Abstract
The human body makes free radicals all the time for useful purposes. The antioxidant levels and control systems recover any injury occurring. The enzymes that repair DNA damage by free radicals are especially important. Inhaled reactive oxidant species (ROS) and those endogenously formed by inflammatory cells constitute an increased intrapulmonary oxidative burden. In many diseases, the balance between oxidants and antioxidants (redox balance) is altered causing severe consequences. He proceeds to review the pathophysiological mechanisms by which free radicals generate various types of stress, such as oxidative stress, nitrative, carbonyl, inflammatory and stress of the endoplasmic reticulum. Later we discuss how these different types of stress affect respiratory functions in diseases such as chronic obstructive pulmonary disease (COPD), bronchial asthma (BA), bronchiectasis, and idiopathic pulmonary fibrosis (IPF). Finally, the role that various strategies to increase lung antioxidants and potentially beneficial effects of antioxidant therapy in the course of the disease are discussed.
Key words
antioxidants, chronic obstructive pulmonary disease, bronchial asthma, bronchiectasis, idiopathic pulmonary fibrosis
Introduction
Humans have evolved complex circulatory, respiratory, and neuroendocrine systems to ensure that oxygen level are precisely maintained, since a deficiency or excess may result in death of cells, tissue, or the organism [1].
What is a free radical? An atom contains a nucleus, around which electrons move-usually in pairs. A free radical is any atom or molecule that contains one or more unpaired electrons. The unpaired electrons alter the chemical reactivity of an atom or molecule, usually making it mores reactivity than the corresponding nonradical [2]. A superscript dot is used to denote free radicals. The hydrogen radical H• (the same as a hydrogen atom), contains one proton and one electron (obviously unpaired). This is the simplest free radical. Free-radical chain reactions often are started by removing H• from other molecules-for example, during lipid peroxidation.
Free radicals are made in the human body or are acquired from the environment. Their action is opposed by antioxidant defense systems. Some antioxidants come from food, others, from the body itself. Although free radicals are an essential part of normal body processes, too many are bad for us [2].
A complex interaction between specific causes of disease, host characteristics, type and intensity of inflammation and various types of stress, produce varied clinical presentations and progression of specific diseases [3].
This paper addresses the pathophysiology of some respiratory diseases facing the redox imbalance and as antioxidant therapies impact the natural evolution of the same.
Oxygen and evolution
The atmosphere as we know it today has about 4.5 billion years. At first probably it consisted of volcanic emanations almost no oxygen. Living organisms appeared on Earth were essentially anaerobic. However, their growth is inhibited and usually die from exposure to concentrations of 21% oxygen, the usual atmospheric level [4]. This level was reached because the primitive marine organisms acquired chlorophyll, a pigment able to convert light energy into chemistry, by photosynthetic combination of water with CO2 generating oxygen. Almost all the free oxygen in the air today was formed by this combination in seawater. Approximately 570 million years ago that the content of dissolved oxygen in the oceans allowed marine life can breathe. It will make about 400 million years that the atmosphere acquired enough oxygen in the gas phase for the animals that emerged from the sea could breathe air [5].
Anaerobic today are presumably descendants of those primitive organism, "adapting" to increased atmospheric levels of oxygen for restricted themselves to environments where oxygen does not penetrate. Other organisms have developed defense systems to protect themselves from oxygen toxicity using the same to metabolic transformations (hydroxylase, oxidase and oxygenase enzymes are examples) and for efficient energy production by using electron transport chains with oxygen as the terminal electron acceptor-such as those present in the mitochondria These produce 80% of our needs cellular adenosine triphosphate (ATP) [2].
It is remarkable that we have developed antioxidant defenses against concentrations of 21% oxygen, but not more. All aerobic species suffer injury when exposed to higher concentrations of 21%. For example, humans if breathed pure oxygen and as early as six hours to develop chest tightness, cough, and sore throat [6]. Exposure periods over the alveoli of the lung damage and premature infants exposed to high doses of oxygen develop fibroplasia lenticular (which may occur with blindness) [7].
In 1954, Gerschman and colleagues in the United States (US) proposed that the harmful effect of oxygen was due to the formation of oxygen radicals, superoxide oxygen theory, followed in 1968 the discovery of the enzyme superoxide dismutase (SOD), specific for the removal of a free radical substrate. In its simplest form, this theory states that oxygen toxicity is due to excess formation of superoxide radical (O2•¯) and the SOD enzymes are important antioxidant defenses [8].
Free radicals
No other molecular factor has had a singular influence on the development and progress of animal life as oxygen [9,10]. PO2 falls as the gas moves from the atmosphere in which he lived until the mitochondria where it is used. At sea level the PO2 of inspired air is 150 mm Hg. When systemic arterial blood reaches the capillary tissue, oxygen diffuses into the mitochondria where PO2 is very low. The "tissue" PO2 probably differs considerably through the body, and at least in some cells, is as low as 1 mm Hg [11].
The molecular oxygen is vital for energy that is essential for life [12]. The hypoxia-inducible factor-1 (HIF-1) mediates the adaptive response experienced by cells to survive oxygen deprivation. One way in which the factor promotes cell survival under hypoxic conditions is the transition from oxidative to glycolytic metabolism. Under aerobic conditions, electrons are transferred from NADH, and flavin adenine dinucleotide (FADH2) to mitochondrial complex I or II, then the complex III and finally the compound IV, where they react with oxygen to form water [1].
Under hypoxic conditions (as occur in respiratory diseases discussed later), the electron release is increased before transfer to complex IV, resulting in the production of superoxide anion, which is then converted to hydrogen peroxide (H2O2) and other ROS. The cost is loss of redox homeostasis [13]. The human body generates, therefore, superoxide by adding a single electron to oxygen.
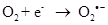
Exposure to high energy radiation (short wavelength) causes splitting of the one of the covalent bonds in water to generate highly reactive radicals called hydroxyl radicals, OH•.
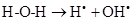
As there is always some degree of exposure to environmental radiation, hydroxyl radicals are always produced in the body. They attack proteins, lipids (damaging cell membranes) and DNA within cells initiating a radical chain reaction, which can be spread over many years. Some free radicals are helpful, as the radical gaseous nitric oxide (NO•), which is produced in almost all tissues. Examples are the walls of blood vessels, where NO• helps to control blood pressure. Nitric oxide and superoxide anion formation carried peroxynitrite that nitrosylates protein amino acid and enzymes, inactivating them [14].
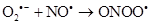
Therefore, the (ROS) and reactive nitrogen species (RNS) are continuously generated at the cellular level by mitochondrial metabolism and inflammatory cells in the case of diseases, by enzymes such as nicotinamide adenine dinucleotide phosphate oxidase (NADP oxidase), xanthine/xanthine oxidase (XO), nitric oxide synthase (NOS) y hem peroxidase. They catalyze the production of radicals.
Antioxidant defences
Endogenous
Endogenous antioxidant defense systems are essential to maintain redox balance. Human tissue has enzymes that protect against free radicals. The SOD enzyme helps convert rid of superoxide to H2O2. Hydrogen peroxide is metabolized by other enzymes such as catalase and glutathione peroxidase. The latter enzyme has H2O2 when used for oxidizing a substrate that is in all cells, the glutathione (GSH) (Figure1). This is a tripeptide composed of glutamic acid, cysteine and glycine is a major cellular antioxidant and thiol redox cycler. Concentrated in the coating fluid has a protective role in maintaining the integrity of the airspace [15,16].
It acts by donating their reducing equivalents/electron transfer in cells leading to the production of oxidized forms (disulfide) which is converted back to GSH by glutathione reductase (Figure1). 20% of the GSH is located within the mitochondria willing to neutralize endogenous ROS (product of metabolism).
