Acute myeloid leukemia (AML) is ranked as the sixth highest cause of cancer-related deaths. Leukemic stem cells (LSCs) are suspected to be the cause of AML’s relapse and resistance to conventional chemotherapy. LSCs and normal hematopoietic stem cells (HSCs) share many properties such as self-renewal capacity, which appears
to be responsible
for the maintenance of leukemia cells in bone marrow and peripheral blood. In recent years, significant achievement in understanding the LSCs’ biology and regulators has been made that opens new windows for targeting LSCs. Some of these strategies are directed against the signaling pathways such as Wnt, and others are directed against transcription factors such as nuclear factor kappa B (NF-κB). Research is also ongoing to look for epigenetic reprogramming of LSCs by targeting DNMT3A mutation. This review highlights studies that target the intrinsic LSC regulators.
acute myeloid leukemia; hematopoietic stem cells; leukemic stem cells; intracellular targeting; self-renewal mechanisms; properties of leukemic stem cells
Cancer stem cell (CSC) was defined by The American Association for Cancer Research Stem Cell Workshop as a cell within the tumor that is able to self-renew and gives rise to the different lineages that comprise the tumor [1]. CSCs, tumor-maintaining cells, or cancer stem-like cells represent malignant stem cells that have recently been thought to be the source of different human cancers [2]. These cells are typically rare and possess specific characteristics that make them different from other tumor cells. The properties of CSCs make the current chemotherapies ineffective. Moreover, targeting CSCs may represent a new and more selective approach in cancer treatment [2].
Acute myeloid leukemia (AML) remains a great hematological challenge for researchers. Every year around 20,000 new cases are diagnosed in the United States alone, with a death rate of more than 50% [3]. AML is defined as a group of genetically and morphologically heterogeneous disorders characterized by the accumulation of blast cells in the bone marrow and blood [4].
AML’s relapse and resistance against conventional chemotherapy might be related to a small clone referred to as Leukemia stem cells (LSCs). LSCs, tumor-maintaining cells in leukemia, can be identified by their unique surface molecules (most LSCs present within CD34+/ CD38- clone), their ability to perform serial replanting in cytokine-enriched methylcellulose media, and their limitless self-renewal ability in vitro. In a xenograft model, they caused leukemia and gave rise to progeny that have no further leukemia-initiating activity [4]. The frequency of LSCs varied between different AML samples and types, ranging from 1 in 104 to 1 in 107 cells, about 0.1 to 1% of the AML cell population [5].
In this review we present the latest therapeutic approaches targeting the intrinsic regulators of the LSCs such as Hedgehog and Wnt pathways and their effect on LSCs [6]. Targeting of the ATP Binding Cassette Transporter and ALDH enzyme also may have a role in increasing cure rates [7]. Additionally, epigenetic reprogramming of LSCs by targeting the DNA (cytosine-5)-methyltransferase 3A (DNMT3A) mutation is another mechanism that increases chemotherapeutic efficacy [8]. A significantly higher expression of apoptotic receptors in LSCs might be suitable for selective eradication of LSCs by targeted apoptosis induction [9].
Two main properties that are the cornerstone for LSCs’ long-term survival are the maintenance of LSCs’ quiescent state and resistance to cytotoxic agents [4]. LSCs can remove potentially toxic agents from the cell through the unique expression of the p-glycoprotein multidrug resistance efflux pump, also known as MDR1. This might play an important role in LSCs’ resistance to chemotherapy [10].
Recent studies regarding the self-renewal activities of LSCs demonstrate the important role of Bmi-1 [11], Wnt/β-catenin [12], and Hedgehog [13] in this phenotype. Also, over-expression of
Hox genes,
especially HoxA9, has been demonstrated to have a critical role in the pathogenesis of MLL-AF9-induced AML [14]. On the other hand, LSCs may evade apoptosis by the upregulation of nuclear factor kappa B (NF-κB) [15].
LSC targeting is the new hope to eradicate AML without harm to normal hematopoietic stem cells (HSCs). It is important to overcome the properties that make LSCs resistant to therapy such as their ability for limitless self-renewal [16].
