Adrenocortical carcinoma in children is a rare cancer that has a poor response to cytotoxic treatment. Approximately half of adrenocortical carcinoma present in the first 4 years of life. Most adrenal tumours in children are hormone secreting and have female preponderance. Our patient was a 15-month-old female and presented with clinical features of Cushing’s syndrome
adrenal cortex, adrenocortical carcinoma, rare tumour, child
Adrenocortical carcinoma is a rare tumour in the paediatric population and it is thought to have a poor prognosis [1,2]. It arises from the adrenal cortex and accounts for less than 0.2% of all paediatric tumours and 6% of all adrenal tumours in children [3]. It has a female preponderance with a ratio of approximately 2:1 [4]. More than 90% of adrenocortical tumors in children are hormone secreting and the clinical features reflect the pattern of the hormones that are secreted [1,3]. They account for approximately 5-10% of cases of Cushing’s syndrome but approximately 40% of patients with both Cushing syndrome and adrenal mass have malignancy [5]. Children with Beckwith Wiedemann syndrome, nephroblastoma and hepatoblastoma tend to have higher incidence of adrenocortical carcinoma than the general population [6] although most occur sporadically.
We present a 15-month-old Ghanaian girl with adrenal carcinoma who presented with clinical features of Cushing’s syndrome.
A 15-month-old girl presented with weight gain over 3 months prior to presentation. The weight gain was gradual, more in the trunk and in the abdomen. The face was said to have become rounded with drooping cheeks. She progressively developed acne on the face and the chest and about a month prior to presentation she developed dark pubic hair and hirsutism on the posterior aspect of the trunk. She had become less active and had developed insomnia but had no bruises or ulcers.
Both the patient and mother were not taking any steroid containing medications. She was still breastfeeding along with supplementary feeds.
On examination, weight = 22kg, (> 99th percentile), length = 83 cm (on 75th percentile), she was apathetic, had extensive facial acne and hirsutism on face and back. There were no ulcers, striae or buffalo hump.
Respiration was 26/min, lungs were clear on auscultation,
Heart rate was 138 beats per minute, regular and blood pressure was 126/82 mmHg, (both systolic and diastolic > 99th percentile).
Abdomen was distended with a palpable mass in the left upper quadrant. There was clitoromegaly with Pubic hair T3 and Breast T1.
Early morning Cortisol 1796 nmol/L (ref: 171-536), testosterone = 8.2 nmol/L (ref: 0.07-0.35).
ACTH 44.7 pg/ml (ref: < 46).
WBC 13.81 x 109/L (ref: 3-15).
Hb 11.5 g/dl (ref: 8-17).
Urea 7.05 mmol/L (ref: 2.5-8.3), creatinine 40 umol/L, (8-36).
Calcium 1.34 mmol/L (ref: 2.24-2.74), Magnesium 0.69 mmol/L (ref: 0.7-1.05), Phosphate 0.35 mmol/L (ref: 0.87-1.45).
Potassium 2.3 mmol/L, Sodium 249 mmol/L.
Histology report
Adrenocortical carcinoma, grade 4.
Imaging
Abdominal ultrasonography revealed left adrenal mass (Figure 1,2,3 and 4).
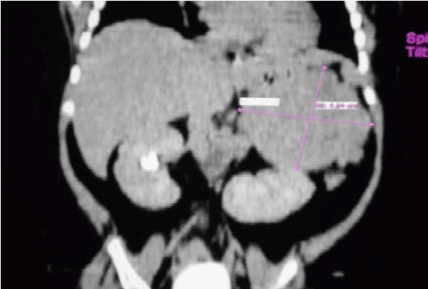
Figure 1. Abdominal CT Scan showing left adrenal mass.
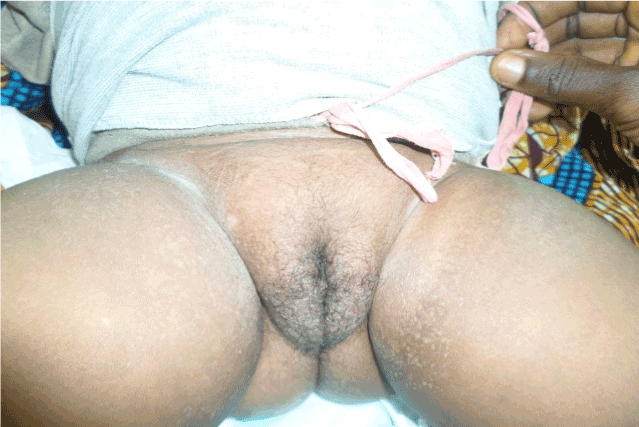
Figure 2. Genital hair.
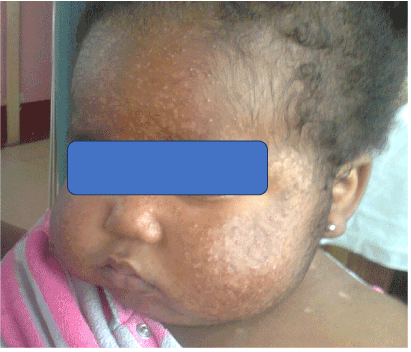
Figure 3. Severe facial acne.