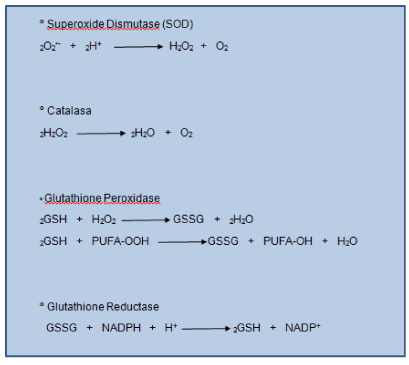
Figure 1. Antioxidant Defence Enzymes. Antioxidant defense mechanisms involve particular substrates (such as glutathione and vitamin E) as well as a set of enzymes. Glutathione peroxidase catalyzes the formation of oxidized glutathione (GSSG show top), in which two GSH molecules are linked by a disulphide bridge. Reduced glutathione (GSH) is a tripeptide (glutamic acid-cysteine-glycine) that has multiple metabolic functions. Glutathione reductase catalyses the recycling of GSSG to GSH, which also involves NADPH (shown above).
Other enzymes with protective role are: the aldheyde deshydrogenase and aldo-ketoreductase that detoxify aldehydes reagents formed by carbonylation of proteins [17], as well as glutathione S transferase and the glutamate cysteine ligase, which attenuate inflammation caused by smoking and elastase induced emphysema [18].
Exogenous
Dietary supplements such as antioxidants vitamin E, vitamin C and beta-carotene amplified antioxidant reserves. When reactive free radicals attack cell membranes, they set off a chain reaction called lipid peroxidation, which destroys cell membrane. This reaction targets the polyunsaturated fats (containing fatty acids with double carbon bonds, PUFAs) in cell membrane (Figure 2).
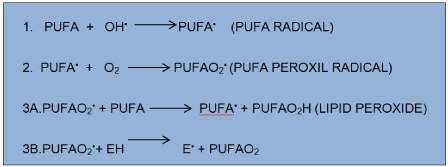
Figure 2. Lipid peroxidation: As shown in the line 3B to the left, the fat-soluble vitamin E (alpha-tocopherol, denoted EH) from food dissolves in cell membranes and can stop the chain reaction.
The poorly reactive residue alpha-tocopherol (E•) is recycled to vitamin E by water-soluble vitamin from food, vitamin C (ascorbic acid). The flavonoid resveratrol a constituent of red wine, has been shown to induce synthesis of GSH [19,20]. Minute amounts of the element selenium are needed in the human diet because, without this element, the enzyme glutathione peroxidase cannot destroy peroxide.
Respiratory diseases
It proceeds to discuss how the redox imbalance produces several pathogenic mechanisms in various respiratory diseases.
Chronic obstructive pulmonary disease
COPD is a chronic inflammatory disease characterized by progressive and partially reversible airway obstruction [21]. The process is initiated by exposure to cigarette smoke or biomass/fuels [22]. COPD affects approximately 200 million people worldwide and is projected in 2020 will be the third leading cause of death worldwide [23-25].
Cigarette smoke and other pollutants/biomass fuels are the initial noxa that stimulates epithelial cells and macrophages to release chemotactic factors that attract T cells, neutrophils and fibroblasts [26-28].
T cells are CD8 + (suppressor/cytotoxic subtype Th1 /Tc1) and release granzymes, perforines and tumor necrosis factor alpha (TNF-α) which cause apoptosis and direct damage to the lung parenchyma, and also release interleukin IL-4 and IL-13 which induce hypersecretion of mucus in the airways [29,30]. Macrophages/neutrophils also play an important role by releasing pro-inflammatory cytokines such as IL-8 and TNF-α and proteases, resulting in inflammation and destruction of lung parenchyma direct [31,32].
There is strong evidence that proteases play critical roles in the pathogenesis of COPD. Alpha-1 protease, matrix metalloproteinase and A desintegrin can cause emphysema [33-35].
Oxidative stress has a pivotal role in the pathogenesis of COPD [36-38] (Figure 3). Exogenous oxidants sources include cigarette smoke, oxidizing gases, ultrafine particulate matter, nanoparticles environmental pollution, exhausted vapors and biomass fuels for cooking and heating homes [39,40]. Endogenous oxidants result mainly from mitochondrial respiration and inflammatory responses to viruses and bacteria. Inflammatory stress mediated by IL-1, TNF-α and interferon-γ generates endogenous ROS. Other sources of intracellular ROS are NADPH oxidase enzyme, XO and hem peroxidases, all of which are increased in the bronchoalveolar lavage (BAL) of COPD patients [41,42].
The inherently unstable free radicals initiate a number of adverse consequences at the cellular level. For example, activate nuclear factor kappa-beta (NF-kβ) increasing the synthesis of IL-8 and TNF-α which recruit neutrophils amplifying the inflammatory stress. Oxidative stress also activates the phosphoinositide 3-kinase (PI3K) which phosphorylates histone deacetylase-2 (HDAC-2) (a key anti-inflammatory enzyme). Oxidants also contribute to the protease/anti-protease imbalance, by reducing anti-protease activity [43].
Expression of transforming growth factor beta (TGF-β) (produced by epithelial cells) is increased in COPD, and inhibits the activity of the enzymes catalase and SOD2. Both are critical to neutralize ROS derived from mitochondria and are under the control of the transcription factor forkhead box class (FOXO3) whose deficiency has been associated with COPD [44]. About 200 antioxidant and detoxifying cellular enzymes are under the control of nuclear erythroid-2 related factor (Nrf2). COPD patients have reduced expression and activity of Nrf2 [45].
The nitrative stress is due to the powerful peroxynitrite radical, which reacts with certain proteins and enzymes (i.e. nitration), reducing their activity and expression. For example, nitration of HDAC-2 inactive it [43]. Carbonyl stress occur when ROS oxidize proteins, lipids, carbohydrates and DNA; producing carbonyl reagents which react with proteins (reactive aldehydes). This is known as protein carbonylation, a non-enzymatic phenomenon [17,46,47]. The endoplasmic reticulum stress can induce mitochondrial apoptosis and cell death [48].
An essential characteristic of COPD is that inflammatory processes and stress continue after stopping exposure to irritants [49]. It is likely that persistent infection and autoimmunity are responsible for this behavior. Carbonyls modified proteins are highly immunogenic, producing autoantibodies which are elevated in the serum of patients with COPD [50]. These auto-antibodies fix complement, and may contribute to emphysema.
Carbonylated proteins are recognized by the innate immune system through PRRs (Pattern Recognition Receptors); expressed by cells that recognize antigens such as dendritic cells and macrophages [51]. In these cells, they are processed and re-expressed in association with the Major Histocompatibility Complex-2 (HLA-2). This promotes the activation of acquired immune response and attracts and accumulates Th1 cells in the lung parenchyma and dendritic cells in small airways [52,53].
Besides producing neo-antigens, the immune response also promotes the influx of immune cells needed to recognize and process them. This stimulus causes the release of CCL2 and CCL20, which recruit dendritic cells, monocytes and lymphocytes. In order to improve the immune response, IL-17 and IL-18 levels are increased. These interleukins activate and mature B cells, and also promote autoimmunity [54].
Respiratory infections may play a role in the development and progression of the disease; and are also the leading cause of acute exacerbations [55]. Tobacco smoke and infections lead to differential activation of multiple PRRs. When it comes to infections, these receptors are activated by PAMPs (Pathogen Associated Molecular Patterns) and DAMPs (Damage Associated Molecular Patterns) [56].
