Extraction of viral nucleic acids from serum samples is widely used in diagnostic pathology tests. However, the heterogeneous nature of non-serum samples may contribute to variations in the yields of viral nucleic acids with different extraction methods and specimen types. Five different nucleic acid extraction methods were compared for optimal extraction of viral DNA or RNA from a prepared cell-free specimen. The viruses used were hepatitis B, hepatitis C. The specimen used was DNase and RNase free normal saline spiked with predefined quantities of viral DNA or RNA. The extraction protocol was carried out according to each manufacturer’s recommendations. The extracted nucleic acids from each of the samples were amplified by PCR and compared to the original un-extracted standard control. The RTA kit as well as the Qiagen kit was shown to yield the highest amounts of viral nucleic acid.
viral DNA, viral RNA, molecular immunology, viral load, qPCR, qRT-PC
The ability to extract high purity viral nucleic acid, either DNA or RNA, is required for many downstream molecular and medical techniques used in research or diagnostic purposes. The efficient extraction of viral nucleic acid is imperative in delivering intact, un-damaged and contamination-free starting materials for the very sensitive method of polymerase chain reaction (PCR) [1,2].
The amplification of viral nucleic acid; either DNA using qualitative real-time PCR (qPCR), or RNA using qualitative revers-transcription real-time PCR (qRT-PCR), is widely used in the field of molecular diagnostics [3]. The procedure determines the copy number, commonly known as the “viral load”, in the blood of patients infected with certain virtues, such as hepatitis B, hepatitis C, human immunodeficiency virus (HIV), and many other viral infections [4-6].
Although many manufacturers of real-time thermal cyclers claim that their instruments are able to detect a single nucleic acid target in samples with a slow as few copy numbers [7]; their claim is usually affected by various factors and conditions. One of the most important factors affecting the instrument’s ability to detect its nucleic acid target is the integrity of the extracted viral nucleic acid [8].
Many commercially available viral nucleic acid extraction kits use the silica-based column extraction methodology. This method relies on the ability of silica particles to adsorb DNA and RNA molecules under certain analytical conditions, including the presence of ethanol, salt concentration and pH [9]. Newer technologies depend on silica-coated magnetic nanoparticles that delivers the same binding capacity, but more developed to use in automated extraction devices [10]. The nucleic acid is then precipitated and ultimately eluted using special buffers or simply nuclease-free water. Each manufacturer, however, modifies their kit components to alter the binding and subsequent release of viral nucleic acid. This creates considerable variability in terms of binding capacity, contaminant removal and overall extraction efficacy [11].
Additionally, viral RNA extraction is different from viral DNA extraction. Not only the physical and biochemical properties and 3D structure of RNA and DNA are different, but also, how these molecules aggregate to form a precipitate that can be successfully eluted. Many modern commercial kits use poly-A carrier RNA molecules to facilitate the aggregation of viral, which is usually present in minute quantities in patients’ samples [12].
In this project, we compared the yield and purity of viral nucleic acid using five different commercially extraction kits and determined their extraction efficiency using PCR amplification followed by Ct value comparison.
Viral samples
Two different solutions were used to prepare the samples from which the viral nucleic acid was extracted. The first solution was normal saline and the second solution was pooled serum extracted from blood samples of healthy donor individuals. 3 µL of 2x105 HBV positive control template from the PCRmax Hepatitis B Virus kit (Cat# PKIT10047, PCRmax, Staffordshire, United Kingdom) as well as 3 µL of 2x105 HCV positive control template from the PCRmax Hepatitis C Virus kit (Cat# PKIT10051, PCRmax, Staffordshire, United Kingdom) were added to 1.5 mL of both solutions. Both un-extracted solutions are then used as controls for viral nucleic acid concentrations as well as in real-time PCR experiments.
Viral nucleic acid extraction
We used five different commercially available viral nucleic acid extraction kits. RTA Viral Nucleic Acid Isolation Kit (Cat# 09029100, RTA Laboratories, Gebze, Turkey). Magnetic Beads Viral DNA/RNA Extraction Kit (Cat# MV096, Geneaid, New Taipei City, Taiwan). AccuPrep Viral RNA Extraction Kit (Cat# K-3033R, Bioneer, Daedeok-gu, Republic of Korea). QIAamp DSP Virus Kit (Cat# 60704, Qiagen, Hilden, Germany). HigherPurity Viral DNA/RNA Kit (Cat# AN0605, Canvax, Córdoba, Spain). In this project, the kits were named: R, G, B, Q and C respectively.
