This review on amyloidosis aims to give a simple and concise practical paper on the vast and complex theme that is amyloidosis. Research has done using PubMed with the terms “amyloid” and “amyloidosis” and selecting review articles. The references of the selected articles were also analysis for more appropriated information. This review methodically makes an overview of the main aspects of the disease (incidence, pathogenesis, clinical features, diagnosis and treatment). This is an expanding field that affects a lot of different specialties and is an important pathology that still needs investigation on the diverse causes, as well as in new therapeutic targets and available therapies.
amyloid, amyloidosis, review, systemic
The word amyloid was its origin in the Latin word amylum, meaning starch. It was first used in 1838 by Matthias Schleiden, a botanic, to describe an amylaceous constituent of plants. In medicine, it was Rudolph Virchow in 1854 that used the term to describe a reaction of cerebral corpora amylacea (nowadays AA amyloid) with iodine [1,2]. Today with more advanced technology, amyloid can be identified by its apple-green birefringence in polarized light when stain with Congo red. In 1959 it was showed by Cohen and Calkins that the amyloid is composed by fibrils with 10nm in width with variable length [3] The stability of these fibrils derives from hydrogen bonds between the backbone of the polypeptide chains and the contribution of side chains [4]. In 1968 Glender discovered that the fibrils were aligned in cross β-sheets [5]. Another major discovery was that a variety of polypetides can aggregate in vivo to form amyloid and that in each patient the derived from a single protein precursor.
Amyloidosis, the pathologic manifestation of amyloid deposits, can be systemic or localized. The fibril constituent determines the disease properties, like persistence and seeding behavior [1]. Besides amyloid fibrils, the deposits can also form by serum amyloid P-component (SAP), heparan sulfate proteoglycan (HSPG) and apolipoproteins. These components are important to some manifestations of the disease. Amyloid is defined as extracellular deposit of a fibrillary protein. There are 36 human protein identified (Table 1).
Table 1. Types of human amyloid fibrils protein and precursors
Adapted from Benson et al.
CNS: Central nervous system; PNS: Peripheral nervous system; ANS: Automonic nervous system; CJD: Creutzfeldt-Jakob disease; GSS: Gerstmann-Sträussler-Scheinker syndrome
*Also called amylin
†Not proven by amino acid sequence analysis
‡Full amino acid sequence to be established
There are few studies on the true incidence of amyloidosis. According to death certificates on the United Kingdom, amyloidosis affects 1 in 100 000 people and it is cause of death in 0,58 per 1000 people [6]. An analysis of a Swedish hospital reports an incidence of 8,29 per million people for non-hereditary amyloidosis and 3,2 per million people for hereditary AL amyloidosis. The National Amyloidosis Center of the United Kingdom states that the most frequent type is AL amyloidosis (68%), following by AA amyloidosis (12%) and in third ATTR amyloidosis (9,8%). The most common forms of systemic amyloidosis are AL, AA, ATTR and Aβ2M [5]. Among hereditary amyloidosis, the most common sub-type is the familial amyloid polyneuropathy (FAP), affecting less than 1:100,000 in Europe. Besides this, a specific TTR mutation (Val30Met variant of TTR) is higher in some countries (as in northern Portugal), where about 1:538 of people have the mutated gene [7].
The primary cause of the pathogenicity of amyloid protein is the misfolding of said protein that leads to its accumulation on the affected tissues. The misfolding could be due to multiple causes, one of them being a genetic mutation that codes a different amino-acid that alter the protein conformation.
Several factors determine the tissue deposition of amyloid (tissue tropism), such as molecular structure, low pH, excess of protein, presence of specific proteolytic activity and the interaction of fibrils with glicosaminoglicans (GAG) and specific receptor on the cell surface (receptor for advanced glycation end products) [7]. The exposure of the polypeptide chain to extracellular denaturation stimuli causes the unfolding of the chain and consequence exposure of hidden hydrophobic residues making the protein susceptible to auto aggregation. Besides that, even a partial unfolded protein can become a target to endopeptidases leading to proteolytic cleavage and destabilization of the protein structure. Amyloid proteins are particularly vulnerable to these fluctuations and a have a high tendency to aggregation without a major energetic barrier to unfolding. Another important factor is that some fragmentation events can expose the surface of growing fibrils and increase the number of growth competent aggregates causing an acceleration of aggregation rate. This last mechanism has also implied in the cell to cell transmission and spreading and the disease [7-13]. Accumulation of amyloid aggregates may disrupt the tissue architecture leading to organ failure. The cytotoxic effects of soluble fibrils may allow membrane permeabilization, with alteration of the permeabilization of membrane to calcium ions, the production of reactive oxygen species and mitochondrial dysfunction [14]. The oxidative stress, that activates the apoptotic pathway, and the mechanical disruption of the cell membrane leads to cell death [15,16]. The interaction of amyloid fibrils with cellular receptors been demonstrated in Aβ amyloidosis, and has been implied as the principal effector of the synaptic dysfunction in Alzheimer’s disease [17].