There are at least 5 PRRs clearly defined groups that receive and process these signals: Tool-Like Receptors (TLRs), NOD-Like Receptors (NLRs), cytosolic DNA sensors, RIG-I-Like Receptors (RLRs) and C-type lectin-like receptors (CLRs) [57]. Cells that express PRRs generate cytokines, interferons and chemokines that recruit macrophages, neutrophils and activate epithelial cells; the innate immune response. Dendritic cells, stimulated by PRRs ligands and associated to HLA-2, initiate signals that attract T cells; the adaptive immune response.
COPD patients have increased bacterial colonization with H. influenza, S. pneumoniae, P. aeruginosa and M. catarrhalis, this contributes to chronic inflammation and the airway dysfunction [54].
It is important to emphasize the PRRs role in COPD, since they mediate autoimmune and infectious response: the two events that have been involved in the persistence of the inflammatory process. Process that becomes autonomous and does not disappears, even after exposure to irritants ceases.
Bronchial asthma
BA is the most common chronic inflammatory disease [58]. GINA defined as a heterogeneous disease, in which the cardinal symptoms (wheezing, breathlessness, chest tightness and coughing) change over time and intensity, along with a variable expiratory airflow obstruction. These changes are triggered by factors such as exercise, exposure to allergens or irritants, changes in climate or viral respiratory infections [59] (Figure 4). This variability distinguishes COPD. BA is a common disease that affects 1-18% of the population in different countries. The global number of people affected is approximately 300 million people [60].
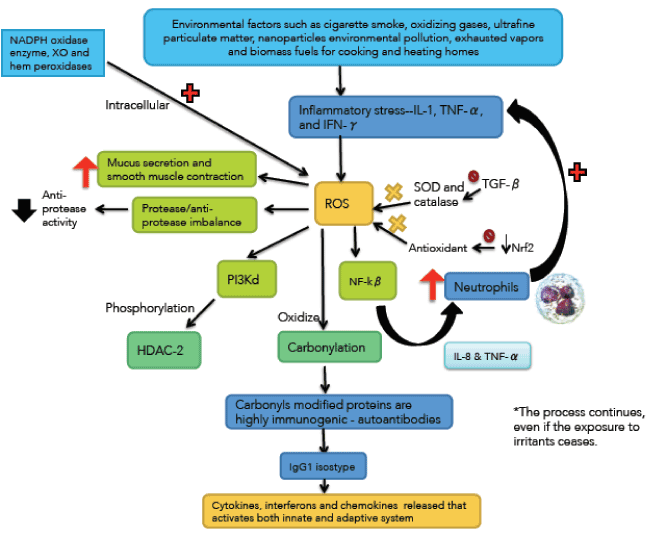
Figure 3. Immunological and inflammatory profile in COPD: Environmental factors produce different types of stress that generates small airways lesion and emphysema; activation of inflammatory cells and autoimmunity. Neutrophils generate more inflammatory stress and autoimmunity activates immune responses that amplify and perpetuate the inflammatory process. The negative sign means repression and the positive sign activation. Straight arrows mean increase or decrease. Curves arrows mean activation and / or expression. See text for description.
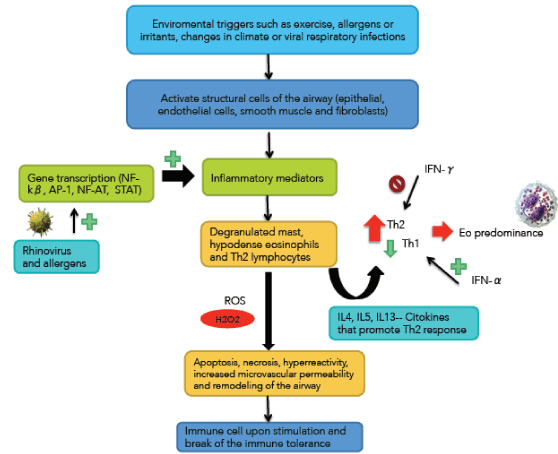
Figure 4. Immunological and inflammatory profile in Bronchial Asthma: Ambient factors activate airway structural cells, and rhinovirus and allergens activate pro-inflammatory factors. Both phenomena produce inflammatory factors that recruit the typical cellular profile of bronchial asthma. These cells generate Th2 cytokines and INF-α that lead to eosinophilic infiltration of the mucosa. Also, these cells, through various types of stress, induce mucosal changes and immunological phenomena. The negative sign means repression and that positive sign activation. Straight arrows mean increase (red) or decrease (green). Curves arrows mean activation and / or expression. See text for description.
These triggers activate structural cells of the airway (epithelial, endothelial cells, smooth muscle and fibroblasts) releasing about 100 inflammatory mediators, within which they are lipid mediators, inflammatory peptides, cytokines, chemokines, inflammatory enzymes, growth factors and adhesion molecules [58,61].
These mediators recruit cells that make up the specific inflammatory pattern of airway of patients with BA: degranulated mast, hypodense eosinophils and Th2 [62] lymphocytes. This inflammatory infiltration is responsible for the clinical manifestations of the entity.
The increased number of Th2 over Th1 carries the eosinophilic inflammation [63]. Interleukin-4 (IL-4) plays a role in Th2 differentiation and the synthesis of immunoglobulin-E (Ig-E) [64]. Interleukin-5 (IL-5) is a factor in the differentiation and survival of eosinophils [65]. Interleukin-13 (IL-13) promotes airway hyperreactivity, hypersecretion of mucus, eosinophilia and airway remodeling [66]. These are Th2 cytokines [67,68].
The expression of inflammatory mediators is regulated by gene transcription, which in turn is controlled by pro-inflammatory factors such as NF-kβ, AP-1 (activator protein-1), NF-AT (nuclear factor of T cells activated) and STAT (signal transducers and activators of transcription) [69,70]. Some of these factors can be activated by rhinovirus and allergens, potentially enhancing asthmatic inflammation [71]. This inflammation creates oxidative stress, which in turn activate AP-1 and NF-kβ and these in turn generate inflammatory response generating a complex interplay between stress and inflammation that perpetuates both phenomena [72-74].
There is strong evidence that increased levels of ROS act as a critical contributor to induce allergic airway inflammation [75-78]. Genes and oxidative stress in BA is an area that needs further research [79,80].
The different response of T cells and other inflammatory cells to various kinds of stress in BA and COPD cells possibly lies in a genetic basis.
It is controversial to where eosinophils are elevated in the sputum of patients with stable COPD, although they are elevated during exacerbations [81]. 10% of patients with COPD respond to inhaled glucocorticoids, these patients have a great numbers of eosinophils in the airways and greater reversibility to bronchodilators. It has been suggested that may have concomitant BA [58]. Recently, in an effort of GINA and GOLD the term ACOS was developed, as a syndrome overlap between asthma and COPD. Definition of the syndrome is mainly descriptive and refers to a subgroup of patients with persistent airflow obstruction, concomitantly they show changes associated with BA and COPD. These patients have frequent exacerbations, poor quality of life, rapid decline in lung function and increased mortality and also consume more health resources than patients who only have BA or COPD. The point is to identify and treat them properly as they have a poor prognosis compared with either of the two components separately [82]. In the sputum of these patients are neutrophils and eosinophils and there is an urgent need to investigate whether the syndrome is the result of a changing pathogenesis of BA into COPD over time or has a different molecular biology and immunopathogenesis [83].
