infection is a manifestation of the post-COVID-19 condition or not. Reviewing the literature, though, only a few data from small studies or case reports support the increased risk of PE after COVID-19 infection. In a database review the minimum reported days after diagnosis is 7 days and the maximum is 180 days (mean 35.1 days), but most of the cases the time of clinical manifestation of PE is during the first month following the COVID-19 infection [108,109]. Studies in post-Covid-19 discharged patients have shown elevation of D-dimers up to 4 months after discharge when all other inflammatory and coagulation parameters have normalized. Patients who underwent CTPA did not show PE despite this chronic elevation of convalescent D-dimers [110]. This exposes the lack of specificity of this parameter in the evaluation of chronic Covid-19 [111]. The European Society of Cardiology is clear in stating that the risk of VTE after hospital discharge is like the risk of other hospitalized patients without Covid-19, and that VTE in non-hospitalized patients is rare. VTE and arterial thromboembolism were the same between 14,000 Covid-19 positive patients and 15,000 covid-19 negative controls. It has not been shown that micro and macro-thrombosis play an etiologic and pathogenic role in chronic Covid-19. In fact, after discharge, serial coagulation tests are not recommended [112]. For this reason, prophylactic anticoagulation is not recommended in the outpatient setting [113]. In conclusion, there is no hard evidence proving that a medical history of Covid-19 will increase the risk for PE in chronic Covid-19 but some indications that need to be further investigated. Further studies are needed to question a potential causal link between “post-COVID-19 PE” and preceding SARS-CoV-2 infection and to outline the high-risk period after the COVID-19 convalescence phase regardless of the severity of the infection.
Anticoagulation
Conventional anticoagulants (oral or injected) are strictly preventative. If used prophylactically, they aim to prevent clot formation. If it is administered therapeutically, it is because the clot or embolism is already present and it is used to prevent the propagation and amplification of the thrombus, but they do not lyse the clot as such. Either vitamin K antagonists (vitamin K is a cofactor in the synthesis of coagulation factors II, VII, IX and XI) and activated factor X inhibitors (direct oral anticoagulants [DOACs]) or unfractionated (UFH), fractionated (hence low molecular weight) heparins (LMWH), and Fondaparinux are included in that statement.Native heparin is a polymer with a molecular weight ranging from 3 to 30 kDa, although the average molecular weight of most commercial heparin preparations is in the range of 12 to 15 kDa. This UFH—from the Greek ηπαρ (epar), liver— is a highly sulfated glycosaminoglycan which, when fractionated, produces low molecular weight fractionated heparins (LMWH) of around 4.5 KD [114]. Heparins work by stimulating antithrombin III (a natural anticoagulant) which inhibits thrombin from converting fibrinogen to fibrin. They also inhibit factor Xa , a common factor in the extrinsic and intrinsic pathways of coagulation. Fondaparinux is a synthetic heparin pentasaccharide with a sequence identical to that found in anticoagulant heparin and that accelerates the suicidal inactivation of factor Xa. It´s used in patients for whom heparin-based anticoagulation is not adequate due to its long half-life or reversibility problems [115].Figure 4 is a very simple diagram of coagulation to understand the level of preventive action of anticoagulants and figure 5 is a simple diagram of fibrinolysis to understand the site of action of plasminogen activator (which is used to lyse already formed clots) and where the plasminogen activation inhibitor (PAI-1) acts induced by the SARS-CoV-19 virus, which reduces fibrinolysis.
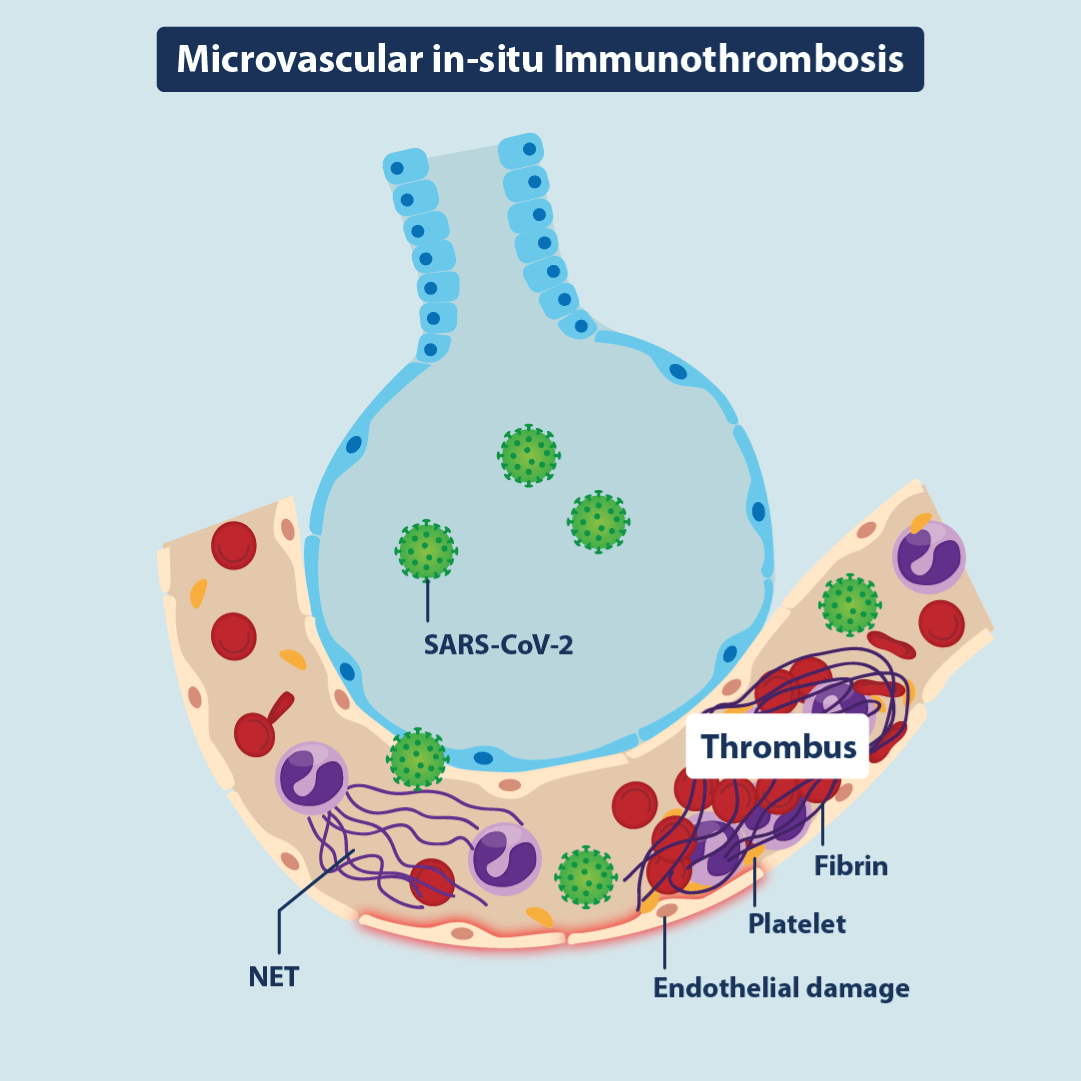
Figure 3. In situ immunothrombosis
Illustration demonstrating microvascular “in situ”immunothrombosis resulting from direct vascular and endothelial injury. NET = neutrophil extracellular trap.