Amyloidosis can potentially affect any organ, making the symptoms have a low specificity to a particular type of amyloidosis. Soft tissue involvement with macroglossia and periorbital purpura are typical of AL amyloidosis, and are present in about 30% of cases. Muscular pseudohypertrophy, swelling of the salivary glands and submandibular soft-tissue infiltration of the neck are also signs of AL amyloidosis. Isolated periorbital purpura is less specific and can be present in other types [6,7].
Cardiac involvement is the main cause of morbidity and mortality in amyloidosis with a 5-year survival of <10% [18,19]. It is characterized by restrictive cardiomyopathy with signs of elevated right ventricular failure (raised jugular venous pressure, edema, congestive hepatomegaly). In more advanced stages of the disease there can be a low cardiac output and hypotension. The cardiac involvement is more common in AL amyloidosis (50% of cases), ATTR amyloidosis and AApoAI and its rare in AA amyloidosis [20,21].
Renal involvement is the most common in affected organ in AA, AL, fibrinogen A α-chain (AFib), ALect2, and AApoAI amyloidosis. Renal dysfunction can be asymptomatic for long periods of time. Albuminuria is typical and can progress to nephrotic syndrome. Albuminuria and kidney disfunction are characteristic of AA amyloidosis and its absence is rare [20]. The hallmark of hereditary AApoAI and lysozyme (ALys) amyloidosis is involvement of the kidneys and liver but the progression is slow [22].
Nervous system involvement is also common in amyloidosis. The two main forms of neuropathies are the compression neuropathy and the generalized neuropathy that can be associated with autonomic dysfunction. AL amyloidosis (50% cases) and some hereditary types of ATTR, ApoAI and A β2M (more frequent carpal tunnel syndrome) are the main types with neurologic symptoms. Peripheral neuropathy is mainly axonal and involves the small and large fibers. It begins with loss of temperature perception and can be painful (small fibers) [23]. The occurrence of autonomic neuropathy is an early manifestation in men, with postural hypotension, impotence and digestive symptoms (early satiety, diarrhea, constipation). Cranial neuropathy can occur in AGel) amyloidosis [6].
Soft tissue involvement in almost exclusive of AL amyloidosis. Carpal tunnel syndrome is a common early symptom in wild-type and hereditary ATTR amyloidosis (found in 1/3 of elderly people undergoing carpal tunnel decompression) [24].
Localized amyloidosis is linked to in-situ production of amyloidogenic light chains by clonal B cells. The most common sites include the respiratory tract, bladder, eyelids, and skin. This is a more indolent form and rarely evolves systemically, but it can have solemn consequences (mass lesion can occupy space) [25].
The correct identification of the protein is vital for the appropriated treatment and prognosis, with the assessment of the extent and severity of amyloidotic organ involvement. The diagnostic is done by biopsy and stain coloration, like Congo red. The final diagnosis is done by the histologic identification of the precursor protein [6,8].
First of all, some common blood and urine test should be requested. Among them are full blood count, hepatic and renal functions tests, clotting tests, serum and urine free chain immunoelectrophoresis and cardiac markers (troponin and N-terminal pro-brain natriuretic peptide). An electrocardiogram (ECG) should be completed, as well as bone marrow aspirate to exclude myeloma. If there is cardiac involvement, serum cardiac markers are important for risk stratification and staging of AL amyloidosis [6,7] (Figure 1 - A stepwise approach to the diagnosis).