Non-cystic bronchiectasis
Bronchiectasis is defined clinically by the symptoms of persistent or recurrent bronchial infection related to dilated bronchi and irreversibly damaged. The condition is persistent or progressive and the bronchial wall is not only long but also thickened [84]. The true incidence remains unknown in many populations. The prevalence increases with age [85]. The general belief is that the incidence is decreasing. This is attributed to the introduction of antibiotics and immunization for children and for these reasons is not considered a major health problem and some consider it an “orphan disease”.
The underlying disease process is damage to the airways that results from an event or series of events where inflammation is central to this process. This is the hypothesis of "vicious circle". Continuous exposure to inhaled pathogens and environmental pollution bankruptcy sophisticated systems of primary and secondary defenses that maintain the sterility of the normal lung. The lung becomes susceptible to infection and colonization (persistence of bacteria in the lower respiratory tract) with subsequent inflammation that causes damage to the airway altering host defenses [84].
As a result of this inflammatory process various kinds of stress are generated. Plasma biomarkers as TAC (Total Antioxidant Capacity) and SOD were decreased. CAT (Catalase Activity), 8-iso-PF2alfa (8-iso-prostaglandine F2alpha) TBAR (Thiobarbituric acid reactive substance), were elevated in patients with bronchiectasis compared with controls [86]. These levels are due to a chronic inflammatory syndrome neutrophil activation.
In turn, inflammation exacerbates ROS induce the production of cytokines and chemokines by stimulating inflammatory genes, to activate NF-kβ and AP-1 [87].
Regardless of the underlying cause, this chronic airway bacterial infection (which is what happens in bronchiectasis) active and recruit phagocytic cells and generates oxidative stress in a scenario where there is a dysregulation of the innate and adaptive immune responses. Cytokines and their receptors may be therapeutic targets [88].
The interrelationship between the three entities (COPD, Asthma and bronchiectasis) is complex and unclear. The prevalence of obstructive diseases in general is increasing in recent decades and increases with age. In the beginning bronchiectasis they are also an obstructive phenomenon probably begins in the small airways [89]. With the evolution of the disease, disorder restrictive added due to the progressive destruction of parenchyma lung [90].
Statistically powerful and excluding Allergic Bronchopulmonary Aspergillosis (ABPA) studies have found varicose bronchiectasis in 60% of patients with non-severe allergic asthma and 50% of severe allergic asthmatics using HRCT scan [91]. Unlike ABPA can affect all lobes and both proximal and distal airways. Therefore the recommendation of the guidelines is that asthma in adults should be considered an etiology of bronchiectasis if another cause can't be identified [84]. However, there is no robust evidence that COPD is a cause of bronchiectasis. This relationship requires further studies [92].
What does seem clear is that the three are inflammatory conditions that cause oxidative stress that damages cells and subcellular compartments [93].
Idiopathic pulmonary fibrosis
IPF is a chronic, progressive and fatal interstitial lung disease characterized by irreversible loss of lung function [94]. Although periods of clinical stability may be observed, it is inevitable progression [95]. The progressive IPF is of unknown etiology. The most common histopathological pattern in biopsies of patients with IPF is the usual interstitial pneumonia. It is characterized for heterogeneous areas of dense fibrosis, the presence of fibroblast foci and "honeycombing" with parenchymal distortion architecture [94].
The prevalence varies around 1.6-1.7 /100,000 inhabitants [96]. Clinically, IPF affects over 500,000 people in US alone, and the average survival is 2-3 years after diagnosis [97]. The 5-year survival time is similar to that of various types of cancer [98-100].
IPF is characterized by progressive fibrosis without substantial swelling [101]. Several proposed mechanism can act sequentially, in parallel or in series and being part of a whole that explain the phenomenon. There is an increased oxidative stress in the lungs of patients with pulmonary fibrosis and decreased antioxidant defenses. The source of ROS is exogenous (such as snuff in IPF) and endogenous epithelial, mesenchymal and endothelial cells within the tissue [102]. ROS cause apoptosis of alveolar epithelial cells and activation of the stress response of endoplasmic reticulum (ER) of these cells. Normally a crosstalk between ER and mitochondria prevent epithelial cell apoptosis. A loss of this "fine" communication favors apoptosis and fibrosis [103].
Furthermore, ROS mitochondrial DNA damage and repair mechanisms aimed at the 8-oxoguanine DNA glycosylase (Ogg1), mitochondrial aconitase (ACO2) and p53 (genome gatekeeper) having a protective role for the regulating thousands of genes. New knowledge of the molecular basis of ROS induced pulmonary disease, in particular death of lung epithelium, could the development of specific therapeutic targets to manage IPF [104].
Research suggests that pulmonary fibrosis is a disease of "two-hit": apoptosis of alveolar cells combined with environmental stress and a genetic predisposition to abnormal cell cycle alveolar epithelial cells regulation [105]. Mutations in genes encoding protein C and A2 surfactant (SFTPC and SFTPA2 respectively), telomerase reverse transcriptase (TERT) and telomerase RNA component (TERC) described in familial pulmonary fibrosis [106]. Later on chromosome 11p were identified gene polymorphism gel-forming mucin (MUC5B) associated with IPF [107]. Genetic studies have resulted in a better understanding of this disease [108].
Another significant pathogenic aspect is the generation, from dysfunctional alveolar epithelial cells, of fibrogenic factors such as TGF-β1, the platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), the fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF). These factors produce signals through tyrosine kinases to produce collagen from myofibroblasts. Block the corresponding receptors is a current therapeutic alternative [109].
Finally the high penetrance susceptibility allele MUC5B and parallelism of idiopathic fibrosis with asbestosis suggests environmental factors that reach the alveoli, and identify them should be a research priority. Concealment chronic infection bacteria, fungi or viruses-are on the list of suspects. The hypothesis of a bacterial component is supported in a recent study where courses trimethoprim-sulfamethoxazole reduced mortality in idiopathic pulmonary fibrosis [110].
Antioxidant treatments
As alternatives, may be common to the various respiratory diseases, this item was developed by therapeutic groups.
Dietary supplements
Dietary antioxidant supplements such as vitamin E, C and beta-carotene, could have a role in the treatment of patients with COPD. Previous epidemiological studies suggest that high income of antioxidants in the diet protects lung function (FEV1 and FVC) and result in a lower prevalence of chronic bronchitis and dyspnea [111-113]. In a cross-sectional study in general population, an increase of 20 mmol /L in plasma concentrations of vitamin C was associated with a 13% reduction in the risk of COPD (odds ratio 0.87), indicating that vitamin C has a role protector COPD [114]. Moreover, polyunsaturated fatty acids n-3, which interferes with the body's inflammatory response, was inversely related to the risk of chronic bronchitis, emphysema diagnosed by the doctor, and COPD detected by spirometry [114].
In an epidemiological study, polyphenols, catechins, flavonol and flavone were positively associated with FEV1 and inversely with symptoms of COPD, suggesting a beneficial effect of a high-income catechin or fruit high in the same in COPD [115]. Furthermore, resveratrol flavonoid, a constituent of red wine, has been shown to induce synthesis of GSH and inhibits macrophage inflammatory cytokines COPD patients [19]. Another polyphenol, curcumin has been shown to inhibit the inflammatory response by inhibiting the activity of NF-kβ and neutrophil recruitment into the lung [20]. Other studies have not found enough evidence of benefit from dietary supplements in COPD and nutritional support suggesting GOLD is for malnourished patients with COPD in order to gain weight and fat free mass, but without any specific recommendation of antioxidants in the diet [16,21].