Eliminating LSCs’ self-renewal mechanisms
There are four main signaling pathways that allow HSCs to self-renew: Hedgehog (Hh), Wnt/β-catenin, homeobox (HOX), and Notch. Recent studies reported that LSCs also depend on these pathways for their self-renewal [6].
Hedgehog pathway is initiated by binding of one of the Hh ligands (Sonic Hedgehog (Shh), Desert Hedgehog (Dhh), or Indian Hedgehog (Ihh)) to the Patched receptor, resulting in relief of the inhibitory effect on Smoothened (Smo) [17]. Smo through the Gli transcription family activates Hh target genes (self-renewal genes) such as B cell-specific Moloney murine leukemia virus insertion site 1 (Bmi-1) and
Cyclin D/E (Figure 1).
Cyclopamine can stabilize Smo in its inactive condition, resulting in the inhibition of Hh pathway and blocking of LSCs’ self-renewal [6]. Hegde et al. tested cyclopamine in vitro and it completely inhibited LSC colony formation compared to control [18]. They also demonstrated that cyclopamine did not increase apoptosis, but the cells’ morphology changed on treatment [18].
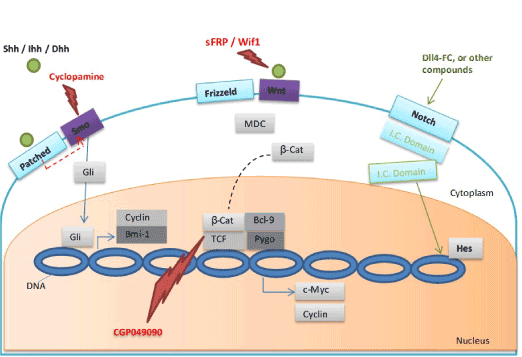
Figure 1. LSC self-renewal mechanisms showing three pathways: Hedgehog, Wnt, and Notch
For the Hedgehog pathway, under normal processes Smo receives inhibitory signal from the cell surface receptor Patched. Binding of a ligand (Shh, Ihh, or Dhh) to Patched leads to release of Smo due to the inhibitory signal from Patched receptors, and through the Gli transcription factors family enhances the expression of several genes including Bmi-1 and Cyclin D/E. Cyclopamine can inhibit the Hedgehog pathway by fixing Smo in its inactive form. For the Wnt pathway, binding of Wnt to Frizzled receptor inhibits the multiprotein destruction complex (MDC). β-catenin accumulates and passes to the nucleus where it associates with other transcription factors such as TCF, Bcl-9, and Pygo, enhancing expression of several genes (e.g. Cyclin D, and CD44). Scavengers (sFRP, Wif1, and Cerberus) prevent binding of Wnt to its receptor and in turn inhibit the Wnt pathway. Two natural compounds, CGP049090 and PKF115-584, may have a role in disturbance of the TCF/β-catenin interaction and in turn inhibit the self-renewal capacity of LSCs. The Notch signaling pathway is inactive in blast cells and also in AML LSCs. Notch pathway reactivation using Notch-activating ligands, specific antibodies, small molecule agonists, or fusion molecules (Dll4-Fc) may be effective in the AML LSCs. Small, solid circles = activators, lightning bolts = inhibitors.
Abbreviations: AML: Acute Myeloid Leukemia; β-cat: β-Catenin; Dhh: Desert Hedgehog; Hes: Transcription Factor Hes; Ihh: Indian Hedgehog; I.C. Domain: Intracellular Domain; LSC: Leukemia Stem Cell; MDC: Multiprotein Destruction Complex; Smo: Smoothened; Shh: Sonic Hedgehog; sFRP: secreted Frizzled-Related Proteins; TCF: T Cell Factor (transcription factor); Wif1: Wnt Inhibitory Factor 1.