For the R kit; 20 μL of Proteinase K was added to 250 μL of both solutions into a 2 ml tube. Then, 750 μL of Solution RL and 15 μL of RNA Carrier was added to each tube followed by quick mixing using pulse-vortex. Both tubes were incubated at 56°C and then briefly centrifuged at 1,500 g for 1 minute. Next 750 μL of 97% ethanol was added, mixed by pulse-vortex and incubated again at room temperature for 3 minutes. After that, 900 μL of the mixture was transferred to the spin column and centrifuge at 5,000 g for 1 minute discarding the flow-through; this step was repeated twice. Then 700 μL of solution W1 was added before centrifuging the tubes at 5,000 g for 1 minute, and the same process was repeated using solution W2. After a final step of washing using 700 μL of 97% ethanol, both tubes were centrifuged at 16,000 g for 1 minute and the tubes caps were left open to dry any residual ethanol at 60°C for 10 minutes. Viral nucleic acid was eventually eluted by adding 50 μL of solution E and centrifuging the tubes at 16,000 g for 3 minutes.
For the G kit; 400 μL of MV1 buffer and 10 μL of RNA Carrier were added to 200 μL of both solutions into a 2 ml tube followed by quick mixing using pulse-vortex and incubation at room temperature for 10 minutes. Then, 450 μL of MV2 buffer and 50 μL of well-mixed MV magnetic beads was added to each tube, mixed by pulse-vortex and the all the supernatant was removed using a magnetic separator. Washing was performed by adding 400 μL of MV3 buffer and two rounds of 600 μL of MV4 buffer followed by heating the tubes at 40ºC for 3 minutes to dry the magnetic beads. Viral nucleic acid was eventually eluted by adding 50 μL of nuclease free water and removing it using the magnetic separator.
For the B kit; 10 μL of Proteinase K was added to 200 μL of both solutions into a 2 ml tube. Then, 300 μL of VB buffer was added to each tube followed by quick mixing using pulse-vortex. Both tubes were incubated at 56°C for 10 minutes. Next 300 μL of 99% isopropanol was added, mixed by pulse-vortex and the centrifuged at 8,000 rpm for 1 minute to discard the flow-through. After that, 500 μL of VW1 buffer was added before centrifuging the tubes at 8,000 rpm for 1 minute, and the same process was repeated using RWA2 buffer. Viral nucleic acid was eventually eluted by adding 50 μL of ER buffer and centrifuging the tubes at 8,000 rpm for 1 minute.
For the Q kit; 75 μL of QP buffer, 500 μL of the AL buffer and 11.2 μg/mL of RNA Carrier were added to 500 μL of each solution into a 2 ml tube. Both tubes were incubated at 56°C and for 15 minutes then centrifuged at 13,000 g for 1 minutes. Next, 600 μL of 97% ethanol was added, mixed by pulse-vortex, incubated at room temperature for 5 minutes and centrifuged at 13,000 g for 1 minute. Then 600 μL of AW1 was added before centrifuging the tubes at 13,000 g for 1 minute, and the same process was repeated using μL 750 of AW2. After a final step of washing using 750 μL of 97% ethanol, both tubes were centrifuged at 16,000 g for 1 minute and the tubes caps were left open to dry any residual ethanol at 56°C for 3 minutes. Viral nucleic acid was eventually eluted by adding 50 μL of AVE buffer and centrifuging the tubes at 16,000 g for 3 minutes.
For the C kit; 25 μL of proteinase K, 200 μL of the BLY buffer and 5.6 μg of RNA Carrier were added to 200 μL of each solution into a 2 ml tube. Both tubes were incubated at 56°C and for 20 minutes then 250 μL of 97% ethanol was added, mixed by pulse-vortex, incubated at room temperature for 5 minutes and centrifuged at 10,000 rpm for 1 minute. Then, 500 μL of WB1 was added before centrifuging the tubes at 12,000 rpm for 1 minute, and the same process was repeated twice using μL 500 of WB2. The tubes caps were left open to dry any residual ethanol at 56°C for 3 minutes. Viral nucleic acid was eventually eluted by adding 50 μL of elution buffer and centrifuging the tubes at 16,000 g for 1 minute.
Real-time PCR reactions
For DNA amplification, 5 µL of eluted viral nucleic acid was added to a total of 15 µL of the master mix. The master mix was prepared by adding 10 µL of the lyophilized OneStep 2X qPCR Master Mix, 1 µL HBV primer/probe mix and 4 µL RNase/DNase free water. The cycling conditions were started by activating the enzyme for 2 minutes at 95°C, followed by 50 cycles of denaturation for 10 seconds at 95°C and then data collection for 60 seconds at 60°C.
For RNA amplification, 5 µL of eluted viral nucleic acid was added to a total of 15 µL of the master mix. The master mix was prepared by adding 10 µL of the lyophilized OneStep 2X RT-qPCR Master Mix, 1 µL HCV primer/probe mix and 4 µL RNase/DNase free water. The cycling conditions were started by a reverse transcription step for 10 minutes at 55°C, followed by activating the enzyme for 2 minutes at 95°C, followed by 50 cycles of denaturation for 10 seconds at 95°C and then data collection for 60 seconds at 60°C.
