Rhabdoid glioblastoma is very rare in clinical Neurosurgery. Herein, we report one case of Rhabdomyoblastoma and summarize its clinical, pathological and imaging characteristics.
Case
Male, fifteen year-old, was admitted to the hospital due to headache for 4 days, accompanied with Paroxysmal convulsions for once at Aug 9th, 2015. Four days ago, this case of patient appeared continuous right frontal and temporal pain without any incentive. The pain turned out to be dull pain, without vomiting. The patient could endure the pain at that moment. Two day ago, sudden involuntary convulsions occurred in his limbs for once time, combined with gaze up in the eyes, trismus, and he could not be awake, above manifestation lasted for two to three minutes and spontaneously relieved. He had not any special history. Nervous system detection did not found positive signs. Head CT (Figure1a) showed an irregular focus with solid and cystic character in the right frontal lobe, with unclear border. The density of this focus was inhomogeneous?and there was punctate calcification in the focus, and flaky edema was found surrounding the focus. Enhanced focus exhibited obviously inhomogeneous strengthening. The right lateral ventricle was slightly compressed and translocated, with middle line structure slightly left deviation. Head MRI (Figure 1b and1c) showed a mass occupying lesion residing in the right frontal lobe, with unclear boundary, turning out to be inhomogeneous and slightly longer T2, T1 signal performance. DWI phase displayed mixed signal, it could be seen patchy and long cystonecrosis of T2 and T1signal. There were patchy and long edematous zone of T2 and T1 signal. The local cerebral sulcus became shallower, the right lateral ventricle slightly got deformation and displacement, accompanied by middle line structure slightly left avertence. Following enhanced scan, the focus turned out to be obviously irregular ring strengthening, the surrounding edema was not strengthened, the size of the focus was in total 3.4cm×4.0cm×4.0cm. The preoperative diagnosis was preliminarily determined as occupying lesion of right frontal lobe, possibly glioma. On Aug 14, 2015, the patient was subjected to the operation of right frontal lobe space occupying resection. During the operation, we found that the boundary between the tumor and the surrounding tissue was unclear, the tumor had rich blood supply, the appearance of the tumor was grayish brown. It was soft, and there were some calcified nodules with slightly hard texture. The tumor was blockly resected, routine pathological examination was carried out. Morphological characteristics of microscope (Figure 1d) revealed Rhabdoid tumor like cells diffusely distributed, with nuclear bias and abundant eosinophilic patina, these cells aggregated in flakiness or showed in stripe arrangement. The tumor cells had apparent heteromorphism and nucleolus, mitotic image could be seen. There was massive necrosis found, accompanied by lymphocytic infiltration. Immunohistochemistry outcomes showedGFAP (+/-),Vimentin(+), EMA(+/-)CK(+/-)S100(+),INI-1(+),Olig2(+/-),Oct4(+/-),HMB45(-),CD1a(-), Ki-67 45%. The pathologic diagnosis is determined as Rhabdoid glioblastoma(WHO? grade) in the right frontal lobe. Two weeks post of the operation, this patient was transferred into the tumor radiotherapy department of our hospital and underwent radiotherapy, once daily, simultaneously he was subjected to chemotherapy by Amines administration. After one month’ treatment, the patient’s condition improved and discharged. Three months after the operation, MRI review indicated that tumor diffused through cerebral pia mater and a new focus of 2.1cm×2.1cm×1.6cm was found in the ipsilateral temporal lobe. This patient did not continue treatment. Finally, five months post of operation, he died because of deterioration of the condition.
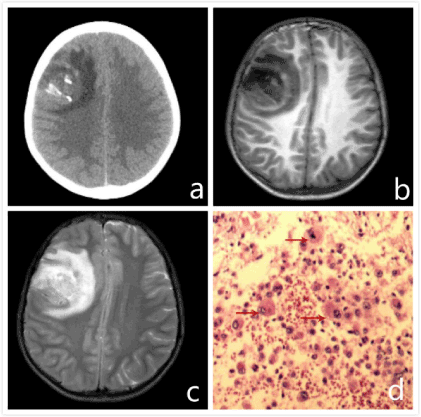
Figure 1.Preoperative CT showed (a)an irregularsolid and cystic focus was found in the right frontal lobe, there was Nodular calcification in the focus, and surrounding with patchy edema. The right lateral ventricle was slightly compressed and translocated, with middle line structure slightly left avertence. Preoperative head MRI (b,c) showed there was a massive occupying lesion in the right frontal lobe, its boundary was unclear, exhibiting inhomogeneity and slightly longer T2, T1 signal performance. Within the focus, there were patchy and long T2,T1cystonecrosis?with patchy and relatively longer T2,T1 signal performance , with edematous zone surrounding the focus. Enhanced scan revealed the tumor as irregular, obvious ring strengthening, surrounding edema was not strengthened. Under the microscope (d): arrow denoted Rhabdoid tumor cells had nuclear bias, cytoplasm was abundant with Eosinophilic substance that diffusely distributed. The tumor cells had obvious heteromorphic and nucleoli. The mitotic image could be seen.