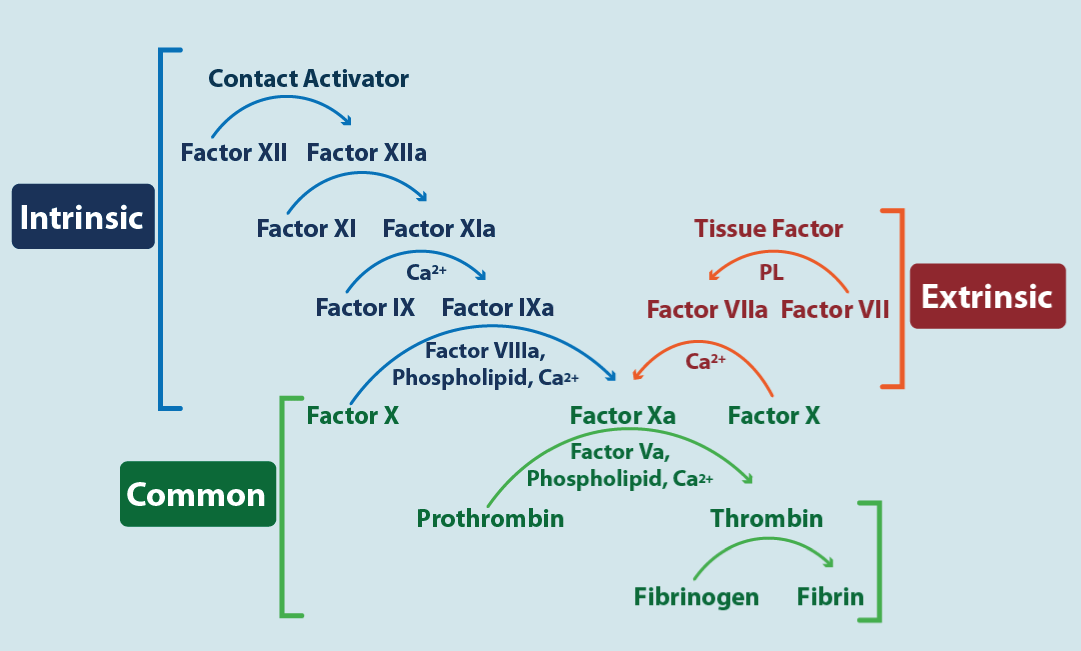
Figure 4. Cascade coagulation
The figure shows the three pathways of coagulation. In blue the intrinsic pathway, in red the extrinsic pathway and in green the common pathway. The image allows to identify (see text) where the various anticoagulants act.
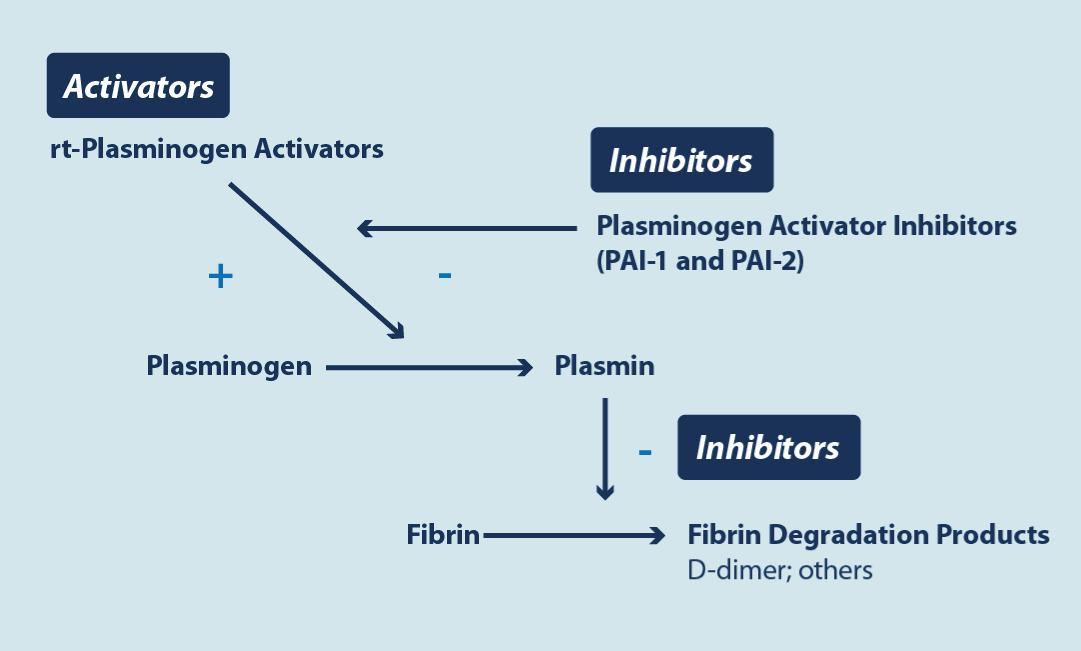
Figure 5. Fibrinolysis cascade
Basically, the image shows where rtPA (recombinant tissue plasminogen activators) act, which are used to lyse the clot and where the plasminogen activator inhibitor (PAI-1) induced by the cytokines of the SARS-CoV-2 virus acts, reducing fibrinolysis, and therefore favoring the persistence of thrombosis.
Elements that carry a high risk of bleeding during anticoagulation are Hemoglobin < 8 g/L; platelet count < 50x109/L; previous use of dual antiplatelet therapy, active bleeding in the last 30 days; bleeding disorders (inherent or acquired).
Prophylactic anticoagulation
In outpatients with Covid-19 (without PE) prophylactic anticoagulation was not significantly associated with a reduction in PE compared with placebo. This occurs with oral anticoagulants or with LMWH, therefore, it is not indicated, ex officio, to prophylactically anticoagulated outpatients with Covid-19 [116-118]. In patients discharged from the hospital due to Covid-19 without PE (and who were prophylactically anticoagulated), outpatient prophylactic anticoagulation or antithrombotic therapy (acetylsalicylic acid or P2Y12 inhibitors [mostly clopidogrel]) is not recommended unless there are other prior indications for its use [119]. In hospitalized noncritically ill patients (without PE) with low doses of oxygen, elevated D-dimers, and low risk of bleeding, who are in the general ward and not in the ICU, anticoagulation with heparins is preferred to prophylactic oral anticoagulation due to less drug-drug interaction, shorter half-lives, allowing its effects to be rapidly reversed, and that DOACs have a higher risk of bleeding. Both heparins can be used although LMWH is preferred as it requires less monitoring and administration occurs at longer intervals which reduces the risk of staff exposure. If there is a risk of bleeding, UFH is preferred. UFH 5,000 subcutaneous units/12 hours is an alternative. LMWH can be administered prophylactically in two schedules. For example, standard enoxaparin (widely used in the Western world) is dosed at 0.5 mg/Kg/once a day (standard prophylaxis) or 1 mg/Kg/once a day (extended, elevated, or increased prophylaxis). “Therapeutic” LMWH refers to double the increased dose, that is, 1 mg/Kg/2 times a day (but is being used prophylactically in this scenario) (for CrCl >30 mL/min). 1 mg equals 100 units in European dosage. Therapeutic dose of heparin is preferred in this type of patient. It should be remembered that the risk of PE in hospitalized patients is high even with prophylactic standard anticoagulation [120]. This scheme must be received during the stay in the general ward until any of 2 events occur; that he be transferred to the ICU or that he be discharged. The use of a prophylactic dose of heparin is recommended for patients who do not meet the criteria for receiving therapeutic heparin or are not receiving a therapeutic dose of heparin for other reasons unless a contraindication exists.