There is evidence that the Wnt signaling pathway plays an important role in LSCs’ self-renewal because it can be activated by several oncogenic fusion proteins (e.g. RUNX1-RUNX1T1) [19]. Recent studies report the role of β-catenin in the survival of Imatinib-resistant chronic myelogenous leukemia (CML)-LSCs in breakpoint cluster gene Abelson (Bcr-Abl)-induced CML [20]. Targeting of the Wnt signaling pathway can be achieved by scavenger proteins such as secreted Frizzled-related proteins (sFRP) and Wnt inhibitory factor 1 (Wif-1), preventing Wnt from binding to its receptors (Figure 1). Two natural compounds that may have a role in disturbance of the T cell factor (TCF)/β-catenin interaction and inhibit the self-renewal capacity of LSCs have been identified, CGP049090 and PKF115-584 [6]; both inhibitors induced cell death in in vitro studies, while normal HSCs were largely unaffected. These results support the role of disturbance of the TCF/β-catenin interaction and its therapeutic values in treatment of AML [21].
In contrast, the Notch signaling pathway is silenced in blast cells and also in AML LSCs. Notch silencing in AML is partially caused by increased levels of H3K27me3, a histone mark associated with transcriptional repression, on Notch target promoters (epigenetic silencing). Notch pathway reactivation via Notch-activating ligands or fusion molecules (Dll4-Fc) efficiently targets both human and mouse AML, leading to growth inhibition, differentiation, and cell apoptosis (Figure 1) [22].
LSCs appear to rely on different survival pathways and transcription factors, described below.
Nuclear factor kappa B
NF-κB is a transcription factor that promotes cell growth and also down regulates apoptotic activity within the cell [23]. The high expression of NF-κB within LSCs might be used for selective LSC eradication that could be regulated by modulation of the NF-κB pathway [24].
Prevention of degradation of inhibitor of κB (IκB) by the proteasome inhibitor bortezomib enhances the anti-NF-κB effect [25]. Also, NF-κB could be inhibited in LSCs by parthenolide (PTL), leading to decreased engraftment of leukemic cells into NOD/SCID mice, but the poor water solubility of PTL remains a problem. Initial in vitro studies were done to compare the effects of PTL against AML versus normal specimens. After 18 hours in culture, the viability of AML CD34 cells was more than 10-fold less than normal CD34 controls. Another NF-κB inhibitor is the parthenolide derivative dimethyl-amino-parthenolide (DMPAT), with similar activity as PTL and with better water solubility, resulting in high bioavailability of orally administrated DMPAT [26].
PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway appears to have links to different transcription factors such as the FOXO family that regulates Reactive Oxygen Species (ROS), the Bcl-2 family (apoptosis regulators), and the Wnt/β-catenin pathway controlling the LSC self-renewal [27]. Several inhibitors were implicated in the PI3K/mTOR pathway inhibition such as temsirolimus and wortmannin and could be used with other agents to eradicate LSCs [27]. There are several in vitro studies in which wortmannin was used to suppress the PI3K/Akt axis of human AML cell. As a result of the treatment, leukemic cells’ apoptosis and sensitivity to chemotherapeutic drugs increased, while the normal hematopoietic progenitors were less affected [28]. Blocking the PI3K/Akt pathway might be a promising approach to treat AML, however the use of wortmannin derivative PX-866 is associated with increased risk of undesirable side effects (e.g. hyperglycemia) because it may target other important enzymes such as lipid kinase [28].
Apoptosis inhibition through the Bcl-2 family
Apoptosis inhibition through the Bcl-2 family is another mechanism used by leukemia cells. This family consists of several members: Bcl-2 and Mcl-1 members are inhibitors for apoptosis, and Bax and Bad are promoters [29]. In AML, the Bcl-2 to Bax ratio is increased and is usually associated with poor prognosis. Several anti-apoptotic Bcl-2 inhibitors now under clinical trials for AML include ABT-737, ABT-263, and obatoclaxmesylate [30]. ABT-737 has been shown to inhibit Bcl-2, Bcl-w and Bcl-XL, but not other anti-apoptotic proteins, especially Mcl-1, that make ABT-737 unable to affect many cell types [31]. Van Delft et al. demonstrated that Mcl-1 overexpression resulted in increased ABT-737 resistance, such that down regulation of Mcl-1 will lead to an increase in sensitivity to the drug [32].