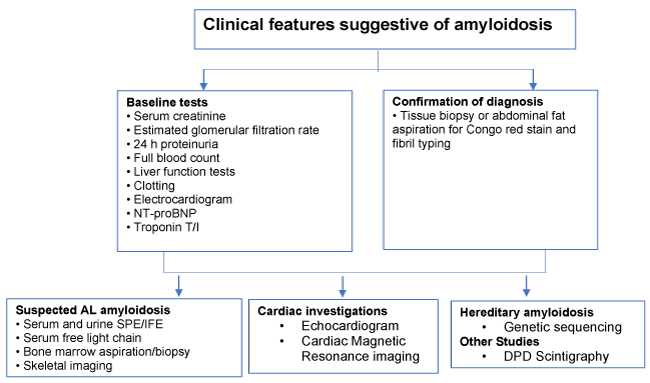
Figure 1. Diagnostic stepwise approach to suspect amyloidosis (AL: Amyloid light-chain; DPD: 3,3-diphosphono-1,2-propanodicarboxylic acid; IFE: Immunofixation electrophoresis; NT-proBNP: N-terminal pro-brain natriuretic peptide; SPE: Serum protein electrophoresis).
When a systemic form is present, the most accessible and common place to biopsy is subcutaneous abdominal fat with fine needle aspiration and do a Congo-red staining (gold standard). It is usually fast and with few side effects, it is an alternative to an organ biopsy and can be done to screen population with suspected diagnosis of Al, AA and ATTR amyloidosis [6,10,11]. The specificity of fat tissue biopsy (if the staining procedure is performed correctly) approaches 100%, sensitivity varies between from 52% to 88% and positive and negative predictive values are 84% and 90%, respectively. The sensitivity is higher with the increased number of samples (1 to 3 samples) and number of observers (1 to 2 observers) [9,10]. In a study done in Amyloidosis Research and Treatment Center (Pavia) with 745 patients, immunoelectron microscopy (IEM) (technique that combines immunohistochemistry and electron microscopy) correctly identified specific form of amyloidosis in 99% of the cases [6].
In cases with cardiac involvement, besides cardiac markers, an ECG and an echocardiogram should be performed. The ECG typically shows reduced QRS voltages for cardiac AL amyloidosis, and the echocardiography shows a concentrically thickened of ventricular walls with normal or small ventricular cavities with thickened valves and dilated atria [26]. Cardiac magnetic resonance imaging can provide additional details about the function and morphology, sub-endocardial late gadolinium enhancement is almost typical of cardiac amyloid [27] The bone-seeking radionuclide tracers ⁹⁹m-technetium- 3,3-diphosphono-1,2-propanodicarboxylic acid (⁹⁹mTc-DPD) and ⁹⁹mTc-pyrophosphate (⁹⁹mTc-PYP) seem to localize with good sensitivity cardiac ATTR deposits, and scans with ⁹⁹mTc-DPD seem to show asymptomatic ATTR cardiac deposits [28].
To evaluate the extent of the disease serum amyloid P component (SAP) scintigraphy detects and identify the distribution of amyloid in systemic amyloidosis, but the heart cannot be visualized with this technique [29]. Sensitivity of the SAP scan for AL and AA amyloidosis is about 90%, but it is only 48% for hereditary ATTR amyloidosis and specificity is about 90% [5]. In hereditary disorders a genetic sequencing should be done [6].
Various variables are powerful clinical indicators of poor outcome in AL amyloidosis, including poor performance status, severe postural hypotension, New York Heart Association functional class 3 or higher, and low systolic blood pressure (SBP; <100 mm Hg).Those in whom troponin I and NT-proBNP are abnormal have median survival of 7–8 months [30].
The treatment option in amyloidosis depends on which type the patient has. A reduction of the supply of precursor proteins is the goal for all treatment, but it is not yet possible in various types. There some drugs for AL and AA amyloidosis but frequently they are poorly tolerated because of organs disfunctions. Orthotopic liver transplant is an option for diminish hepatic source of hereditary forms of ATTR, AApoAI and AFib [6,7]. Assessment of treatment response includes a dual evaluation of a treatment’s suppression of supply of amyloid precursor protein and its effects on the function of amyloidotic organs. Supportive care is fundamental to control symptoms, maintain the organ function and to manage the adverse effects of the medication. These include blood pressure control (renal amyloidosis), fluid restriction (cardiac amyloidosis) and α-agonists (autonomic neuropathy with postural hypotension) and nutritional support [6].