For adults who require ICU-level care (without PE), including those receiving high-flow oxygen it is recommended a prophylactic standard dose of heparin as VTE prophylaxis unless a contraindication exists. For patients who start on a therapeutic dose of heparin in a non-ICU setting due to COVID-19 and then transfer to the ICU, it is recommended switching from the therapeutic dose to a prophylactic standard dose of heparin, unless VTE is confirmed. It is not recommended the use of an intermediate dose (e.g., enoxaparin 1 mg/kg once daily) or a therapeutic dose of anticoagulation for VTE prophylaxis (1mg/kg 2 times a day) [121]. There are two reasons for this behavior. First, these last two regimens have not been shown to be superior to standard doses of prophylactic heparin in ICU patients, and second, there is a high risk of bleeding in this subgroup, so aggressive prophylactic anticoagulation may be potentially harmful. In an older patient, central nervous system bleeding can be devastating.
It is recommended that pregnant patients who are receiving anticoagulant or antiplatelet therapies for underlying conditions continue these medications after they receive a diagnosis of COVID-19, and the use of prophylactic dose of LMWH for pregnant patients who are hospitalized for manifestations of COVID-19, unless a contraindication exists. In pregnant women, LMWH is preferred over vitamin K antagonists and DOACs, since oral anticoagulants cross the placenta and are associated with adverse effects and teratogenicity during pregnancy. As in nonpregnant patients, VTE prophylaxis after hospital discharge is not routinely recommended for pregnant patients. UFH, LMWH, and warfarin do not accumulate in breast milk and do not induce an anticoagulant effect in the newborn; therefore, they can be used by breastfeeding individuals who require VTE prophylaxis or treatment [122]. Therefore, it is recommended prophylactic anticoagulation for children aged ≥12 years who are hospitalized for COVID-19, unless there are contraindications. In a patient with Covid-19 who is going to receive anticoagulation in addition to the coagulation profile (PT, TPT, platelets, fibrinogen, fibrinogen degradation product, and D-dimers), kidney and liver function tests should be requested. Before prescribing ritonavir-boosted nirmatrelvir (Paxlovid) to patients who are receiving anticoagulant or antiplatelet therapy, clinicians should carefully review the patient's concomitant medications to evaluate potential drug-drug interactions. It may be necessary to modify the dosage of the antithrombotic agent, switch to another antithrombotic agent, or prescribe an alternative COVID-19 therapy. For assess the extent of inflammation, baseline CRP, ferritin (30-400 ng/ml [nanograms/milliliter]), D-dimers, and IL-6 (if assay available) is desirable, prior to starting anticoagulation [123]. Anticoagulated patients can be vaccinated. It is recommended to use a number 23 needle and pressure at the injection site for 2 minutes.
Therapeutic anticoagulation
Initial treatment of pulmonary embolism is guided by risk stratification of the pulmonary embolism as high, intermediate, or low risk based on the patient’s clinical presentation. The nomenclature of “massive” and “sub-massive” in describing pulmonary embolism is confusing, given that clot size does not dictate therapy(124). The therapeutic approach in COVID-19 associated PE is like conventional PE (until new information appears). In low or moderate risk patients there is no significant hemodynamic compromise.
High Risk
This risk occurs in patients with shock, end-organ hypoperfusion, hypotension (systolic blood pressure of <90 mm Hg or a decrease in systolic blood pressure of >40 mm Hg that is not caused by sepsis, arrhythmia, or hypovolemia), or cardiac arrest (5%). Intravenous systemic thrombolysis is the most readily available option for reperfusion, and protocols include a weight-based dose of Tenecteplase (fibrinolytic agent, plasminogen activator), a dose of 0.6 mg per kilogram of body weight, or Alteplase at a dose of 100 mg administered over a period of 1 to 2 hours [124-126]. Alteplase (rt-PA) is a recombinant protein of the naturally occurring t-PA. After human t-PA was cloned and expressed in E. coli and in mammalian cells, it became the first commercially available recombinant protein drug that was entirely produced in mammalian cells. Since then, alteplase has been widely used as a fibrinolytic agent. There is insufficient evidence to support one of these agents over the other; however, Tenecteplase can be administered as a bolus in an emergency, and weight-based dosing may be preferable in elderly patients or patients with low body weight. Contraindications are the presence of brain metastases, bleeding disorders, and recent surgery [81]. Alternative reperfusion approaches include surgical thrombectomy and catheter-directed thrombolysis (with or without thrombectomy). Additional supportive measures include the administration of inotropes and the use of extracorporeal life support. Upon completion of the plasminogen-activating factor infusion, a UFH infusion (1000-1200 units per hour) is started and titrated according to the aPTT (activated partial thromboplastin time). aPTT should be maintained between 45-60 seconds if there is a risk of bleeding and between 60-85 seconds if there is not. Clinical and hemodynamic improvement will dictate the UFH infusion time (48-72 hours) and if the patient stabilizes, oral anticoagulants can be started prior to discharge if an invasive procedure is not anticipated and there is no significant drug-drug interaction.
Intermediate Risk
In conventional PE, patients with echocardiographic or CT evidence of right heart strain, elevated cardiac biomarkers (such as troponin or brain natriuretic peptide), or both are considered to have intermediate-risk pulmonary embolism (they are hemodynamically stable) [124]. However, the utility of transthoracic echocardiography in the diagnosis of PE in Covid-19 is not clear, since right ventricular dysfunction is a hallmark of severe disease and elevated troponin (which usually translates to right ventricular stretch) may be elevated by severe virus infection. If the right ventricular dysfunction is severe or there are signs of pulmonary hypertension, the diagnosis of PE is considered [119]. Systemic thrombolysis is not typically recommended for these patients due to increased risk of major bleeding, hemorrhagic stroke, and poor absolute risk reduction. LMWH (once or twice a day) is the preferred immediate anticoagulant for patients with intermediate-risk pulmonary embolism. The therapeutic effects of immediate treatment with direct oral anticoagulants (rivaroxaban and apixaban) as compared with low-molecular-weight heparin have not been studied in patients at intermediate risk for pulmonary embolism, and UFH causes excess bleeding [127]. There is insufficient evidence to support catheter-directed thrombolysis over low-molecular-weight heparin in these patients. Patients with intermediate or high risk for PE should be treated in hospitals. Low-molecular-weight heparin should be administered for 5 to 10 days. After stabilization, discharge with oral anticoagulants is considered.