ABT-263, which is structurally related to ABT-737, has shown strong cytotoxicity against different human tumor cell lines including hematological malignancies. Recent preclinical trials showed in vitro activity of ABT-263 against acute lymphoblastic leukemia’s (ALL) cell lines but with less activity against solid tumor cell lines. Similar to ABT-737, it has low affinity for Mcl-1. ABT-263 is currently in a phase I/II clinical trial [31].
Obatoclax has been shown to inhibit the pro-apoptotic/anti-apoptotic proteins’ interaction and also increase the pro-apoptotic protein Bim. Obatoclax is currently in a clinical phase I/II trial [31]. A recent study demonstrated the synergism between obatoclax and ABT-737 to induce apoptosis, such that obatoclax might have an important role in augmentation of the activity of chemotherapy or other apoptosis inhibitors [31].
SALL4
SALL4 is another transcription factor that appears to protect HSCs and LSCs by inhibiting different apoptotic pathways [33]. The effect of SALL4 is mediated mainly through the Bmi-1 gene. Several observations illustrate the role of SALL4 in leukemia including the reduced ability of leukemic cell lines with a defect in SALL4 to initiate leukemia in NOD-SCID mice [25]. Using SALL4 as a target to initiate apoptosis of LSCs is attractive, but the high risk of multi-organ involvement raises doubts about this strategy. Therefore, Bmi-1 targeting may be a better alternative to inhibit SALL4’s effect and down regulated cell growth [25]. Rizo et al. demonstrated that down regulation of Bmi-1 resulted in marked impairment in the proliferative capacity of CD34+ cells (decreased by more than 10-fold) [34].
Hepatic leukemic factor
Hepatic leukemic factor (HLF) appears to be involved in drug resistance and survival of LSCs [35]. Cantharidins (derived from Chinese conventional medicine) showed a good efficacy in inhibition of HLF and promoted leukemic cell apoptosis in vitro without notable effects on normal HSCs, but their activity in vivo is disappointing [25].
Farnesylation
Another strategy depends on inhibition of farnesylation by tipifarnib and lonafarnib, which are responsible for the post-translational modification of several proteins implicated in LSC proliferation, especially RAS [36]. However, the clinical trial of farnesylation inhibitors did not improve survival or remission rates in comparison with conventional chemotherapies [25].
Aurora A kinases
A family of mitotic serine/threonine kinases that is essential for cytokinesis during cell division, Aurora A kinase (AurA), has been reported as an AML-LSC target [16]. The development of specific AurA inhibitors may be effective to reduce AML-LSCs [37]. An AurA inhibitor was tested against AML cell lines (NB4 and KG1) and also CD34+/CD38- populations containing LSCs, obtained from AML patients. AurA inhibitor was associated with increased apoptosis (more than 3-fold) [37]. Further clinical trials are needed to evaluate the effect of AurA inhibitors.
Phosphatase and tensin homologue
Deletion of phosphatase and tensin homologue (PTEN) in 19 mice resulted in the development of myeloproliferative disorder in 17 mice and progressed to leukemia. PTEN deletion was associated with increased cell cycle progression and impaired HSC replenishment. It was also noted that the role of the mammalian target of rapamycin (mTOR) can mediate the effects of PTEN mutations through the success of the mTOR inhibitor rapamycin to reverse the PTEN mutation effects [38]. Therefore, rapamycin seems to be a new approach for selective eradication of LSCs.
Activation of apoptosis-inducing receptors
The tumor necrosis factor (TNF) related receptor (TRAIL-R) family plays a key role in cell fate and survival. The most important members of the TRAIL-R family are TRAIL-R1 and TRAIL-R2 [39]. Binding of ligands to TRAIL-R1 or TRAIL-R2 activates and initiates different caspases, leading to cell apoptosis [39]. The high expression of both TRAIL-R1 and TRAIL-R2 on LSCs has been reported [40]. Recent studies demonstrated that normal cells appear to be more resistant to TRAIL-R ligation than malignant cells [6]. There have been several clinical trials to activate TRAIL-R1 and TRAIL-R2 on LSCs to induce selective LSC elimination using specific antibodies and recombinant preparations of soluble TRAIL (sTRAIL), but the results were variable and further clinical trials are needed to evaluate the effect of TRAIL activation [41]. Trebing et al. demonstrated that TRAIL fusion proteins with CD70 antibody, a typical member of the TNF that is present with high frequency in hematological malignancies, triggered cell apoptosis by about 10- to 100-fold [42].