In BA, the effect of many antioxidants in allergic airway inflammation is at best, moderate [72,112] and GINA not suggest therapeutic use [59]. Although by a different mechanism, vitamin D has indicated a subset of asthmatic patients. IL-10 is an important anti-inflammatory and immunoregulatory cytokine secreted by T cells in response to glucocorticoids (GC) [116]. GC resistant asthmatic patients there are not enough secrete IL-10 in response to hormone and when adding D3 (calcitriol), restore interleukin production, suggesting therapeutic implications. Therefore, a low income dietary vitamin or little exposure to sunlight, could contribute to GC resistance in inflammatory diseases [117].
International guidelines make no recommendation of dietary antioxidants in non-cystic bronchiectasis or IPF [84,94].
Small molecules thiol antioxidants
N-acetylcysteine (NAC) has direct and indirect properties as an antioxidant in the treatment of COPD. NAC free-thiol group is capable of interacting with electrophilic groups of ROS. Indirectly, NAC is a precursor of GHS, a neutralizer of ROS. NAC may serve as a protective factor against external and internal ROS [118]. The longest work of an antioxidant in COPD, the BRONCHUS study, failed to show any effect of orally administered NAC in reducing progression of the disease or frequency of exacerbations [119]. The results of the study can be justified by a failure to act in the sub-cellular compartment, but can also be explained by insufficient drug doses or frequency. An Israeli study found beneficial effects of NAC in the gas trap [120]. The recent HIACE study (1200 mg / day oral NAC for one year), showed significant improvement in the function of the small airways and frequency of exacerbations. But these studies need to be validated with larger caseloads [121]. However, it has not been demonstrated effect on symptoms or quality of life of patients receiving NAC [122].
Other thiol antioxidant (carbo-cysteine) increases the content of sialomucin and mucociliary clearance, and decrease the activity of IL-6. The PEACE study showed that patients treated with carbo-cysteine had fewer exacerbations than the placebo group and were not receiving steroids [123]. The EQUALIFE study, using another antioxidant thiol, erdosteine showed similar results [124]. Finally, fudosteine shows better bioavailability than NAC and reduces mucus secretion by inhibiting the expression of MUC5AC gene [125].
GOLD not recommends regular use of these drugs, while acknowledging that inhaled steroids with or whitout, the use of high doses of NAC significantly reduces exacerbations but especially in patients with stage 2 spirometry [21].
In BA it has not shown a significant effect of antioxidant treatment [72,112]. In fact the latest edition of GINA does not provide this therapy in either adults or children alternative [59]. However, it is necessary to clarify the role of oxidative stress in the development of asthma and to control the intracellular oxidative stress with proper timing, as opposed to simply reduce oxidative stress, could be important for effective treatment [72]. In non-cystic bronchiectasis only carbo-cysteine has shown reduction in gas trapping small trials, but the data are insufficient to justify clinical use. Should be randomized controlled trials to establish clinical efficacy [84].
In IFIGENIA study, researchers found that a regimen of three drugs (prednisone, azathioprine and acetylcysteine) preserved the better lung function than the two-drug regimen (azathioprine more prednisone) in IPF [126].
The PANTHER-IPF study with a similar three-drug regimen had to be suspended because an alert was generated by clinical safety reasons on 14 October 2011 [127]. After redirecting the study N-acetylcysteine was compared against placebo in moderate idiopathic pulmonary fibrosis with relatively little casuistry (133 and 131 patients, respectively). The work showed no significant difference with the antioxidant in the preservation of the FVC, compared to placebo (p=0.77) [128].
Pirfenidone is not a thiol molecule but is a synthetic derivative of pyridone with anti-fibrotic, anti-inflammatory and antioxidant properties, and is the only approved in EU for the treatment of IPF (2,403 mg/day) and one of the two approved in US for IPF. It is associated with a reduction in the decline of FVC in patients with mild to moderate IPF in some studies (CAPACITY study 004 and ASCEND, but not CAPACITY study 006), prolongs progression-free survival and has a manageable tolerability profile, with gastrointestinal and skin-related events occurring most commonly [129].
Nintedanib (originally known as BIBF 1120) is intracellular inhibitor which targets block receptor tyrosine kinases which form the signaling for the fibrogenic factors generated in IPF induce collagen production of myofibroblasts [130]. The two studies of 52 weeks (150 mg BID), randomized, double-blind, Phase 3 (INPULSIS 1 and INPULSIS 2) with a total of 1066 patients, showed significant difference in both (p < than 0.001) in the rate annual decline in FVC. The most common side effect in the group with Nintedanib was diarrhea (61.5% compared with 18.6% in the placebo group) [131].
Both drugs (pirfenidone and nintedanib) were approved by FDA in October 2014 for IPF using the absolute change in percentage of FVC as primary endpoint, but this variable has approval and disapproval as a measure of efficacy in the literature [132]. The threshold of decline, clinically significant FVC was uncertain and had not been validated as a substitute for other clinical variables or death, to assess effectiveness. In recent work it has been a relationship between FVC and mortality trend which has strengthened the reliability of FVC as clinically relevant measure of efficacy. It remains to determine whether the combined use of both drugs may increase the return on monotherapy [133].
IPF being a devastating disease of unknown cause, and that for years the only treatment available was lung transplant, availability of drugs that impact the various types of stress and / or molecular fibrogenic signals opens an avenue of research and potential drugs for these patients.
Nrf2 Activators
In COPD, Nrf2 activators can prevent oxidative stress induced autoimmunity [134]. Activators are within the sulforaphanes, phytodrugs present in the broccoli and cruciform vegetables, but are not always very powerful. New activators are more potent than the sulforaphanes but present safety issues [135].
In bronchial asthma, in animal models, it has been shown that pharmacological activation of lung epithelial cells Nrf2 is a potential therapeutic target to reduce susceptibility to asthma [136]. The mechanism of action would increase the integrity of the epithelial barrier airway rather than inhaled steroids (137). There are no reports of clinical use of activators factor in bronchiectasis or IPF.
Oxidative enzyme inhibitors
XO level is 4 times higher in the sputum and BAL of patients with COPD, than in healthy controls. The use of XO inhibitors, such as allopurinol and febuxostat, may offer some benefit [138]. Celastrol inhibits the four isoforms of NOX (NADP oxidase), and has a potential use in inflammatory diseases [139].
AZ1 (2-thioxanthone an MPO inhibitor) appears to downregulate the inflammatory response induced tobacco smoke, with the advantage that the inhibition is irreversible (i.e., suicide inhibitors) [140]. Use of enzyme inhibitors requires confirmatory, well-designed, large scale studies to be considered as a therapeutic intervention in COPD [138].
While there have been studies in asthma and bronchiectasis with various antioxidants, AZ1, coenzyme Q10 (or ubiquinone, a key component of the mitochondrial electron transport chain) transfer to important clinical outcomes is uncertain [141]. In IPF small molecules have been identified that inhibit NOX4/NOX1 (GKT137831) with potent anti-fibrotic activity in models of pulmonary fibrosis induced in mice [142]. It is possible that future concert academia, industry, funding agencies and patient groups, more effective therapies for IPF and other forms of fibrosis is located [142].
Radical scavengers
Edaravone is a powerful free radical neutralizer, it reduces carbonyl stress and lipid peroxidation; therefore, it has a potential future in COPD treatment. Lazaroids are non-glucocorticoid-methylprednisolone analogue that penetrate hydrophobic membrane regions and inhibit lipid peroxidation. Their protective effects have been reported in animal models of lung injury, both require studies to assess their potential in respiratory diseases. Spin traps are compounds which can stabilize free radicals, to form stable end products. Spin traps have been widely used for in vitro studies and their therapeutic effects have also been investigated in vivo models of lung disease, some have shown to have benefits [143].