Risk stratification in patients with AL amyloidosis is critical for optimum selection of therapy. A consensus criterion to define hematological and organ responses (strong predictors of survival) have been studied. Those who achieve a complete response (no detectable monoclonal immunoglobulin) or very good partial response (VGPR) have the best outcomes [31]. The treatment for AL amyloidosis is chemotherapy targeting clonal plasma cells dyscrasia with consequent decrease in light chains and reduce organ damage [32]. The clinical array of symptoms can vary from monoclonal gammopathy of undetermined significance (MGUS, the majority of patients) to multiple myeloma. The poorer the functional status, the greater is treatment toxicity, so frequent evaluations of hematological response are needed. The patient can be divided in low, intermediate and high risk. Low-risk patients are those with excellent performance status; NT-proBNP lower than 5000 ng/mL; troponin lower than 0,06 ng/mL, no significant pleural effusions, autonomic neuropathy or amyloid-related gastrointestinal bleeding and good renal function. The classical treatment for low risk patients was melphalan followed by autologous stem cell transplantation (ASCT) [33,34]. Bortezomib can be used for induction and consolidation to achieve VGPR in >90% of these patients [35,36]. Most of the patients with AL amyloidosis have an intermediate risk should be treated with combinate chemotherapy. Some options include melphalan with dexamethasone (MDex) or cyclophosphamide, thalidomide and dexamethasone (CTDa) [37]. A combination of bortezomib with cyclophosphamide and dexamethasone (CyBorD) gives high response rates > 90% for patients treated upfront, with 60% achieving complete response or VGPR [38]. Bortezomib should be used with caution in the patients with neuropathy symptoms because it can have neurotoxicity. Second-generation proteasome inhibitor ixazomib, provide good hematological response (> 50% of patients) with good cardiac and renal responses and it’s given orally and are usually well tolerated [39]; and carfilzomib (intravenous) has a potential cardiotoxicity, but in a phase I study involving patients with both relapsed and refractory AL showed to be an effective treatment [40]. Lenalidomide and pomalidomide in combinations with alkylators showed a good response, but lower than those with bortezomib [41,42]. There are useful in refractory disease after the use of bortezomib or in patients with neuropathic symptoms. In high risk patients no particular regimen has yet been shown to be superior and chemotherapy should be started at low doses with frequent evaluations [43].
In patients with AA amyloidosis the treatment is to control the underlying inflammatory disease, therefor, the specific treatment depends on the cause. Antimicrobials are the first choice for patients in infectious diseases, like tuberculosis or bronchiectasis; and colchicine is effective in familiar Mediterranean fever [44,45]. In those patients with rheumatological diseases, tumor necrosis factors (TNF) inhibitors (etanercept, infliximab, adalimumab) are effective [46]. In auto-inflammatory disorders like “TNF-related periodic fever syndrome” (TRAPS) and cryopyrin-associated periodic fever syndromes (CAPS), interleukin-1 blockage with anakinra or canakinumab showed efficacy [47-49]. Inhibition of interleukin IL-1 or IL-6 improved the symptoms in patients with AA amyloidosis with an unknown origin, but the treatment regimen has to be carefully adapted [50]. The reduction of the interactions between GAG and amyloid fibrils with eprodisate constitutes a hypothesized novel approach, but the clinical efficacy is still uncertain [51].
In hereditary amyloidosis, organs transplant remains as the main approach, the principal transplant being the liver as producer of amyloid precursors, such as transthyretin, apolipoprotein AI and fibrinogen. This transplant allows the substitution of the mutant protein for a normal one [52]. It is the treatment of choice in younger patients with ATTR amyloidosis with the Val30Met variant and early in the disease course [53]. Cardiac transplant can be considered in selected young patients with ATTRwt and V122I-related ATTR [54]. AFib amyloidosis has had some treatment success with renal transplant even though recurrent amyloid deposition has been reported [55]. A double kidney and liver transplant have been proposed for AFib but the mortality is high [56]. AApoAI amyloidosis (depending on the mutation) have reported good long-term outcomes of liver, kidney and heart transplantation in spite of recurrent deposition [57].