ATP binding cassette transporter
The ATP binding cassette transporters (ABC) family consists of several members (e.g. multidrug resistance proteins (MRPs/ABCC) and P-glycoprotein (P-gp/ABCB1)) [43], with variable expression from one cell type to another. Stem cells express high levels of these transporters, which may play an important role in protection of stem cells against cytotoxic agents [23]. Verapamil, methylene blue MS-209, and tariquidar are examples of ABC inhibitors. Use of the ABC inhibitors with other cytotoxic agents has shown promising effects because they caused marked accumulation of the cytotoxic agents within the malignant cells, resulting in enhanced tumor cell death [44]. Preclinical trials of verapamil and valspodar (first and second generation of ABC transport inhibitors) were not successful due to the non-specificity and the need of high concentrations to inhibit activity. On the other hand, phase I trials of third generation inhibitors (e.g. tariquidar) revealed great effects, but the unexpected drug interactions, involvement of other drug transporters and variability in the expression of drugs’ transporters among individuals restricts the sensitivity of such agents in clinic [45]. Some researchers seek to target the regulators of ABC protein expression by targeting ABC transporter mRNA using siRNA [23].
2021 Copyright OAT. All rights reserv
Aldehyde dehydrogenase (ALDH)
ALDH is a cytosolic enzyme that has an important role in detoxification of alkylating drugs such as cyclophosphamide and the synthesis of retinoic acid [6]. The normal bone marrow consists of two populations: CD34+CD38-ALDH low and CD34+CD38-ALDH high. The CD34+CD38-ALDH high population consists of a higher proportion of long-term colony initiating cells and was capable of initiating normal hematopoiesis in a mouse model. In AML, additional populations with intermediate ALDH activity (CD34+CD38-ALDHint) could be detected with the ability to generate AML in a mouse model [46]. Other studies reported higher relapse rates in patients with an increased proportion of CD34+CD38-ALDHint [27]. ALDH inhibitors such as diethylaminobenzaldehyde and all-trans retinoic acid may be effective to sensitize AML-LSCs to chemotherapy, especially to cyclophosphamide [27]. An in vitro study was performed by incubating ALDH+ cells with BODIPY aminoacetaldehyde (in the presence of verapamil), which converted the BODIPY aminoacetaldehyde in cytoplasm into a fluorescent substance. In the presence of diethylaminobenzaldehyde, ALDH activity was decreased by more 90% with no fluorescent subpopulation detected [47].
Epigenetic reprograming of LSCs
DNA (cytosine-5)-methyltransferase 3A (DNMT3A) is an enzyme that is responsible for the DNA methylation process by transfer of methyl groups to specific CpG sequences present in high concentrations upstream of genes. Increased methylation of these CpG islands is often associated with reduced expression of the downstream gene [48]. Human DNMTA3 enzyme is encoded by the DNMT3A gene, which is represented as a target of different mutations. DNA hypermethylation, especially CpG islands in the promoters of tumor-suppressor genes, has been hypothesized to be involved in the pathogenesis of different malignant tumors [48]. Recent studies reported an association between DNMT3A gene mutations and AML, especially at amino acid R88. These mutations were highly linked to patients with an intermediate-risk cytogenetic profile [8]. Many trials have been designed to inhibit DNA methyltransferase, but the response rate has not been satisfactory [49]. Recent trials with a specific microRNA to target DNMT3A mRNA were associated with improvement in the response rates [50]. Garzon et al. demonstrated that infection of AML cell line, Kasumi-1, with miR-29b lentivirus decreased the endogenous mRNA levels of DNMT3A by about 6.1 fold within 24 hours [51].