Low risk
Patients with pulmonary embolism whose conditions are hemodynamically stable and who have no right ventricular strain and normal cardiac biomarkers are considered to have low-risk pulmonary embolism. Most of these patients can be treated with DOACS (based on high-quality trial data) and assessed for outpatient treatment [128]. Unlike conventional PE, where the Pulmonary Embolism Severity Index (PESI) or the HESTIA score can guide the decision to treat a patient at home or not, in Covid-19 associated PE there is no checklist of criteria that preclude treatment at home, or that predicts the risk of death [129,130]
In the subsequent management DOACs are the first-line treatment for most patients. Randomized trials have shown that direct oral anticoagulants, which do not necessitate monitoring, are as effective at reducing the risk of recurrent venous thromboembolism as vitamin K antagonists and result in a lower risk of major bleeding [128]. Because comparisons of direct oral anticoagulants are lacking, the choice of agent is guided by pharmacologic properties and patient characteristics and preferences [131]. In patients with cancer, DOACs are alternatives to treatment with low-molecular-weight heparin [132,133]. In conventional PE the standard duration is 3-6 months. In COVID-19 associated PE the minimum time established is 3 months. The maximum is not known and therefore the extension beyond 3 months will depend on the evolution of the patient, the possibility of recurrences and the clinical judgment of the physician. The decision to continue treatment indefinitely depends on whether the associated reduction in the risk of recurrent venous thromboembolism outweighs the increased risk of bleeding and should consider patient preferences, although the risk factors for each type of PE were different. If a decision to continue anticoagulation indefinitely was made at the time of diagnosis of pulmonary embolism, this decision should be reassessed annually or more often; anticoagulation may need to be discontinued if the risk of bleeding increases, a major bleeding event occurs, or the patient prefers to stop treatment. Adverse effects of anticoagulation are bleeding, hyperkalemia, and mucosal hypersensitivity. Patients should be followed longitudinally after an acute pulmonary embolism to assess for dyspnea or functional limitation, which may indicate the development of post–pulmonary-embolism syndrome or chronic thromboembolic pulmonary hypertension [80]. DOACs and low-molecular-weight heparin are contraindicated in patients with severe renal impairment (CrCl < 30 ml/m) or severe liver disease and in patients with antiphospholipid syndrome who are triple-positive (i.e., positive for lupus anticoagulant, anticardiolipin, and anti–β2-glycoprotein I antibodies), have very high antibody titers, or have a history of arterial thrombosis [134]. Tables 1 and 2 of the excellent and recent work by Kahn contain anticoagulant treatment regimens for PE and summary of key guideline recommendations for treatment of PE [80]. About use of direct oral anticoagulants in patients with obesity, post hoc analyses of phase 3 trials, observational data, and pharmacokinetic and pharmacodynamic data suggest that DOACs and vitamin K antagonists have similar effectiveness and safety in patients with body weight up to 120 kg or a body-mass index (BMI; the weight in kilograms divided by the square of the height in meters) of up to 40. For patients who weigh more than 120 kg or have a BMI higher than 40, standard doses of rivaroxaban or apixaban are among appropriate anticoagulant options; fewer supportive data exist for apixaban than for rivaroxaban. Other options include vitamin K antagonists, weight-based low-molecular-weight heparin (administered according to manufacturer recommendations), and fondaparinux [135].
VITT (vaccine-induced immune thrombotic thrombocytopenia) has a risk of 1/1 million vaccinees and can cause both arterial and venous thrombosis, with very atypical venous thrombosis (including cerebral venous sinus) and generally occurs 2-3 weeks after the vaccine. Treatment based on IVIG (intravenous immune globulin= 1 g/Kg current weight/24 hours 2 doses) and methylprednisolone (1 mg/Kg current weight and the de-escalation to oral steroids) is usually very effective in reversing the process. It is recommended to use non-heparin anticoagulants such as fondaparinux or oral anticoagulants if VTE with PE occurs. This is because VITT and HIT (heparin-induced thrombocytopenia) share similar pathogenic mechanisms. There are antibodies (induced by the vaccine or heparin) against platelet phospholipid 4, causing platelet lysis and the release of membrane phospholipids that activate coagulation [136-138].
Conclusions
COVID-19-associated PE has some specific clinical characteristics compared with conventional non–COVID-19-associated PE, suggesting it can include a different disease phenotype. Further research is needed to clarify its true prognostic significance on fatal and nonfatal outcomes. Major scientific organizations provide conditional recommendations in favor of full-intensity over standard-intensity prophylactic anticoagulation in noncritically ill hospitalized patients with COVID-19 who have a low risk of bleeding. Although treatment of acute COVID-19-associated PE is generally like pre-COVID-19 management, careful attention must be given to potential drug–drug interactions between COVID-19 therapies and oral anticoagulants. Whether a particular direct oral anticoagulant is preferable for the treatment of pulmonary embolism is not known. The optimal type and duration of anticoagulation, long-term effects of COVID-19-associated PE on mortality or CTEPH, and the relationship with long COVID will require future investigation.
Source of economic support
No.
Conflict of interest
No.
Authorship
This work was only carried out by the author. Author AA contributed on the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
References
- Huang C, Wang Y, Li X, Ren L, Zhao J, et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497-506. [Crossref]
- Zhou F, Yu T, Du R, Fan G, Liu Y, et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 395:1054-1062.[Crossref]
- Dong E, Du H, Gardner L(2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 20:533-534.[Crossref]
- Ortega-Paz L, Capodanno D, Montalescot G, Angiolillo DJ (2021)Coronavirus disease 2019-associated thrombosis and coagulopathy: review of the pathophysiological characteristics and implications for antithrombotic management. J Am Heart Assoc 10: e019650.[Crossref]
- Bahraini M, Dorgalaleh A (2022) The impact of SARS-CoV-2 infection on blood coagulation and fibrinolytic pathways: a review of prothrombotic changes caused by COVID-19. Semin ThrombHemost48:19-30.
- Bikdeli B, Madhavan MV, Gupta A, Jiménez D, Burton JR, et al (2020) Global COVID-19 Thrombosis Collaborative Group. Pharmacological agents targeting thromboinflammation in COVID-19: review and implications for future research. ThrombHaemost120:1004-1024.
- Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermattet R, et al (2020) Endothelial cell infection and endotheliosis in COVID-19. Lancet395:1417-1418.[Crossref]
- Arévalos V, Ortega-Paz L, Rodríguez-Arias JJ, Calvo-López M1, Castrillo-Golvanoal L (2021) Acute and chronic effects of COVID-19 on the cardiovascular system. J Cardiovasc Dev Dis8:128.[Crossref]
- Poredos P, Poredos P (2022) Involvement of inflammation in venous thromboembolic disease: an update in the age of COVID-19. Semin ThrombHemost 48:93-99.
- Talasaz AH, Sadeghipour P, Kakavand H, Aghakouchakzadeh M, Kordzadeh-Kermani E, et al. (2021) Recent randomized trials of antithrombotic therapy for patients with COVID-19: JACC state-of-the-art review. J Am Coll Cardiol77:1903-1921.[Crossref]
- Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med383: 120-128.[Crossref]
- Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, et al (2020)Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two Centre descriptive study. LancetInfect Dis20:1135-1140.