Among new therapies, has been some developments in the treatment of ATTR. A novel molecule, tafamidis, nowadays standard of care to treat hereditary ATTR, makes a kinetic stabilization by binding TTR in the plasma and maintain the naïve tetrameric structure [58], leading to a slower disease progression in 18 months [59]. Diflunisal, a non-steroidal anti-inflammatory drug, can bind TTR, although the binding is moderate, it has a high bioavailability and good clinical activity in vivo [60]. Doxycycline and tauro-ursodeoxycholic acid (TUDCA) may interfere with TTR fibrillogenesis and slow the disease progression [6]. Two RNA-inhibiting therapies for TTR (small interfering RNA therapy and anti-sense oligonucleotide therapy), patisiran and inotersen already approved by the European medicines Agency, don´t seem to have major side effects so far [61,62].
Immunotherapy with antibodies are another new option for the treatment of amyloidosis. The chimeric antibody, Mu11–1F4, has the ability to bind AL fibrils in amyloid deposits [63]. Another monoclonal antibody, mAb2A4, has the ability to identify human AL/AA aggregates, it immunoreacts in vitro with insoluble light-chain aggregates and normal folded light chain aggregates were spared [64]. Another new combined option is using a monoclonal antibody, a specific IgG1 antibody identifying SAP (reduce amyloid aggregates in systemic amyloidosis) [65]. This uses R)-1-[6-[(R)- 2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2- carboxylic acid (CPHPC) to reduce SAP and then they are recognized by IgG anti-SAP antibodies [66]. This approach appears to show promising results with reduction in liver amyloid deposits [67].
Amyloidosis is a complex disease with multiple organs affects and different manifestations. The clinicians need to have a high degree of suspicion to make an early and correct diagnosis and initiate the adequate treatment. There have been an efforts from the scientific community to improve the treatment option for this multifactorial disease, but it still will be a few years before those are available to the patients.
I would like to thank to the support given by the Loulé county for the writing of this article.
No funding.
No competing interest.
- Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, et al. (2018) Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee.Amyloid25: 215-219. [Crossref]
- Kyle RA (2001) Amyloidosis: a convoluted story.Br J Haematol114: 529-538. [Crossref]
- Cohen AS, Calkins E (1959) Electron microscopic observations on a fibrous component in amyloid of diverse origins.Nature183: 1202-1203. [Crossref]
- Bellotti V, Nuvolone M, Giorgetti S, Obici L, Palladini G, et al. (2007) The workings of the amyloid diseases.Ann Med39: 200-207. [Crossref]
- Eanes ED, Glenner GG (1968) X-ray diffraction studies on amyloid filaments. J Histochem Cytochem 16: 673-677. [Crossref]
- Wechalekar AD, Gillmore JD, Hawkins PN (2016) Systemic amyloidosis.Lancet387: 2641-2654. [Crossref]
- D'Aguanno V, Ralli M, Artico M, Russo FY, Scarpa A, et al. (2019) Systemic amyloidosis: A contemporary overview.Clin Rev Allergy Immunol. [Crossref]
- Hazenberg BP (2013) Amyloidosis: A clinical overview.Rheum Dis Clin North Am39: 323-345. [Crossref]
- Fernández de Larrea C, Verga L, Morbini P, Klersy C, Lavatelli F, et al. (2015) A practical approach to the diagnosis of systemic amyloidoses. Blood 125: 2239. [Crossref]
- van Gameren II, Hazenberg BP, Bijzet J, van Rijswijk MH (2006) Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum 54: 2015-2021. [Crossref]
- Dhingra S, Krishnani N, Kumari N, Pandey R (2007) Evaluation of abdominal fat pad aspiration cytology and grading for detection in systemic amyloidosis. Acta Cytol 51: 860. [Crossref]
- Westermark P (2012) Subcutaneous adipose tissue biopsy for amyloid protein studies. Methods Mol Biol 849: 363-371. [Crossref]
- Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu Rev Biochem 86: 27-68. [Crossref]
- Glabe CG, Kayed R (2006) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66: S74-S78. [Crossref]
- Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis.N Engl J Med349: 583-596. [Crossref]