Ebselen (an organic selenium-based compound) neutralizes oxidative and nitrative stress, by resembling glutathione peroxidase activity. No reports are available as the beneficial effect of Ebselen against cigarette smoke-induced lung inflammation, although it has been shown to prevent LPS-induced lung inflammation in animal models (143). Inhibition of inducible nitric oxide-synthase (iNOS) by various chemical complexes may provide a strategy in COPD and bronchial asthma. Inhaled and orally administered iNOS inhibitors would be useful for the treatment of numerous conditions, including asthma and other pulmonary inflammatory disorders [144].
While there are anecdotal reports of the use of selenium in bronchial asthma with positive results in improving lung function, there are insufficient clinical trials to recommend its use in asthma, bronchiectasis or IPF [145].
Enzymatic redox sensors
Thioredixin (Trx) and redox effector factor-1 (Ref-1), belong to the oxidoreductase family of redox sensors. Trx inhibition resulted in diminished neutrophil influx and TNF-α production in an animal model. Activation of Trx can attenuate oxidative stress. The antioxidant actions of ambroxol at physiological concentrations are, at least partially, mediated by TrxR and/or Trx system. Trx effects in respiratory diseases remain to be investigated [143,146].
It is obvious that the conventional treatment of these chronic respiratory diseases impacting little the natural evolution of the same. For example, COPD is an increased prevalence and accepted therapy is ineffective in reducing disease progression. Indeed searches the molecular biology of the entity and therapeutic alternatives are sought [147]. Up to 50% of patients in the first world are uncontrolled bronchial asthma [148]. GINA adds drugs to basic therapy LABA-IGCS trying to gain control. In fact, in May 2015 he joined the strategy tiotropium bromide as an aggregate patients who remain symptomatic and have frequent exacerbations despite LABA-IGCS (step 4 and 5) [59].
While it is true in non-cystic bronchiectasis prevalence reduced, symptoms are persistent, evolution is chronic, with frequent exacerbations. IPF has the worst prognosis of all.
Conclusions
The molecular oxygen is vital for life, but their excess or deficit can be harmful and even more deadly; through toxic oxygen radicals.
ROS can be acquired from the environment or generated by cellular metabolism by altering the fine balance of redox titration.
These radicals produce various types of stress generated intracellular, structural and functional damage, producing an inflammatory process that is self-perpetuating and amplified. This process continues, at times, although the initial stimulus has been suspended.
The entities described above conventional treatment fails to reduce the natural evolution of most patients and progressive deterioration.
More and better primary and clinical research is needed to find new powerful drugs that impact the sub-cellular compartments and metabolic routes for more effective therapy in these diseases. It is likely that more drug combination requires monotherapy. Pharmacology and medical science have to integrate basic scientific research to design these molecules and to demonstrate its efficacy and safety in powerful, controlled and randomized clinical trials.
References
- Semenza GL (2011) Oxygen sensing, homeostasis, and disease. N Engl J Med 365: 537-547. [Crossref]
- Halliwell B (1996) The Radical View: Free radicals, health and disease. Odyssey 2: 10-15.
- Dekhuijzen PN, Van Beurden WJ (2006) The role for N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis 1: 99-106. [Crossref]
- Herman JR, Newman PA, McPeters R, Krueger AJ, Bhartia PK, et al. (1995) Meter 3/total ozone mapping spectrometer observation of the 1993 ozone hole. J Geophys Res 100: 2973-2983.
- Alvarado A (1998) El Enos, el efecto invernadero, la capa de ozono y los inhaladores. Una revisión. Rev Med Costa Rica y Centroamérica (544): 125-133.
- Bryan CL and Jenkinson SG (1988) Oxygen toxicity. Clin Chest Med 9: 141-152. [Crossref]
- Tobin MJ (1986) Update on strategies in mechanical ventilation. Hosp Pract 21: 69-84. [Crossref]
- Gerschman R, Gilbert DL, Nye SW, Dwyer P, Fenn Wo (1954) Oxygen poisoning and x-irradiation: a mechanism in common. Science 119: 623-626. [Crossref]
- Chapman DJ, Schopf JW (1983) Biololgical and biochemical effects of the development of an aerobic environment. In: Earths Earliest Biosphere: Its origin and Evolution Schopf JW [Ed] Princenton Univ. Press pp: 302-320.
- Reeves JT, Groves BM, Cymerman A, Sutton Wagner PD, Turkevich DJR, et al. (1990) Operation Everst II: cardiac filling pressure during cycle exercise at sea level. Respir Physiol 80: 147-154.
- West JB (2000) Respiratory Physiology: the essentials (6th edn) Lippincott Williams & Wilkins, USA.
- Maina JN, West JB (2005) Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev 85: 811-844. [Crossref]
- Hamanaka RB, Chandel NS (2009) Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol 21: 894-899. [Crossref]
- Hansel TT, Barnes PJ (2003) An atlas of chronic obstructive pulmonary disease: COPD. A resource for reference, teaching and lecturing (1st edn) London: Taylor & Francis.
- Cantin AM, North SL, Hubbard RC, Crystal RG (1987) Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol 63: 152-157.
- Tsiligianni IG, Van der Molen T (2010) A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir Res 11: 171. [Crossref]
- Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A (2003) Protein carbonylation in human diseases. Trends Mol Med 9: 169-176. [Crossref]
- Yaho H, Arunachalam G, Hwang JW (2010) Extracelular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Nat/Acad Sci USA 107: 15571-15576.
- Kode A, Rajendrasozhan S, Caito S, Yang SR, Megson IL (2008) Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 294: L478-L488.
- Biswas SK, McClure D, Jimenez LA, Megson IL, Rahaman I (2005) Curcumin induces glutathione biosynthesis and inhibits NF-kappa β activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radicals scavenging activity. Antioxid Redox Signal 7: 32-41.
- (GOLD) (2015) Global strategy for the diagnosis, management, and prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease.
- Salvi S, Barnes PJ (2010) Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest 138: 3-6. [Crossref]
- Jha P, Ramasundarahettige C, Landsman V, Rostron B, Thun M, et al. (2013) 21st-century hazards of smoking and benefits of cessation in the United States. N Engl J Med 368: 341-350. [Crossref]
- Thun MJ, Carter BD, Feskanich D, Freedman ND, Prentice R, et al. (2013) 50-year trends in smoking-related mortality in the United States. N Engl J Med 368: 351-364. [Crossref]
- Lopez AD, Murray CC (1998) The global burden of disease, 1990-2020. Nat Med 4: 1241-1243. [Crossref]
- Hunninghake GM, Cho2021 Copyright OAT. All rights reservla L, et al. (2009) MMP12, lung function, and COPD in high-risk populations. N Engl J Med 361: 2599-2608. [Crossref]
- Castaldi PJ, Cho MH, Cohn M, Langerman F, Moran S, et al. (2010) The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Hum Mol Genet 19: 526-534. [Crossref]
- Wilk JB, Chen TH, Gottlieb DJ, Walter RE, Nagle MW, et al. (2009) A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet 5: e1000429. [Crossref]
- Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM (2001) Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 1304-1309. [Crossref]
- Barnes PJ, Shapiro SD, Pauwels RA (2003) Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 22: 672-688. [Crossref]
- Daheshia M (2005) Therapeutic inhibition of matrix metalloproteinases for the treatment of chronic obstructive pulmonary disease (COPD). Curr Med Res Opin 21: 587-594. [Crossref]
- Macnee W, Rahman I (1999) Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 160: S58-65. [Crossref]
- Mocchegiani E, Giaconni R, Costarelli L (2011) Metalloproteases/antimetalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr Opin Pulm Med 17: S11-S19.