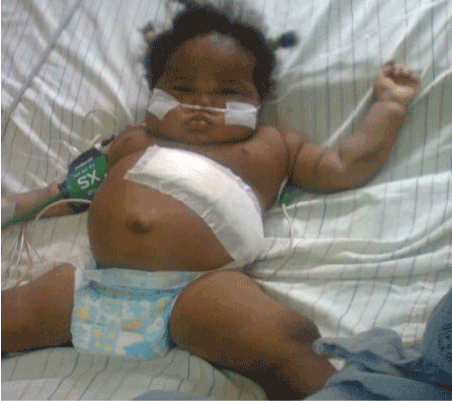
Figure 4. After left adrenalectomy.
CT Scan
Revealed huge left adrenal mass
Although Adrenal carcinoma in childhood constitutes less than 1% of all malignancies in the pediatric age population, it accounts for 12% of all primary carcinomas in children [7]. It has a bimodal age distribution with case clustering in children under 6 years and in adults 30 to 40 years of age [8]. The general incidence in children ranges from 0.5 to 2.0 cases per million. However, a higher incidence of 4.7 cases per million has been reported in girls under 4 years old in Southern Brazil [9]. No specific reason has been found for the high incidence in Brazil compared to the other parts of the world. There is no published data on incidence of adrenal carcinoma among children in Ghana.
The actual cause of adrenal carcinoma is not known. However, certain genetic syndromes have high incidence rate of adrenal carcinoma. In Li-Fraumeni syndrome (LFS) adrenal carcinoma occurs 100 times more frequent than in the general population. In most cases of adrenal carcinoma in LFS the germ line mutation has been found to be TP53 gene [10]. There is also increased incidence of adrenal carcinoma in patients with isolated hemihypertrophy and Beckwith-Widermann Syndrome [4]. Children with congenital adrenal hyperplasia (CAH) have been reported to have developed adrenal carcinoma. Mutation in CYP21 gene is responsible for this tumorogenesis [4,6]. Genetic studies are not available in Ghana and so were not done in our patient.
Clinical features reflect the pattern of adrenal excess [1,3] consistent with Cushing’s syndrome, virializing or feminizing features. Our patient presented with features of Cushing’s syndrome and of virilization such as weight gain, hirsutism, acne vulgaris, apathy, hypertension, electrolyte imbalance, pubic hair and clitoromegaly. These are features of cortisol, aldosterone and androgen excesses. In this patient cortisol was more than three times the upper limit and testosterone was more than 20 times the upper limit for females and this accounted for hirsutism, pubic hair and clitoromegaly. These are features of cortisol, aldosterone and androgen excesses. In this patient cortisol was more than three times the upper limit and testosterone more than 20 times the upper limit for females. Conversely, Estrogen secreting adrenal tumours in males lead to testicular atrophy and gynaecomastia and are usually malignant [11]. Nonfunctional tumours may still produce appreciable amounts of adrenal hormones detectable on laboratory evaluation but not in significant quantities to produce accompanying clinical features [3].
Adrenal carcinoma should be staged so as to direct the management including follow up. Stage 1 has greatest dimension of less than 5 cm, tumour cells are confined to the adrenal glands and hormone levels are normal after surgery. Stage II has greatest dimension greater than 5 cm, tumour cells are confined to the adrenal glands and hormone levels are persistently high after surgery. In Stage III disease there is local invasion of the adrenal gland without the involvement of adjacent organs or regional lymph nodes. This is also known as inoperable primary tumour. In stage IV disease, there is distant metastasis, invasion of adjacent regional lymph nodes and /or adjacent organs [1,2,11]. Histologically, our patient had stage IV disease, which implies that she had distant metastasis with likely poor prognosis.
Diagnosis began with clinical suspicion in a patient with Cushingoid features with an abdominal mass on palpation or revealed by imaging. Abdominal ultrasonography is effective in detecting adrenal carcinoma. Ultrasonography is cheaper and can be afforded by many patients in resource limited countries such as Ghana. Computed tomography (CT) scan is preferred in evaluating adrenal masses and can detect nodules as little as 3 cm. Magnetic resonance imaging (MRI) improves tumour detection and extent of spread and can provide prognostic help in terms of staging [12].
Management of a patient with adrenal carcinoma is multidisciplinary requiring coordination between pediatric endocrinologist, pediatric surgeon and pediatric oncologist. Complete resection of the tumor is the treatment of choice and can be curative especially for stages I and II where the tumour is restricted to the adrenal gland [2]. This may have good prognosis and in some cases, they may have survival rate of 70% but in cases of distant metastasis survival rate can be as poor as 7% [1]. In patients with stages III and IV disease who have metastasis beyond the adrenal gland, the best treatment is combination of surgery and chemotherapy. The chemotherapy of choice is mitotane. Mitotane selectively inhibits the activity of the adrenal cortex by causing adrenal necrosis. A combination of surgery and mitotane gives a survival benefit in children [1,12]. It is effective in controlling the endocrine symptoms and may cause tumour regression. Other effective chemotherapeutic agents include cisplantinum, etoposide, doxorubicin, 5-florouracil and cyclophosphamide[13].