Real-time PCR reactions were carried out on the Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Massachusetts, United States) without activating the fast cycling option.
Nucleic acid concentration
Yield and purity of extracted viral nucleic acid were measured from 1 µL of eluted samples, from each of the five kits, using the NanoDrop ND-1000 UV-Vis Spectrophotometer (Thermo Fisher Scientific, Massachusetts, United States).
Viral nucleic acid yield
Eluted viral nucleic acid from each of the five different extraction kits was assessed by NanoDrop (Table 1). It is expected that the concentration of viral DNA or RNA in patient samples are hardly detectable by using spectrophotometry. Alternatively, viral loads are assessed using real-time PCR amplification to accurately determines the quantity of viral nucleic acid in biological samples. Spectrophotometric measurements of extracted DNA concentrations showed considerably variable quantities ranging from to 0.2 to 377.2 ng/µL with purity, as determined by assessing the 260/280, ranging from 0.06 to 22.6. Also, RNA concentrations showed even lower quantities ranging from to 0.06 to 1.86 ng/µL with purity, as determined by assessing the 260/230, ranging from 0.01 to 1.86.
Table 1. Viral nucleic acid concentration and purity as assessed by NanoDrop form each extractions kit as compared to the normal saline and serum controls
|
|
STD |
Kit C |
Kit B |
Kit R |
Kit G |
Kit Q |
|
DNA concentration |
446.7 |
3.3 |
1.1 |
377.2 |
0.2 |
171.0 |
|
DNA purity: 260/280 |
2.08 |
0.86 |
1.16 |
3.30 |
0.22 |
3.15 |
|
RNA Concentration |
41.4 |
0.88 |
0.66 |
22.6 |
0.06 |
16.8 |
|
RNA purity: 260/230 |
1.66 |
0.47 |
0.91 |
1.86 |
0.01 |
0.89 |
PCR amplification of viral targets
The extracted samples from each of the extraction kits were amplified using qPCR, for hepatitis B, and by qRT-PCR for hepatitis C. The amplification reactions for all three targets were carried out using similar materials and consumables to prevent inter-experimental variations. After the PCR amplification cycles are finished, the software of the ABI 7500 fast real-time instrument was able to choose an optimized threshold based on the amplification curves of all reactions for each viral target. The Ct value is then calculated for each extraction kit that have been used (Table 2). To try and compensate for threshold settings by other real-time PCR instrument (Figure 1), we introduced two additional threshold points: one that is lower than the default (or optimized) threshold and one that is higher than the default (or optimized) threshold. The three Ct values for each extraction kit were plotted as a single box, with the high and low Ct values serving as error bars (Figure 2).
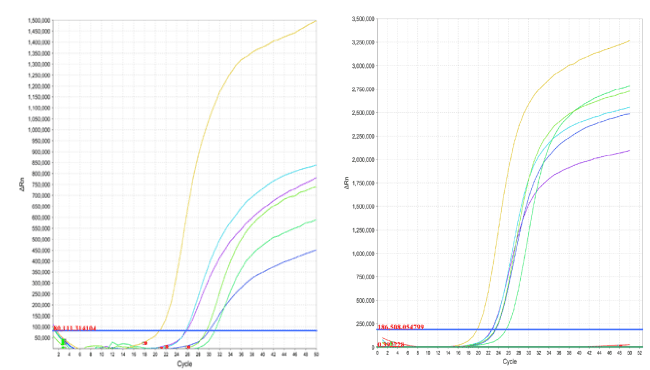
Figure 1. qPCR amplification curves form HBV DNA samples (left) and qRT-PCR amplification curves from HCV RNA samples (right)

Figure 2. Ct values of the HBV PCR reaction (left) and HCV PCR reaction (right) from each of the viral nucleic acid extraction kits. STD: the standard control; R: RTA kit; G: Geneaid kit; B: Bioneer kit; Q: Qiagen kit; C: Canvax kit
Table 2. Ct values after the amplification of HBV and HCV targets using qPCR
|
Ct threshold |
STD |
Kit R |
Kit G |
Kit B |
Kit Q |
Kit C |
HBV |
High: 174,195 |
22.520 |
27.157 |
32.462 |
32.625 |
27.638 |
30.944 |
Auto: 80,111 |
20.784 |
25.606 |
29.969 |
31.008 |
25.791 |
29.472 |
Low: 57,389 |
19.912 |
25.034 |
29.215 |
30.518 |
25.100 |
29.008 |
HCV |
High: 502,764 |
21.611 |
24.737 |
25.363 |
27.480 |
24.712 |
25.266 |
Auto: 366,753 |
20.972 |
24.043 |
24.693 |
26.836 |
23.963 |
24.612 |
Low: 186,508 |
19.778 |
22.763 |
23.426 |
25.525 |
22.634 |
23.323 |
The effective quantification of viral nucleic acid in biological samples is of great medical and scientific value. Researchers and medical professionals use different approaches and methodologies to extract viral nucleic acid from cell free samples such serum, plasma, pleural effusions, gastric lavage, urine, etc. the extraction process does not come without a cost. The cost is in terms of viral nucleic acid integrity and purity. The sole purpose of many downstream diagnostic medical processes, after viral nucleic acid extractions, is the efficient quantification of target regions in the viral DNA or RNA using real-time PCR. The successful amplification of these target regions require that the template viral nucleic acid is intact and not fragmented, sheared or contaminated. Moreover, the extracted viral nucleic acid should be of sufficient quantities for the PCR instrument to successfully detect and amplify.