- Churg A, Zhou S, Wright JL (2012) Series "matrix metalloproteinases in lung health and disease": Matrix metalloproteinases in COPD. Eur Respir J 39: 197-209. [Crossref]
- Malemud CJ (2006) Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci 11: 1696-1701. [Crossref]
- Yao H, Rahaman I (2011) Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol 254: 72-85.
- Hassett DJ, Borchers MT, Panos RJ (2014) Chronic obstructive pulmonary disease (COPD): evaluation from clinical, immunological and bacterial pathogenesis perspectives. J Microbiol 52: 211-226.
- Ahmad A, Shameem, Usain Q (2013) Altered oxidant-antioxidant levels in the disease prognosis of chronic obstructive pulmonary disease. Int J Tuberc Lung Dis 17: 1104-1109.
- Salvi SS, Barnes PJ (2009) Chronic obstructive pulmonary disease in non-smokers. Lancet 374: 733-743. [Crossref]
- Köksal H, Saygi A, Sarıman N, Alici E, Yurtlu S, et al. (2013) Evaluation of clinical and functional parameters in female subjects with biomass smoke exposure. Respir Care 58: 424-430. [Crossref]
- Pinamonti S, Leis M, Barbieri A, Leoni D, Muzzoli M, et al. (1988) Detection of xanthine oxidase activity products by EPR and HPLC in bronchoalveolar lavage fluid from patients with chronic obstructive pulmonary disease. Free Radic Biol and Med 25: 771-779.
- Aaron SD, Angel JB, Lunau M, Wright K, Fex C, et al. (2011) Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 349-355.
- Marwick JA, Adcock IM, Chung KF (2010) Overcoming reduced glucocorticoid sensitivity in airway disease: molecular mechanisms and therapeutic approaches. Drugs 70: 929-948.
- Hwang JW, Rajendrasozhan S, Yao H, Chung S, Sundar IK, et al. (2011) FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol 187: 987-998.
- Kobayashi M, Yamamoto N (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul 46: 113-140.
- Kirkham P (2007) Oxidative stress and macrophage function: a failure to resolve the inflammatory response. Biochem Soc Trans 35: 284-287. [Crossref]
- Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, et al. (2011) Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184: 796-802. [Crossref]
- Obeng EA, Boise LH (2005) Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem 280: 29578-29587. [Crossref]
- Hogg JC (2006) Why does airway inflammation persist after the smoking stops? Thorax 61: 96-97. [Crossref]
- Kheradmand F, Shan M, Xu C, Corry DB (2012) Autoimmunity in chronic obstructive pulmonary disease: clinical and experimental evidence. Expert Rev Clin Immunol 8: 285-292. [Crossref]
- Allison ME, Fearon DT (2000) Enhanced immunogenicity of aldehyde-bearing antigens: a possible link between innate and adaptive immunity. Eur J Immunol 30: 2881-2887.
- Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, et al. (2007) Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 175: 998-1005. [Crossref]
- Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, et al. (2008) Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177: 156-163. [Crossref]
- Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, et al. (2009) T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol 157: 316-324.
- Sethi S, Murphy TF (2008) Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 359: 2355-2365. [Crossref]
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossamo BT, et al. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674-677.
- Opitz B, Van Laak V, Eitel J, Suttorp N (2010) Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med 181: 1294-1309. [Crossref]
- Barnes PJ (2008) Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183-192. [Crossref]
- Bozek A, Jarzab J (2011) Improved activity and mental function related to proper antiasthmatic treatment in elderly patients with Alzheimer's disease. Allergy Asthma Proc 32: 341-345. [Crossref]
- Mortaz E, Masjedi MR, Barnes P (2011) Identification of Novel Therapeutic Targets in COPD. Tanaffos 10: 9-14. [Crossref]
- Busse WW, Lemanske RF Jr (2001) Asthma. N Engl J Med 344: 350-362. [Crossref]
- Barnes PJ (2011) Biochemical basis of asthma therapy. J Biol Chem 286: 32899-32905. [Crossref]
- Tattersfield AE, Knox AJ, Britton JR, Hall IP (2002) Asthma. Lancet 360: 1313-1322. [Crossref]
- Romagnani S (2004) Immunologic influences on allergy and the TH1/TH2 balance. J Allergy Clin Immunol 113: 395-400. [Crossref]
- Simon HU (2000) Eosinophil apoptosis--pathophysiologic and therapeutic implications. Allergy 55: 910-915. [Crossref]
- Wills-Karp M1, Luyimbazi J, Xu X, Schofield B, Neben TY, et al. (1998) Interleukin-13: central mediator of allergic asthma. Science 282: 2258-2261. [Crossref]
- Rogge L, D'Ambrosio D, Biffi M, Penna G, Minetti LJ, et al. (1998) The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol 161: 6567-6574. [Crossref]
- Simon HU (2006) Cytokine and anti-cytokine therapy for asthma. Curr Allergy Asthma Rep 6: 117-121. [Crossref]
- Barnes PJ, Chung KF, Page C (1998) Inflammatory mediators in asthma: an update. Pharmacol Rev 50: 515-526.
- Adcock IM1 (2001) Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther 14: 211-219. [Crossref]
- Barnes PJ, Karin M (1997) Nuclear factor-kappaβ: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336: 1066-1071.
- Cho YS, Moon HB (2010) The role of oxidative stress in the pathogenesis of asthma. Allergy Asthma Immunol Res 2: 183-187. [Crossref]
- Sugiura H, Ichinose M (2008) Oxidative and nitrative stress in bronchial asthma. Antioxid Redox Signal 10: 785-797. [Crossref]
- Stone JR, Yang S (2006) Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal 8: 243-270. [Crossref]
- Park CS, Kim TB, Lee KY, Moon KA, Bae YJ, et al. (2009) Increased oxidative stress in the airway and development of allergic inflammation in a mouse model of asthma. Ann Allergy Asthma Immunol 103: 238-247. [Crossref]
- Yang IA, Fong KM, Zimmerman PV, Holgate ST, Holloway JW (2008) Genetic susceptibility to the respiratory effects of air pollution. Thorax 63: 555-563.
- Fatani SH (2014) Biomarkers of oxidative stress in acute and chronic bronchial asthma. J Asthma 51: 578-584. [Crossref]
- Sahiner UM, Birben E, Erzurum S, Sackesen C, Kalayci O (2011) Oxidative stress in asthma. World Allergy Organ J 4: 151-158. [Crossref]
- Kinnula VL, Lehtonen S, Koistinen P, Kakko S, Savolainen M (2004) Two functional variants of the superoxide dismutase genes in Finnish families with asthma. Thorax 59: 116-119.
- Juul K, Tybjaerg-Hansen A, Marklund S, Lange P, Nordestgaard BG (2006) Genetically increased antioxidative protection and decreased chronic obstructive pulmonary disease. Am J Respir Crit Care Med 173: 858-864.
- Zhu J, Qiu YS, Majumdar S, Gamble E, Matin D, et al. (2001) Exacerbations of bronchitis: Bronchial eosinophilia and gene expression for interleukin-4, interleukin-5 and eosinophil chemoattractans. Am J Respir Crit Care Med 164:109-116.
- Diagnosis of Asthma (2015) COPD and Asthma-COPD overlap syndrome (ACOS). A joint project of GINA and GOLD.
- Postma DS, Rabe KF (2015) The Asthma-COPD Overlap Syndrome. N Engl J Med 373: 1241-1249. [Crossref]
- Pasteur MC, Bilton D, Hill AT (2010) British Thoracic Society guideline for non-CF bronchiectasis. Thorax 65 Suppl 1: i1-58. [Crossref]
- Wyecker D, Edelsberg J, Oster G (2004) Prevalence and economic burden of bronchiectasis. Am J Respir Crit Care Med 169: A330.