- Woodard PK(2021) Pulmonary Thromboembolism in COVID-19. Radiology 298: E107-E108.[Crossref]
- Wu C, Liu Y, Cai X, Zhang W, Li Y (2021)Prevalence of Venous Thromboembolism in Critically Ill Patients with Coronavirus Disease 2019: A Meta-Analysis. Front Med8: 603558. [Crossref]
- Virchow R. (1856)GesammelteAbhandlungen Zur WissenschaftlichenMedizin. Frankfurt Am Taf20:1-1024.
- Bagot CN, Arya R (2008) Virchow and His Triad: A Question of Attribution. Br J Haematol143:180-190.[Crossref]
- Sakuma M, Nakamura M, Yamada N, Ota S, Shirato K, et al (2009)Venous Thromboembolism Deep Vein Thrombosis with Pulmonary Embolism, Deep Vein Thrombosis Alone, and Pulmonary Embolism Alone.Circ J 73:305-309.[Crossref]
- Benns M, Reilly P, Kim P (2014) Early Pulmonary Embolism after Injury: A Different Clinical Entity? Injury 45:241-244.[Crossref]
- Madani M, Ogo T, Simonneau G(2017) The Changing Landscape of Chronic Thromboembolic Pulmonary Hypertension Management. Eur Respir Rev26:170105.[Crossref]
- Zerangian N, Erabi G, Poudineh M, Monajjem K, Diyanati M, et al (2023) Venous thromboembolism in viral diseases: A comprehensive literature review. Health Sci Rep6: e1085.[Crossref]
- Alvarado A, Arce I (2016) Immune recognition in lung diseases: basic research and clinical application. Clinic Infect Immunity1:31-40.
- Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, et al (2020) Global COVID-19 Thrombosis Collaborative Group, endorsed by the ISTH, NATF, ESVM, and the IUA, supported by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC state of-the-art review. J Am Coll Cardiol75:2950-2973.[Crossref]
- Vityala Y, Krishna A, Pandla D, Kanteti K P, Sadhu J, et al (2022) Pathophysiology of thromboembolism in patients with COVID-19. Eur J Clin Exp Med2: 10.
- Sriram K, Insel PA (2021) Inflammation and thrombosis in Covid-19 pathophysiology: proteinase-active and purinergic receptors as drivers and candidate therapeutic targets. Physiol Rev101:545-567.[Crossref]
- Koupenova M (2020) Potential role of platelets in Covid-19: implications for thrombosis. Res PractThrombHaemost4:737-740.[Crossref]
- Saleh J, Peyssonraux C, Singh KK, Eder M (2020) Mitochondria and microbiota dysfunction in Covid-19 pathogenesis. Mitochondrion 54:1-7.[Crossref]
- Poor HD (2021) Pulmonary thrombosis and thromboembolism in Covid-19. Chest160:1471-1480.[Crossref]
- Jiménez D, García-Sanchez A, Rali P, Muriel A, Bikdeli B, et al (2021)Incidence of VTE and bleeding among hospitalized patients with coronavirus disease 2019: a systematic review and meta-analysis. Chest159:1182-1196.[Crossref]
- Nopp S, Moik F, Jilma B, Pabinger I, Ay C (2020) Risk of venous thromboembolism in patients with COVID-19: a systematic review and meta-analysis. Res PractThrombHaemost4:1178-1191.[Crossref]
- Ortega-Paz L, Galli M, Angiolillo DJ (2022) Updated meta-analysis of randomized controlled trials on the safety and efficacy of different prophylactic anticoagulation dosing regimens in non-critically ill hospitalized patients with COVID-19. Eur Heart J Cardiovasc Pharmacother8: E15-E17.[Crossref]
- Law N, Chan J, Kelly C, Auffermann WF, Dunn DP (2022) Incidence of pulmonary embolism in COVID-19 infection in the ED: ancestral, delta, omicron variants and vaccines. EmergRadiol29:625-629.[Crossref]
- Katsoularis I, Fonseca-Rodríguez O, Farrington P. Jerndal H, Lundevaller EH, et al (2022)Risks of deep vein thrombosis, pulmonary embolism, and bleeding after COVID-19: nationwide self-controlled cases series and matched cohort study. BMJ 377: e069590.
- Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J ThrombHaemost18: 844-847.[Crossref]
- Sakr Y, Giovini M, Leone M, Pizzilli G, Kortgen A, et al (2020)Pulmonary embolism in patients with coronavirus disease-2019 (COVID-19) pneumonia: a narrative review. Ann Intensive Care10:124.[Crossref]
- Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, et al. (2020)High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med46:1089-1098.[Crossref]
- Fauvel C, Weizman O, Trimaille A, Mika D, Pommier T, et al (2020)Critical COVID-19 France Investigators. Pulmonary embolism in COVID-19 patients: a French multicentre cohort study. Eur Heart J 41:3058-3068.[Crossref]
- Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, et al (2020)Thrombosis in hospitalized patients with COVID-19 in a New York City health system. JAMA 324:799-801.[Crossref]
- Zuin M, Engelen MM, Barco S, Zuin M, Spyropoulos AC, et al (2022)Incidence of venous thromboembolic events in COVID-19 patients after hospital discharge: a systematic review and meta-analysis. Thromb Res209:94-98.[Crossref]
- Connors JM, Brooks MM, Sciurba FC, Krishnan JA, Bledsoe JR, et al (2021) ACTIV-4B Investigators. Effect of antithrombotic therapy on clinical outcomes in outpatients with clinically stable symptomatic COVID-19: the ACTIV-4B Randomized Clinical Trial. JAMA326:1703-1712.[Crossref]
- Ananworanich J, Mogg R, Dunne MW, Bassyouni M, David CV, et al (2022)Randomized study of rivaroxaban vs placebo on disease progression and symptoms resolution in high-risk adults with mild coronavirus disease 2019. Clin Infect Dis75: e473-e481.[Crossref]
- Chan KH, Lin SL, Shaban H, Guron G, Slim J (2021) Persistent hypercoagulable state in Covid-19: a case series of Covid-19 associated pulmonary embolism. J Glob Infect Dis13:38-41. [Crossref]
- George PM, Barratt SL, Condliffe R, Desai SR, Devaraj IF, et al (2020) Respiratory follow-up of patients with Covid-19 pneumonia. Thorax 75:1009-1016.
- Xie Y, Xu E, Bowe B, Al-Ally Z (2022) Long-term cardiovascular outcomes in Covid-19. Nat Med 28:583.