The rising interest in studying LSCs’ biology and signal transduction has led to the development of new powerful tools for targeting acute myeloid leukemia. Until now data from in vivo and in vitro studies has led to development of agents directed against specific components in the survival pathways and epigenetic reprograming, but still the most important question is: does inhibition of the survival pathways and transcription factors also affect normal cells?
In vitro findings demonstrate that inhibitors commonly kill leukemic cells that show significant increase in the intrinsic regulators (e.g. increasedPI3K/Akt activity). It may be related to the increased dependence of the LSCs on the intrinsic regulators promoting cellular survival and resistance to the different forms of stress (e.g. chemotherapy). Also, LSCs might be more sensitive to those inhibitors than normal cells because LSCs grow in microenvironments that are deprived of nutrients, which lead to up regulation of the signaling pathways during the progression of the disease for their survival. Thus, even a partial inhibition of these intrinsic regulators might be sufficient to affect the LSCs’ survival and proliferation without damaging HSCs.
To date there have been many trials with different levels of success because of the difference in expression of each target from one patient to another. Therefore, development of personalized therapies may be a promising way to design an ideal strategy for each patient.
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, et al. (2006) Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 66: 9339-9344. [Crossref]
- Jordan CT (2007) The leukemic stem cell. Best Pract Res Clin Haematol 20: 13-18. [Crossref]
- Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, et al. (2009) Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 5: 31-42. [Crossref]
- Lane SW, Williams DA (2012) In: Scatena R, Mordente A, Giardina B. Advances in Cancer Stem Cell Biology. New York: Springer: 85-103.
- Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, et al. (2011) Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest 121: 384-395. [Crossref]
- ten Cate B, de Bruyn M, Wei Y, Bremer E, Helfrich W (2010) Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr Drug Targets 11: 95-110. [Crossref]
- Lutz C, Hoang VT, Buss E, Ho AD (2013) Identifying leukemia stem cells--is it feasible and does it matter? Cancer Lett 338: 10-14. [Crossref]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, et al. (2014) Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506: 328-333. [Crossref]
- Majeti R (2011) Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 30: 1009-1019. [Crossref]
- Mahadevan D, List AF (2004) Targeting the multidrug resistance-1 transporter in AML: molecular regulation and therapeutic strategies. Blood 104: 1940-1951. [Crossref]
- Lessard J, Sauvageau G (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255-260. [Crossref]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, et al. (2010) The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327: 1650-1653. [Crossref]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, et al. (2009) Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458: 776-779. [Crossref]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, et al. (2006) Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442: 818-822. [Crossref]
- Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, et al. (2005) The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105: 4163-4169. [Crossref]
- Al-Mawali A (2013) Leukemic stem cells show the way for novel target of acute myeloid leukemia therapy. J Stem Cell Res Ther 3: 151.
- Rohatgi R, Scott MP (2007) Patching the gaps in Hedgehog signalling. Nat Cell Biol 9: 1005-1009. [Crossref]
- Hegde S, Hankey P, Paulson RF. (2012) Self-renewal of leukemia stem cells in Friend virus-induced erythroleukemia requires proviral insertional activation of Spi1 and hedgehog signaling but not mutation of p53. Stem Cells 30: 121-130. [Crossref]
- Mikesch JH, Steffen B, Berdel WE, Serve H, Müller-Tidow C (2007) The emerging role of Wnt signaling in the pathogenesis of acute myeloid leukemia. Leukemia 21: 1638-1647. [Crossref]
- Hu Y, Chen Y, Douglas L, Li S (2009) beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 23: 109-116. [Crossref]
- Minke KS, Staib P, Puetter A, Gehrke I, Gandhirajan RK, et al. (2009) Small molecule inhibitors of WNT signaling effectively induce apoptosis in acute myeloid leukemia cells. Eur J Haematol 82: 165-175. [Crossref]
- Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, et al. (2013) Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med 210: 301-319. [Crossref]
- Chen K, Huang YH, Chen JL (2013) Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin 34: 732-740. [Crossref]
- Burnett AK, Knapper S (2007) Targeting treatment in AML. Hematology Am Soc Hematol Educ Program. 429-434. [Crossref]
- Annaloro C, Onida F, Saporiti G, Lambertenghi Deliliers G (2011) Cancer stem cells in hematological disorders: current and possible new therapeutic approaches. Curr Pharm Biotechnol 12: 217-225. [Crossref]
- Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, et al. (2007) An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 110: 4427-4435. [Crossref]
- Felipe Rico J, Hassane DC, Guzman ML (2013) Acute myelogenous leukemia stem cells: from Bench to Bedside. Cancer Lett 338: 4-9. [Crossref]
- Martelli AM, Nyåkern M, Tabellini G, Bortul R, Tazzari PL, et al. (2006) Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 20: 911-928. [Crossref]
- Reed JC (2008) Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood 111: 3322-3330. [Crossref]
- Kang MH, Reynolds CP (2009) Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15: 1126-1132. [Crossref]
- Azmi AS, Mohammad RM (2009) Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol 218: 13-21. [Crossref]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, et al. (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10: 389-399. [Crossref]
- Yang J, Chai L, Gao C, Fowles TC, Alipio Z, et al. (2008) SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood 112: 805-813. [Crossref]
- Rizo A, Olthof S, Han L, Vellenga E, de Haan G, et al. (2009) Repression of BMI1 in normal and leukemic human CD34(+) cells impairs self-renewal and induces apoptosis. Blood 114: 1498-1505. [Crossref]
- Dorn DC, Kou CA, Png KJ, Moore MA (2009) The effect of cantharidins on leukemic stem cells. Int J Cancer 124: 2186-2199. [Crossref]
- Harousseau JL, Lancet JE, Reiffers J, Lowenberg B, Thomas X, et al. (2007) A phase 2 study of the oral farnesyltransferase inhibitor tipifarnib in patients with refractory or relapsed acute myeloid leukemia. Blood 109: 5151-5156. [Crossref]
- Kim SJ, Jang JE, Cheong JW, Eom JI, Jeung HK, et al. (2012) Aurora A kinase expression is increased in leukemia stem cells, and a selective Aurora A kinase inhibitor enhances Ara-C-induced apoptosis in acute myeloid leukemia stem cells. Korean J Hematol 47: 178-185. [Crossref]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, et al. (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441: 475-482. [Crossref]
- Sarhan D, D'Arcy P, Lundqvist A (2014) Regulation of TRAIL-receptor expression by the ubiquitin-proteasome system. Int J Mol Sci 15: 18557-18573. [Crossref]
- Majeti R, Becker MW, Tian Q, Lee TL, Yan X, et al. (2009) Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci U S A 106: 3396-3401. [Crossref]
- Ashkenazi A (2008) Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 7: 1001-1012. [Crossref]
- Trebing J, El-Mesery M, Schäfer V, Weisenberger D, Siegmund D, et al. (2014) CD70-restricted specific activation of TRAILR1 or TRAILR2 using scFv-targeted TRAIL mutants. Cell Death Dis 5: e1035. [Crossref]
- Deeley RG, Westlake C, Cole SP (2006) Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev 86: 849-899. [Crossref]
- Khdair A, Chen D, Patil Y, Ma L, Dou QP, et al. (2010) Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J Control Release 141: 137-144. [Crossref]
- Fox E, Bates SE (2007) Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther 7: 447-459. [Crossref]
- Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, et al. (2012) A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood 119: 3571-3577. [Crossref]
- Alison MR, Guppy NJ, Lim SM, Nicholson LJ (2010) Finding cancer stem cells: are aldehyde dehydrogenases fit for purpose? J Pathol 222: 335-344. [Crossref]
- McLellan M, Hasserjian R, Emerick K (2010) Marginal zone B-cell lymphoma of the head and neck: a rare case and review. Laryngoscope 120: S165. [Crossref]
- Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, et al. (2009) MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114: 3448-3458. [Crossref]
- Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, et al. (2010) Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A 107: 7473-7478. [Crossref]
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, et al. (2009) MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113: 6411-6418. [Crossref]