- Williams TL, Serpell LC (2011) Membrane and surface interactions of Alzheimer’s Abeta peptide-insights into the mechanism of cytotoxicity. FEBS J 278: 3905-3917. [Crossref]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, et al. (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: 535-539. [Crossref]
- Falk RH (2005) Diagnosis and management of the cardiac amyloidoses.Circulation112: 2047-2060. [Crossref]
- Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, et al. (2010) Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via anon-canonical p38alpha MAPK pathway. Proc Natl Acad Sci USA 107: 4188-4193. [Crossref]
- Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, et al. (2007) Natural history and outcome in systemic AA amyloidosis.N Engl J Med356: 2361-2371. [Crossref]
- Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, et al. (2013) Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2: e000098. [Crossref]
- Sattianayagam PT, Gibbs SD, Rowczenio D, Pinney JH, Wechalekar AD, et al. (2012) Hereditary lysozyme amyloidosis—phenotypic heterogeneity and the role of solid organ transplantation. J Intern Med 272: 36-44. [Crossref]
- Reilly MM, Staunton H (1996) Peripheral nerve amyloidosis. Brain Pathol 6: 163-177. [Crossref]
- Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, et al. (2011) High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol 42: 1785-1791. [Crossref]
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, et al. (2005) Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th international symposium on Amyloid and Amyloidosis. Am J Hematol 79: 319-328. [Crossref]
- Gillmore JD, Wechalekar A, Bird J, Cavenagh J, Hawkins S, et al. (2015) Guidelines on the diagnosis and investigation of AL amyloidosis.Br J Haematol168: 207-218. [Crossref]
- Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, et al. (2005) Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 111: 186-193. [Crossref]
- Hutt DF, Quigley AM, Page J, Hall ML, Burniston M, et al. (2014) Utility and limitations of 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardiovasc Imaging 15: 1289-1298. [Crossref]
- Hawkins PN, Lavender JP, Pepys MB (1990) Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med 323: 508-513. [Crossref]
- Mollee P, Renaut P, Gottlieb D, Goodman H (2014) How to diagnose amyloidosis.Intern Med J44: 7-17. [Crossref]
- Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, et al. (2012) New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 30: 4541-4549. [Crossref]
- Comenzo RL, Reece D, Palladini G, Seldin D, Sanchorawala V, et al. (2012) Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis.Leukemia26: 2317-2325. [Crossref]
- Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Buadi FK, et al. (2011) Trends in day 100 and 2-year survival after auto-SCT for AL amyloidosis: outcomes before and after 2006. Bone Marrow Transplant 46: 970-975. [Crossref]
- Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Dingli D, et al. (2013) Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis.Bone Marrow Transplant48: 557-561. [Crossref]
- Landau H, Hassoun H, Rosenzweig MA, Maurer M, Liu J, et al. (2013) Bortezomib and dexamethasone consolidation following risk-adapted melphalan and stem cell transplantation for patients with newly diagnosed light-chain amyloidosis. Leukemia 27: 823-828. [Crossref]
- Sanchorawala V, Brauneis D, Shelton AC, Lo S, Sun F, et al. (2015) Induction therapy with bortezomib followed by bortezomib-high dose melphalan and stem cell transplantation for AL amyloidosis: results of aprospective clinical trial. Biol Blood Marrow Transplant 21: 1445-1451. [Crossref]
- Palladini G, Perfetti V, Obici L, Caccialanza R, Semino A, et al. (2004) Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation.Blood103: 2936-2938. [Crossref]
- Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, et al. (2012) Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival.Blood119: 4387-4390. [Crossref]
- Sanchorawala V, Palladini G, Kukreti V, Zonder JA, Cohen AD, et al. (2017) A phase 1/2 study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis.Blood130: 597-605. [Crossref]
- Cohen AD, Scott EC, Liedtke M, Kaufman JL, Landau H, et al. (2014) A phase I dose-escalation study of carfilzomib in patients with previously treated systemic light-chain (AL) amyloidosis. Blood 124: 4741.