- Roberts LN, Whyte MB, Georgiou L, Giron G, Czupryuska J, et al (2020) Post discharge venous thromboembolism following hospital admission with Covid-19. Blood136:1347-1350.[Crossref]
- Xie J, Prats-Uribe A, Feng Q, Wang Y, Gillet D, et al (2022) Clinical and Genetic Risk Factors for Acute Incident Venous Thromboembolism in Ambulatory Patients With COVID-19. JAMA Intern Med182:1063-1070.[Crossref]
- Fang, MC, Reynolds K, Tabada GH, Prasad PA, Sung SH, et al (2023) Assessment of the Risk of Venous Thromboembolism in Non-hospitalized Patients With COVID-19. JAMA Network Open6: e232338.[Crossref]
- Klock FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommerset D, et al (2020)Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res191:148-150.[Crossref]
- Song WJ, Hui CKM, Hull JH, Birring SS, McGarvey L, et al (2021) Confronting COVID-19-associated cough and the post-COVID syndrome: role of viral neurotropism, neuroinflammation, and neuroimmune responses. LancetRespir Med 9:533-544.[Crossref]
- Donina ZA (2022) Causes of Hypoxemia in COVID-19. J EvolBiochemPhysiol58:73-80.[Crossref]
- Koriyama N, Moriuchi A, Higashi K, Kataoka T, Arimizu T, et al (2022)COVID-19 with Rapid Progression to Hypoxemia Likely due to Imbalance between Ventilation and Blood Flow: A Case Report. Med Insights Circ Respir Pulm Med16: 11795484211073273.[Crossref]
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, et al (2020)Pathological findings of COVID-19 associated with acute respiratory distress syndrome. LancetRespir Med8:420-422.[Crossref]
- Inui S, Yoon SH, Doganay O, Gleeson FV, Kim M (2022) Impaired pulmonary ventilation beyond pneumonia in COVID-19: A preliminary observation. PLoS ONE17: e0263158.[Crossref]
- Ortega-Paz L, Tabsaz AH, Sadeghipour P, Potpara TS, Aronou HD, et al (2022)COVID-19-associated pulmonary embolism: review of the pathophysiology, epidemiology, prevention, diagnosis, and treatment. Semin ThrombHemost.[Crossref]
- Di Stasi V, Rastrelli G (2021) The Role of Sex Hormones in the Disparity of COVID-19 Outcomes Based on Gender. J Sex Med18:1950-1954.[Crossref]
- Miró Ò, Jiménez S, Mebazaa A, Freund Y, Burillo-Putze G, et al (2021)Spanish Investigators on Emergency Situations TeAm (SIESTA) network. Pulmonary embolism in patients with COVID-19: incidence, risk factors, clinical characteristics, and outcome. Eur Heart J42: 3127-3142.[Crossref]
- Sabbula B, Akella J (2022) Chronic thrombo-embolic pulmonary hypertension (Updated 2022. June. 27) In: Stat pearls (Internet) Treasure Island (FL): Stat Pearls Publishing. 2022 June. [Crossref]
- Wang L, Chen F, Bai L, Yi Q, Peng Y (2020)In situ pulmonary thrombosis in patients with COVID-19 pneumonia: different phenotypes may exist. Thromb Res196:541-542.[Crossref]
- Cueto-Robledo G, Graniel-Polafox LE, Roldan-Valadez E, Garcia-Cesar M, Torres-Rojas MB, et al (2022)Chronic thromboembolism pulmonary hypertension (CTEPH): a review of another sequel of severe post-Covid-19 pneumonia. Curr Prob Cardiol2022:101187.[Crossref]
- The Task Force for the management of Covid-19 of the European Society of Cardiology (2022) European society of cardiology guidance for the diagnosis and management of cardiovascular disease during the Covid-19 pandemic: part 1-epidemiology, pathophysiology, and diagnosis. Eur HearthJ 43:1033-1058.[Crossref]
- Kearon C, de Wit K, Parpia S, Schulman S, Afilalo M, et al (2019) Diagnosis of Pulmonary Embolism with d-Dimer Adjusted to Clinical Probability Diagnosis of Pulmonary Embolism. N Engl J Med381:2125-2134.[Crossref]
- Quinn DA, Fogel RB, Smith CD, Laposata MB, Thompson T, et al (1998) d-Dimers in the Diagnosis of Pulmonary Embolism.Am J Respir Crit Care Med159:1445-1449. [Crossref]
- Halaby R, Popma CJ, Cohen A, Chi G, Zacarkim MR, et al (2015) d-Dimer elevation and adverse outcomes.ThrombThrombolysis39:55-59.
- Silva BV, Rigueira J, Ricardo A, Mendonca C, Da Silva PA, et al (2021)Best approach in d-dimer algorithm to exclude pulmonary thromboembolism: a comparative study. Eur Heart J - Cardiovascular Imaging22: jeaa356.250.
- Geersing GJ, Takada T, Klok FA, Büller HR, Courtney D, et al (2022)Ruling out pulmonary embolism across different healthcare settings: a systematic review and individual patient data meta-analysis. PLoS Med19: e1003905-e1003905.[Crossref]
- Ramadan L, Koziatek CA, Caldwell JR, Pecoriello J, Christopher Kuhner C, et al (2021) Pulmonary thromboembolism in COVID-19: Evaluating the role of D-dimer and computed tomography pulmonary angiography results. Am J Emerg Med46:786-787.[Crossref]
- Brem FL, Asmae B, Amane Y, Bouazzaoui MA, Chaymae M, et al (2021) Diagnostic Accuracy of D-Dimers for Predicting Pulmonary Embolism in COVID-19-Patients. Clin Appl ThrombHemost27:1-7. [Crossref]
- Lehmann A, Prosch H, Zehetmayer S, Gysan MR, Bernitzky D, et al (2021) Impact of persistent D-dimer elevation following recovery from COVID-19. PLoS ONE16: e0256351.[Crossref]
- Elberts SJ, Bateman R, Koutsoubis A, London KS, White JL, et al (2021) The impact of COVID-19 on the sensitivity of D-dimer for pulmonary embolism. AcadEmerg Med28:1142-1149.[Crossref]
- Houghton DE, Wysokinska E, Casanegra AI, Padrnos L, Shah S, et al (2022) Accuracy and Prediction of D-Dimers for Pulmonary Embolism in Patients with COVID-19 Infection. Blood 140:5647-5648.[Crossref]
- Eljilany I, Elzouki A-N (2020) D-Dimer, Fibrinogen, and IL-6 in COVID-19 Patients with Suspected Venous Thromboembolism: A Narrative Review. Vasc Health Risk Manag16: 455-462.
- Močibob L, Šušak F, Šitum M, Višković K, Papić N, et al (2022) COVID-19 and Pulmonary Thrombosis—An Unresolved Clinical Puzzle: A Single-Center Cohort Study. J Clin Med11:7049.[Crossref]
- Rostami M, Mansouritorghabeh H (2020) D-dimer level in COVID-19 infection: a systematic review. Expert Rev Hematol13:1265-1275.[Crossref]
- Dong M, Chen S, Lin S, Hang F, Zhong M (2023) Insights into COVID-19-associated critical illness: a narrative review. Ann Transl Med11: 220. [Crossref]
- Khan MZ, Jamal Y, Sutton B, Rauf F (2020) Venous thromboembolism in patients with COVID-19 and correlation with D-dimers: a single-centre experience. BMJ Open Res7: e000779.[Crossref]
- Zhao R, Su Z, Komissarov A A, Liu S-L, Yi G. et al (2021) Associations of D-Dimer on Admission and Clinical Features of COVID-19 Patients: Review, Meta-Analysis, and Meta-Regression. Front Immunol12:691249.[Crossref]
- Lim W, Le Gal G, Bates SM, Righini M, Haramati LB, et al (2018) American Society of Hematology 2018 guidelines for management of venous thromboembolism: diagnosis of venous thromboembolism. Blood Adv2:3226-3256.[Crossref]
- Germini F, Zarabi S, Eventov M, Turcotte M, Li M, et al (2021)Pulmonary embolism prevalence among emergency department cohorts: a systematic review and meta-analysis by country of study. J ThrombHaemost19:173-185.[Crossref]
- Espallargas I, Rodríguez-Sevilla JJ, Rodríguez-Chiaradía DA, Salar A, Guillem Casamayor G, et al (2021)CT imaging of pulmonary embolism in patients with COVID-19 pneumonia: a retrospective analysis. EurRadiol31:1915-1922.[Crossref]
- van Dam LF, Kroft LJM, van der Wal LI, Cannegieter SC, Eikenboom J, et al (2020)Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: a different phenotype of thrombotic disease? Thromb Res193: 86-89.
- Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, et al (2020)Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med173:268-277. [Crossref]
- Kahn SR, de Wit K (2022) Pulmonary embolism. N Engl J Med388:45-47. [Crossref]
- Miller WT Jr, Marinari LA, Barbosa E Jr, Litt HI, Schmjit JE, et al (2015)Small pulmonary artery defects are not reliable indicators of pulmonary embolism. Ann Am Thorac Soc12:1022-1029.[Crossref]
- Davenport MS, Perazella MA, Yee J, Dillman JR, Derek Fine D, et al (2020) Use of Intravenous Iodinated Contrast Media in Patients with Kidney Disease: Consensus Statements from the American College of Radiology and the National Kidney Foundation. KidneyMed2: 85-93.[Crossref]
- El-Qutob D, Alvarez-Arroyo L, Barreda I, Nieto M, Pin M, et al (2022)High incidence of pulmonary thromboembolism in hospitalized SARS-CoV-2 infected patients despite thrombo-prophylaxis. Heart Lung 53: 77-82.[Crossref]
- Kline JA, Courtney DM, Kabrhel C, Moore CL, Smithline HA, et al (2008) Prospective multicenter evaluation of the pulmonary embolism rule-out criteria. J ThrombHaemost6:772-780.[Crossref]
- Zarabi S, Chan TM, Mercuri M, Kearon C, Turcotteet M, et al (2021) Physician choices in pulmonary embolism testing. CMAJ 193: E38-E46.
- van der Hulle T, Cheung WY, Kooij S, Beenen LFM, van Bemmelet T, et al (2017)Simplified diagnostic management of suspected pulmonary embolism (the YEARS study): a prospective, multicentre, cohort study. Lancet 390:289-297.[Crossref]
- Kirsch B, Aziz M, Kumar S, Burke M, Webster T, et al (2021) Wells Score to Predict Pulmonary Embolism in Patients with Coronavirus Disease 2019. Am J Med134:688-690.[Crossref]
- Girardi AM, Bettiol RS, Garcia TS, Ribeiro GLH, Rodrigues EM, et al (2020) Wells and Geneva Scores Are Not Reliable Predictors of Pulmonary Embolism in Critically Ill Patients: A Retrospective Study. J Intensive Care Med35:1112-1117.[Crossref]
- Quezada-Feijoo M, Ramos M, Lozano-Montoya I, Sarró M, Cabo Muiños V, et al (2021)Elderly Population with COVID-19 and the Accuracy of Clinical Scales and D-Dimer for Pulmonary Embolism: The OCTA-COVID Study. J Clin Med10:5433.[Crossref]
- Porfidia A, Mosoni C, Talerico R, Porceddu E, Lupascu A, et al (2021)Pulmonary Embolism in COVID-19 Patients: Which Diagnostic Algorithm Should We Use?Front Cardiovasc Med8:1-5. [Crossref]
- Bellou V, Tzoulaki I, van Smeden M, Moons KGM, Evangelou E, et al (2022) Prognostic factors for adverse outcomes in patients with COVID-19: a field-wide systematic review and meta-analysis. Eur Respir J59:2002964.[Crossref]
- Harrison SL, Buckley BJR, Rivera-Caravaca JM, Zhang J, Lip GYH (2021) Cardiovascular risk factors, cardiovascular disease, and COVID-19: an umbrella review of systematic reviews. Eur Heart J Qual Care Clin Outcomes7:330-339.[Crossref]
- No authors listed (2021) Update to living systematic review on prediction models for diagnosis and prognosis of COVID-19. BMJ 372: n236.[Crossref]
- Li Z, Wang Y, Li L, He H, Lin L, et al (2022) A bibliometric analysis of the cause of sudden unexplained death in forensic medicine: Research trends, hot spots, and prospects. Comp Biol Med144:105330.[Crossref]
- Markwerth P, Bajanowski T, Tzimas I, Dettmeyer R (2021)Sudden cardiac death—update. Internat J Leg Med135: 483-495. [Crossref]
- Keane G, Dorman T (2022) Fatal pulmonary thromboembolism in asymptomatic COVID-19. Ir J Med Sci 191:1777-1783.[Crossref]
- Polat V, Bostanci GI (2020) Sudden death due to acute pulmonary embolism in a young woman with COVID-19. J Thromb Thrombolysis50:239-241.[Crossref]
- Romanova ES, Vasilyev VV, Startseva G, Karev V, Rybakova MG, et al (2021) Cause of death based on systematic post-mortem studies in patients with positive SARS-CoV-2 tissue PCR during the COVID-19 pandemic. J InternMed290:655-665. [Crossref]
- Sarfraz Z, Sarfraz A, Barrios A, Garimella R, Dominari A, et al (2021)Cardio-pulmonary sequelae in recovered Covid-19 patients: consideration for primary care. J Prim Care Communit Health12:1-14.[Crossref]
- Wostyn P (2021) COVID-19 and chronic fatigue syndrome: Is the worst yet to come? Med Hypotheses146:1-5.[Crossref]
- Karoly M, Pawelka E, Kelani H, Maden T, Baumgartner S, et al (2021) Late onset pulmonary embolism in young male otherwise healthy. Eur J Clin Microbiol Infect Dis40:633-635.[Crossref]
- Roach A, Chikwe J, Catarino P, Rampolla R, Noble PW, et al (2022) Lung Transplantation for Covid-19-Related Respiratory Failure in the United States. N Engl J Med386:1187-1188.[Crossref]
- Cypel M, Keshavjee S (2020) When to consider lung transplantation for COVID-19. LancetRespir Med8:944-946.[Crossref]
- Holm AM, Mehra MR, Courtwright A, Teuteberg J, Sweet S, et al (2020)Ethical considerations regarding heart and lung transplantation and mechanical circulatory support during the COVID-19 pandemic: an ISHLT COVID-19 Task Force statement. J Heart Lung Transplant39:619-626. [Crossref]
- Hawkins RB, Mehaffey JH, Charles EJ, Mannem HC, Roeser M (2021)Lung transplantation for severe post-Coronavirus disease 2019 respiratory failure. Transplantation 105:1381-1387.[Crossref]
- Valapour M, Lehr CJ, Skeans MA, Smith JM, Miloler E, et al (2021) OPTN/SRTR 2019 annual data report: Lung Am J Transplant21:441-520.[Crossref]
- Richeldi L, Azuma A, Cottin V, Hesslinger C, Stowasser S, et al (2022) Trial of a preferential phosphodiesterase 4B inhibitor for Idiopathic Pulmonary Fibrosis. N Engl J Med386:2178-2187.[Crossref]
- Mouzarou A, Ioannou M, Leonidou E, Chaziri I (2022) Pulmonary Embolism in Post-Covid-19 Patients, a Literature Review: Red Flag for Increased Awareness? SN Compr Clin Med4: 190.[Crossref]
- Halawa S, Pullamsetti SS, Bangham CRM, Stenmark KR, Dormuller P, et al (2022) Potential long-term effects of SARS-CoV-2 infection on the pulmonary vasculature: a global perspective. Nat Rev Cardiol19:315-331.[Crossref]
- Gómez CA, Sun CK, Tsai IT, Chang YP, Lin MC, et al (2021) Mortality and risk factors associated with pulmonary embolism in coronavirus disease 2019 patients: a systematic review and meta-analysis. Sci Rep11:16025.