Preoperatively, the patient had hypertension and hypokalemia. She was put on tablet amlodipine 5 mg daily and tablet lisinopril 5 mg daily. Hypokalaemia was corrected parenterally before surgery.
Intra-operatively, left extended Kocher’s incision was made and incision deepened with electrocautery. A highly vascularized left adrenal was identified. It was carefully separated from the spleen, left colon, left kidney and pancreas. The left adrenal vessels were sutured and ligated and the left adrenal was totally excised. Bleeding points were secured. The tumour mass measured 1.1 cm x 8 cm x 8 cm and weighed 10.85g. The estimated blood loss of was 400 ml. She was hemotransfused, postoperatively, 350 mls of blood. She was given 50mg hydrocortisone intraoperatively and repeated immediately after the surgery.
Postoperatively, there was persistent hypokalemia and hypertension. She was admitted to the intensive care unit (ICU) and put on intravenous (IV) ringer’s lactate, IV cefuroxime, IV hydrocortisone, IV Potassium chloride (KCl) and suppository paracetamol. She became stable after 72 hours and she was then transferred to the paediatric surgical ward for further management. Investigations were ordered to ascertain cortisol and electrolytes levels to guide management but the parents could not afford to do most of the requested laboratory tests. The few electrolytes checked showed persistent hypokalemia ranging between 1.3 to 2.8 mmol/L (Table 1) despite continuous intravenous KCl. However, ease of correction was limited by access to frequent and timely potassium results from the laboratory.
Table 1. Serum electrolyte.
Day |
Potassium (mmol/L) |
Sodium (mmol/L) |
1 |
2.3 |
149 |
2 |
- |
- |
3 |
2.0 |
150 |
4 |
1.3 |
148 |
5 |
1.7 |
140 |
6 |
- |
- |
7 |
2.8 |
145 |
8 |
- |
- |
9 |
- |
- |
10 |
2.1 |
143 |
11 |
1.9 |
155 |
2021 Copyright OAT. All rights reserv
From the histological report our patient had stage IV disease, which implied there was distant metastasis. It was also most likely that adrenal hormonal secretion persisted as the patient remained hypokalemic and hypertensive. She probably would have benefitted from mitotane. However, mitotane is not available in Ghana and the parents could not afford other chemotherapy agents.
She died eleven days postoperatively on the ward. The last potassium checked before death was 1.9 mmol/L.
This research did not receive any financial support from any funding agency.
No conflict of interest
- Teinturier C, Pauchard MS, Brugières L, Landais P, Chaussain JL, et al. (1999) Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol 32: 106-111. [Crossref]
- Stewart JN, Flageole H, Kavan P (2004) A surgical approach to adrenocortical tumors in children: the mainstay of treatment. J Pediatr Surg 39: 759-763. [Crossref]
- Kebebew E, Reiff E, Duh QY, Clark OH, McMillan A (2006) Extent of disease at presentation and outcome for adrenocortical carcinoma: have we made progress? World J Surg 30: 872-878. [Crossref]
- Mayer SK, Oligny LL, Deal C, Yazbeck S, Gagné N, et al. (1997) Childhood adrenocortical tumors: case series and reevaluation of prognosis--a 24-year experience. J Pediatr Surg 32: 911-915. [Crossref]
- Hemmady S (2009) Adrenal carcinoma: a case study. N Z J Med Lab Sci 63: 48-50.
- Lack EE, Mulvihill JJ, Travis WD, Kozakewich HP (1992) Adrenal cortical neoplasms in the pediatric and adolescent age group. Clinicopathologic study of 30 cases with emphasis on epidemiological and prognostic factors. Pathol Annu 27 Pt 1: 1-53. [Crossref]
- Dehner LP, Hill DA (2009) Adrenal cortical neoplasms in children: why so many carcinomas and yet so many survivors? Pediatr Dev Pathol 12: 284-291. [Crossref]
- Mahmood M, Akhtar N, Ismail S, Uqman S, Owiwii Y (2012) Adrenocortical Tumor in Children- Report of a Case. Int J of Pathology 10: 82–84.
- Custódio G, Komechen H, Figueiredo FR, Fachin ND, Pianovski MA, et al. (2012) Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol Cell Endocrinol 351: 44-51. [Crossref]
- Sorrell AD, Espenschied CR, Culver JO, Weitzel JN (2013) Tumor protein p53 (TP53) testing and Li-Fraumeni syndrome: current status of clinical applications and future directions. Mol Diagn Ther 17: 31-47. [Crossref]
- Fassnacht M, Kenn W, Allolio B (2004) Adrenal tumors: how to establish malignancy? J Endocrinol Invest 27: 387-399. [Crossref]
- Westra SJ, Zaninovic AC, Hall TR, Kangarloo H, Boechat MI (1994) Imaging of the adrenal gland in children. Radiographics 14: 1323-1340. [Crossref]
- Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, et al. (2007) Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 356: 2372-2380. [Crossref]