Discussion
In 2001, Wyatt-Ashmead [1] found under the microscope that glioma had the structure of Rhabdoid cells. Based on this, they proposed the first case report and named this kind of disease as Rhabdoid glioblastoma. Because the few number reported, in 2016,it was not got involved in the classification of Central Nervous System Tumor of WHO [2].Rhabdoid glioblastoma is a kind of highly malignant tumor that is common in young patients. The predilection site locates at frontal and temporal lobe, with the characteristic of early recurrence and leptomeningeal spread [3-7]. The symptom of Rhabdoid glioblastoma is mainly craniofacial symptoms resulted from intracranial space occupying effect, such as headache, vomiting, visual disturbance and epilepsy, etc. MRI of Rhabdoid glioblastoma generally expresses as inhomogeneous and mixed T1, T2 signals, after enhancement the focus turns out to be ring strengthening, commonly accompanied with extensive necrotic foci, occasionally there is hemorrhage, cystic degeneration occurred in the focus [6]. So far there are not any distinct imaging features found for Rhabdoid glioblastoma. On the aspect of Pathology, Rhabdoid glioblastoma displayed as: under the microscope it can be seen that tumor cells show round or oval shape, with cytoplasmic rich eosinophilic, nucleus large deviation, and have obvious nucleoli. Immunohistochemistry reveals that this kind of tumor shows GFAP and Vimentin positive, focal expresses EMA, CK and INI-1 positive or special expression absence [3-8]. Although Atypical teratoma/ rhabdomyosarcoma also has Rhabdoid tumor cells in the field of vision, the INI-1 expression is completely absent. While for Rhabdoid glioblastoma, INI-1 expression is positive or special expression absence, giving concrete evidence to distinguish with above two diseases [8]. In 2016, Epithelioid glioblastoma, newly added in the CNS tumor classification of WHO shows character of rich in Eosinophilic cytoplasm, chromatin vacuoles, Epithelioid cells with large nucleolus. Parts of tumor cells have nuclear bias and para inclusion body, similar to Rhabdoid cells [2]. It has some morphological features that overlaps with those of Rhabdoid glioblastoma, but these two tumors are hard to be distinguished from each other on immunohistochemical and genetic level. Some scholars found that Epithelioid glioblastoma, unlike Rhabdoid glioblastoma, has fewer expression of immunological markers, shows INI-1 positive, without INI-1 focal expression absence [8]. Additionally, Epithelioid glioblastoma commonly accompanies by BRAF V600 mutation [2], whereas in the Rhabdoid glioblastoma, BRAF V600 is generally negative [7]. The treatment of Rhabdoid glioblastoma is commonly predominant on operation, in which the tumors are required to be totally resected. Following operation, radiotherapy and chemotherapy, serving as adjunctive therapy, would be performed. Even though the strategies above mentioned been done, it has been reported that [3-8] most cases recurred or committed Leptomeningeal spread, with poor prognosis. The mean life span of Rhabdoid glioblastoma is about 4.1 months [4]. This case in our report died five months after the operation from Leptomeningeal spread. The number of Rhabdoid glioblastoma cases is limited, based on literature retrieval, there are only 20 cases of this kind of tumor [1, 3-8]. It’s clinical diagnosis and therapeutic methods remain elucidation.
Acknowledgement
This clinical case research has been endowed by the Yunnan Province Science and Technology Agency –Kunming Medical University Joint Special Fund (2015FB008).
References
- Wyatt-Ashmead J, Kleinschmidt-DeMasters BK, Hill DA, Mierau GW, McGavran L, et al. (2001) Rhabdoid glioblastoma. ClinNeuropathol20: 248-255.[Crossref]
- Louis DN,Perry A,Reifenberger G (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary.Acta Neuropathol131:803-820.
- Momota H,Iwami K,Fujii M (2011) Rhabdoid glioblastoma in a child:case report and literature review.Brain TumorPathol28:65-70.
- He MX,Wang JJ (2011) Rhabdoid glioblastoma:case report and literature review.Neuropathology31:421-426.
- Chen SC,Lin DS,Lee CC (2013) Rhabdoid glioblastoma:a recently recognized subtype of glioblastoma.Acta Neurochir(Wien)155:1443-1448.
- Gelal MF,Rezanko TA,Sarp AF (2014) Magnetic Resonance Imaging Features of Rhabdoid Glioblastomas. ClinNeuroradiol03:66-67.
- Li Yang, Zeng Tao, Li Bin (2015) Clinicopathologic characteristics of unusual rhabdoid glioblastoma. Chinese Journal of Pathology44: 772-777.
- K1einschmidt-DeMasters BK,Alassiri AH,Birks DK (2010) Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI-1 but not claudin 6.Am J SurgPathol34:341-354.