- Townsend L, Fogarty H, Dyer A, Martin-Loeches I, Bannan C, et al (2021) Prolonged elevation of D-dimers levels in convalescent Covid-19 patients is independent of the acute phase response. J ThrombHaemost19:1064-1070.[Crossref]
- The Task Force for the management of Covid-19 of the European Society of Cardiology (2022) European society of cardiology guidance for the diagnosis and management of cardiovascular disease during the Covid-19 pandemic: part 2-Care pathways treatment, and follow-up. Eur Hearth J43:1059-1103.[Crossref]
- Schreck K, Carlin C, Ricco J (2022) Risk of thromboembolic events following Covid-19 diagnosis. J Am Board Fam Med35:1163-1167.[Crossref]
- Bentolila A, Vlodavsky I, Haloun C, Domb AJ (2000) Synthesis and heparin-like biological activity of amino acid-based polymers. PolymAdvan Technol11:377-387.
- Zhang Y, Zhang M, Tan L, Pan N, Zhang L, et al (2019) The clinical use of Fondaparinux: A synthetic heparin pentasaccharide. Prog Mol Biol Transl Sci163:41-53.[Crossref]
- Connors JM, Brooks MM, Sciurba FC, Krishnan JA, Bledsoe JR, et al (2021)Effect of antithrombotic therapy on clinical outcomes in outpatients with clinically stable symptomatic COVID-19: The ACTIV-4B Randomized Clinical Trial. JAMA 326:1703-1712.
- Cools F, Virdone S, Sawhney J, Lopes RD, Jacobson B, et al (2022)Thromboprophylactic low-molecular-weight heparin versus standard of care in unvaccinated, at-risk outpatients with COVID-19 (ETHIC): an open-label, multicentre, randomized, controlled, phase 3b trial. LancetHaematol 9: e594-e604.[Crossref]
- Barco S, Voci D, Held U, Sebastian T, Bingisser R, et al (2022)Enoxaparin for primary thromboprophylaxis in symptomatic outpatients with COVID-19 (OVID): a randomized, open-label, parallel-group, multicentre, phase 3 trial. LancetHaematol 9: e585-e593.[Crossref]
- Cam Anh Tran Phan (2022)Management of venous thromboembolism in Covid-19. An overview of the current guidelines on anticoagulation management of Covid-19 associated thrombosis. Pharmac J.
- Tang N, Bai H, Chen X, Gong J, LI D, et al (2020) Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J ThrombHaemost18:1094-1099.[Crossref]
- Moores LK, Tritschler T, Brosnahan S, Carrier M, Collen JF, et al (2022)Thromboprophylaxis in patients with COVID-19: a brief update to the CHEST guideline and expert panel report. Chest 162:213-225.[Crossref]
- NIH (2022)Antithrombotic Therapy in Patients With COVID-19.COVID-19 Treatment Guidelines.
- Cuker A, Tseng EK, Nieuwlaat R, Angchaisuksiri P, Blair C, et al (2021) American Society of Hematology 2021 guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19. Blood Adv5:872-888.[Crossref]
- Konstantinides SV, Meyer G, Becattini C, Bueno H, Geersin G-J, et al (2020) 2019 ESC guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS).Eur Heart J41:543-603.[Crossref]
- Meyer G, Vicaut E, Danays T, Agnelli G, Becattini C, et al (2014)Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med370:1402-1411.[Crossref]
- Jimenez D, Martin-Saborido C, Muriel A, Zamora J, Morillo R, et al (2018)Efficacy and safety outcomes of recanalization procedures in patients with acute symptomatic pulmonary embolism: systematic review and network meta-analysis. Thorax73:464-471.[Crossref]
- Robertson L, Jones LE (2017) Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for the initial treatment of venous thromboembolism. Cochrane Database Syst Rev2:CD001100-CD001100.[Crossref]
- van Es N, Coppens M, Schulman S, Middeldorp S, Büller HR (2014) Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood124:1968-1975.[Crossref]
- Stevens SM, Woller SC, Kreuziger LB, Bounameaux H, Doerschug K, et al (2021)Antithrombotic therapy for VTE disease: second update of the CHEST guideline and expert panel report. Chest160: e545-e608.[Crossref]
- Roy P-M, Penaloza A, Hugli O, Klok FA, Arnoux A, Lee AYY, et al (2021)Triaging acute pulmonary embolism for home treatment by Hestia or simplified PESI criteria: the HOME-PE randomized trial. Eur Heart J 42:3146-3157.[Crossref]
- Chan N, Sobieraj-Teague M, Eikelboom JW (2020)Direct oral anticoagulants: evidence and unresolved issues. Lancet396:1767-1776. [Crossref]
- Key NS, Khorana AA, Kuderer NM, BohlkeK,Lee AYY,et al (2020)Venous thromboembolism prophylaxis and treatment in patients with cancer: ASCO clinical practice guideline update. J Clin Oncol38:496-520.[Crossref]
- Mulder FI, Bosch FTM, Young AM, Marshall A, McBane RD, et al (2020) Direct oral anticoagulants for cancer associated venous thromboembolism: a systematic review and meta-analysis. Blood 136:1433-1441.[Crossref]
- Dufrost V, Wahl D, Zuily S (2021) Direct oral anticoagulants in antiphospholipid syndrome: meta-analysis of randomized controlled trials. Autoimmun Rev20:102711-102711. [Crossref]
- Martin KA, Beyer-Westendorf J, Davidson BL, Huisman MV, Sandset PM, et al (2021)Use of direct oral anticoagulants in patients with obesity for treatment and prevention of venous thromboembolism: updated communication from the ISTH SSC Subcommittee on Control of Anticoagulation. J ThrombHaemost19:1874-1882.[Crossref]
- Greinacher A, Thiele T, Warkentin TE, W20eisse K, Kyrle PA, et al (2021) Thrombotic thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N Engl J Med384:2092-2101.[Crossref]
- Schultz NH, Sorvol IH, Michelsen AE, Munthe LA, Lund-Johansen F, et al (2021) Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N Engl J Med384:2124-2130.[Crossref]