Here we compared the efficiency of five commercially available viral nucleic acid extraction kits. Each kit had its own, principles, methodology and protocol. Therefore, variations in the overall performance was expected. While their protocol was the most time consuming, we demonstrated that the Q kit was able to deliver abundant quantities of starting material for each of the viral nucleic acid enabling the successful amplification of both HBV and HCV targets. Moreover, possible variations in threshold settings did not affect the overall Ct value when compared to the Ct value of the standard control. Although the nucleic acid quantification, using the NanoDrop, did not provide acceptable evidence of the actual concentration of DNA and RNA in the eluted samples. Real-time PCR provided an adequate quantitative solution to measure the starting material present in the extracted material using each of the kits.
Moreover, the R kit was also successful in delivering adequate viral nucleic acid quantities after the extraction process. In addition, the amplification curves of both HBV and HCV targets started in similar ranges by the real-time PCR from the R kit when compared to the Q extraction kit. The Qiagen extraction kits are considered the gold-standard extraction methodology in the market. This suggests that more starting material was present in the PCR reaction from the R kit, and therefore, would imply the superiority of their extraction protocol and overall methodology of the R kit when compared to other commercially available viral nucleic acid extraction kits.
The author would like to express his gratitude for Ms Areej Al-Johani (BndrGene Medical Lab, Madinah, Saudi Arabia) for helping with the extraction procedures.
- Berger A, Braner J, Doerr HW, Weber B (1998) Quantification of viral load: clinical relevance for human immunodeficiency virus, hepatitis B virus and hepatitis C virus infection. Intervirology 41: 24-34. [Crossref]
- Huang JT, Liu YJ, Wang J, Xu ZG, Yang Y, et al. (2015) Next generation digital PCR measurement of hepatitis B virus copy number in formalin-fixed paraffin-embedded hepatocellular carcinoma tissue. Clinical Chemistry 61: 290-296. [Crossref]
- Klein D (2002) Quantification using real-time PCR technology: applications and limitations. Trends Mol Med 8: 257-260. [Crossref]
- Kok T, Wati S, Bayly B, Devonshire-Gill D, Higgins G (2000) Comparison of six nucleic acid extraction methods for detection of viral DNA or RNA sequences in four different non-serum specimen types. J Clin Virol 16: 59-63. [Crossref]
- Lole KS, Arankalle VA (2006) Quantitation of hepatitis B virus DNA by real-time PCR using internal amplification control and dual TaqMan MGB probes. J Virol Methods 135: 83-90. [Crossref]
- MacKenzie DJ, McLean MA, Mukerji S, Green M (1997) Improved RNA extraction from woody plants for the detection of viral pathogens by reverse transcription-polymerase chain reaction. Plant Dis 81: 222-226. [Crossref]
- Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, et al. (2003) New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 41: 4531-4536. [Crossref]
- Seah C, Chow V, Chan Y, Doraisingham S (1995) A comparative, prospective study of serological, virus isolation and PCR amplification techniques for the laboratory diagnosis of dengue infection. Serodiagnosis and Immunotherapy in Infectious Disease 7: 55-58.
- Shaw KJ, Thain L, Docker PT, Dyer CE, Greenman J, et al. (2009) The use of carrier RNA to enhance DNA extraction from microfluidic-based silica monoliths. Anal Chim Acta 652: 231-233. [Crossref]
- Tian H, Hühmer AF, Landers JP (2000) Evaluation of silica resins for direct and efficient extraction of DNA from complex biological matrices in a miniaturized format. Anal Biochem 283: 175-191. [Crossref]
- Wang J, Ali Z, Wang N, Liang W, Liu H, et al. (2015) Simultaneous extraction of DNA and RNA from Escherichia coli BL 21 based on silica-coated magnetic nanoparticles. Science China Chemistry 58: 1774-1778.
- Zuo X, Xia F, Xiao Y, Plaxco KW (2010) Sensitive and selective amplified fluorescence DNA detection based on exonuclease III-aided target recycling. J Am Chem Soc 132: 1816-1818. [Crossref]