- Kastritis E, Terpos E, Roussou M, Gavriatopoulou M, Pamboukas C, et al. (2012) A phase 1/2 study of lenalidomide with low-dose oral cyclophosphamide and low-dose dexamethasone (RdC) in AL amyloidosis.Blood119: 5384-5390. [Crossref]
- Palladini G, Russo P, Milani P, Foli A, Lavatelli F, et al. (2013) A phase II trial of cyclophosphamide, lenalidomide and dexamethasone in previously treated patients with AL amyloidosis. Haematologica 98: 433-436. [Crossref]
- Wechalekar AD, Schonland SO, Kastritis E, Gillmore JD, Dimopoulos MA, et al. (2013) A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood 121: 3420-3427. [Crossref]
- Lane T, Loeffl er JM, Rowczenio DM, Gilbertson JA, Bybee A, et al. (2013) AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum 65: 1116-1121. [Crossref]
- Pinney JH, Lachmann HJ (2012) Systemic AA amyloidosis.Subcell Biochem65: 541-564. [Crossref]
- Nakamura T, Higashi S, Tomoda K, Tsukano M, Shono M (2010) Etanercept can induce resolution of renal deterioration in patients with amyloid A amyloidosis secondary to rheumatoid arthritis. Clin Rheumatol 29: 1395-1401. [Crossref]
- Pettersson T, Kantonen J, Matikainen S, Repo H (2012) Setting up TRAPS.Ann Med44: 109-118. [Crossref]
- Obici L, Meini A, Cattalini M, Chicca S, Galliani M, et al. (2011) Favourable and sustained response to anakinra in tumour necrosis factor receptor-associated periodic syndrome (TRAPS) with or without AA amyloidosis.Ann Rheum Dis70: 1511-1512. [Crossref]
- Kuemmerle-Deschner JB, Hachulla E, Cartwright R, Hawkins PN, Tran TA, et al. (2011) Two-year results from an open-label, multicenter, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin associated periodic syndrome across different severity phenotypes. Ann Rheum Dis 70: 2095-2102. [Crossref]
- Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN (2001) Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet 358: 24-29. [Crossref]
- Kisilevsky R, Ancsin JB, Szarek WA, Petanceska S (2007) Heparan sulfate as a therapeutic target in amyloidogenesis: prospects and possible complications.Amyloid14: 21-32. [Crossref]
- Saraiva MJ (2002) Sporadic cases of hereditary systemic amyloidosis. N Engl J Med 346: 1818-1819. [Crossref]
- de Carvalho M, Conceição I, Bentes C, Luís ML (2002) Long-term quantitative evaluation of liver transplantation in familial amyloid polyneuropathy (Portuguese V30M).Amyloid9: 126-133. [Crossref]
- Hamour IM, Lachmann HJ, Goodman HJ, Petrou M, Burke MM, et al. (2008) Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant 8: 1056-1059. [Crossref]
- Gillmore JD, Lachmann HJ, Rowczenio D (2009) Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J Am Soc Nephrol 20: 444-451. [Crossref]
- Gillmore JD, Lachmann HJ, Wechalekar A, Hawkins PN (2010) Hereditary fibrinogen A alpha-chain amyloidosis: clinical phenotype and role of liver transplantation.Blood115: 4313. [Crossref]
- Gillmore JD, Stangou AJ, Lachmann HJ, Goodman HJ, Wechalekar AD, et al. (2006) Organ transplantation in hereditary apolipoprotein AI amyloidosis. Am J Transplant 6: 2342-2347. [Crossref]
- Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW (2012) The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug.J Mol Biol421: 185-203. [Crossref]
- Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, et al. (2012) Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial.Neurology79: 785-792. [Crossref]
- Sekijima Y, Dendle MA, Kelly JW (2006) Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 13: 236-249. [Crossref]
- Adams D (2013) Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord 6: 129-139. [Crossref]
- Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, et al. (2013) Safety and efficacy of RNAi therapy for transthyretin amyloidosis.N Engl J Med369: 819-829. [Crossref]
- Wall JS, Kennel SJ, Stuckey AC, Long MJ, Townsend DW, et al. (2010) Radioimmunodetection of amyloid deposits in patients with AL amyloidosis.Blood116: 2241-2244. [Crossref]
- Renz M, Torres R, Dolan PJ, Tam SJ, Tapia JR, et al. (2016) 2A4 binds soluble and insoluble light chain aggregates from AL amyloidosis patients and promotes clearance of amyloid deposits by phagocytosis †.Amyloid23: 168-177. [Crossref]
- Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, et al. (2015) Therapeutic clearance of Amyloid by antibodies to serum Amyloid P component.N Engl J Med373: 1106-1114. [Crossref]
- Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, et al. (2010) Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 468: 93-97. [Crossref]
- Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, et al. (2002) Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 417: 254-259. [Crossref]