- Olveira G, Olveira C, Dorado A, García-Fuentes E, Rubio E, et al. (2013) Cellular and plasma oxidative stress biomarkers are raised in adults with bronchiectasis. Clin Nutr 32: 112-117. [Crossref]
- Fuschillo S, De Felice A, Balzano G (2008) Mucosal inflammation in idiopathic bronchiectasis: cellular and molecular mechanisms. Eur Respir J 31: 396-406. [Crossref]
- Attenburg J, Wortel K, Van der Werf TS, Boersma WG (2015) Non-cystic fibrosis bronchiectasis: clinical presentation, diagnosis and treatment, illustrated by data from Dutch Teaching Hospital. NJM 73: 147-154.
- Athanazio R (2012) Airway disease: similarities and differences between asthma, COPD and bronchiectasis. Clinics (Sao Paulo) 67: 1335-1343. [Crossref]
- Hamutcu R, Rowland JM, Horn MV, Kaminsky C, MacLaughlin EF, et al. (2002) Clinical findings and lung pathology in children with cystic fibrosis. Am J Respir Crit Care Med 165: 1172-1175. [Crossref]
- Paganin F, Séneterre E, Chanez P, Daurés JP, Bruel JM, et al. (1996) Computed tomography of the lungs in asthma: influence of disease severity and etiology. Am J Respir Crit Care Med 153: 110-114. [Crossref]
- NICE (NIFfCE) (2004) Chronic obstructive pulmonary disease: national clinical guideline for management of chronic obstructive pulmonary disease in adult in primary and secondary care. Thorax 59: 1-232.
- Bartoli ML, NovelliF, Costa F, Malagrine L, Melosinil L, et al. (2011) Malondialdheyde in Exhaled Breath Condensate as a Marker of Oxidative Stress in Different Pulmonary Diseases. Mediators of Inflammation.
- Vancheri C, Failla M, Crimi N, Raghu G (2010) Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J 35: 496-504. [Crossref]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, et al. (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788-824. [Crossref]
- Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, et al. (2005) The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 142: 963-967. [Crossref]
- Cottin V (2013) The role of pirfenidone in the treatment of idiopathic pulmonary fibrosis. Respir Res 14 Suppl 1: S5. [Crossref]
- Ley B, Collard HR, King TE Jr (2011) Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183: 431-440. [Crossref]
- Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, et al. (2007) Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med 176: 277-284. [Crossref]
- Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, et al. (2011) The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 66: 462-467. [Crossref]
- Thannickal VJ, Toews GB, White ES, Lynch JP, Martinez FJ (2004) Mechanisms of pulmonary fibrosis. Annu Rev Med 55: 395-417. [Crossref]
- Winterbourn CC (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 4: 278-286. [Crossref]
- Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, et al. (2008) Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 178: 838-846.
- Cheresh P, Kim SJ, Tulasiram S, Kamp DW (2013) Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta 1832: 1028-1040. [Crossref]
- Boucher RC (2011) Idiopathic pulmonary fibrosis--a sticky business. N Engl J Med 364: 1560-1561. [Crossref]
- Lawson WE, Loyd JE, Degryse AL (2011) Genetics in pulmonary fibrosis--familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis. Am J Med Sci 341: 439-443. [Crossref]
- Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, et al. (2011) A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 364: 1503-1512. [Crossref]
- Cookson WO, Moffatt MF (2013) Bedside to gene and back in idiopathic pulmonary fibrosis. N Engl J Med 368: 2228-2230. [Crossref]
- Downey GP (2011) Resolving the scar of pulmonary fibrosis. N Engl J Med 365: 1140-1141. [Crossref]
- Shulgina L, Cahn AP, Chilver ER (2013) Treating idiopathic pulmonary fibrosis with the addition of cotrimoxazole: a randomized controlled trial. Thorax 68: 155-162.
- Britton JR, Pavord ID, Richards KA, Knox AJ, Wisniewski AF, et al. (1995) Dietary antioxidant vitamin intake and lung function in the general population. Am J Respir Crit Care Med 151: 1383-1387. [Crossref]
- Grievink L, Smit HA, Ocké MC, Van 't Veer P, Kromhout D (1998) Dietary intake of antioxidant (pro)-vitamins, respiratory symptoms and pulmonary function: the MORGEN study. Thorax 53: 166-171. [Crossref]
- Rautalahti M, Virtamo J, Haukka J, Heinonen OP, Sundvall J, et al. (1997) The effect of alpha-tocopherol and beta-carotene supplementation on COPD symptoms. Am J Respir Crit Care Med 156: 1447-1452. [Crossref]
- Sargeant LA, Jaeckel A, Wareham NJ (2000) Interaction of vitamin C with the relation between smoking and obstructive airway disease in EPOC Norfolk. European Prospective Investigation into Cancer and Nutrition. Eur Repir J 16: 397-403.
- Santus P, Sola A, Carlucci P (2005) Lipid peroxidation and 5-lipoxygenase activity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 171: 838-843.
- Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, et al. (2008) Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax 63: 784-790.
- Alvarado A, Arce I (2013) Mecanismos de acción y resistencia a glucocorticoides en asma y enfermedad pulmonar obstructiva crónica. AMC 55: 162-168.
- Dekhuijzen PN, van Beurden WJ (2006) The role for N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis 1: 99-106. [Crossref]
- Decramer M, Rutten-van Mölken M, Dekhuijzen PNR, Troosters T, Van Herwaaden C, et al. (2005) Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis randomized on NAC cost-utility study, BRONCUS): A randomized placebo-controlled trial. Lancet 365: 1552-1560.
- Stav D, Raz M (2009) Effect of N-acetylcysteine on air trapping in COPD: a randomized placebo-controlled study. Chest 136: 381-386. [Crossref]
- Tse HN, Raiteri L, Wong KY, Yee KS, Ng LY, et al. (2013) High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 144: 106-118. [Crossref]
- Tze HN, Tzeng CZS (2014) Update on the pathological processes, molecular biology, and clinical utility of N-acetylcysteine in chronic obstructive pulmonary disease. Int J Obstruct Pulmon Dis 9: 825-336.
- Zheng JP, Kang J, Huang SG, Chen P, Yao WZ, et al. (2008) Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study): A randomized placebo-controlled study. Lancet 371: 2013-2018.
- Moretti M, Bottrighi P, Dallari R, Da Porto R, Dolcetti A, et al. (2004) The effect of long-term treatment with erdosteine on chronic obstructive pulmonary disease: the EQUALIFE Study. Drugs Exp Clin Res 30: 143-152. [Crossref]
- Rhee CK, Kang CM, You MB, Yoon HK, Kim YK, et al. (2008) Effect of fudosteine on mucin production. Eur Respir J 32: 1195-1202. [Crossref]
- Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, et al. (2005) High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 353: 2229-2242. [Crossref]
- The Idiopathic Pulmonary Fibrosis Clinical Research Network (2012) Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366: 1968-1977.
- The Idiopathic Pulmonary Fibrosis Clinical Research Network (2014) Randomized Trial of Acetylcysteine in Idiopathic Pulmonary Fibrosis. N Engl J Med 370: 2093-2101.
- Kim ES, Keating GM (2015) Pirfenidone: a review of its use in idiopathic pulmonary fibrosis. Drugs 75: 219-230. [Crossref]
- Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, et al. (2011) Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 365: 1079-1087. [Crossref]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, et al. (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071-2082. [Crossref]
- Raghu G, Collard HR, Anstrom KJ (2012) Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med 185: 1044-1048.
- Karimi-Shah BA, Chowdhury BA (2015) Forced vital capacity in idiopathic pulmonary fibrosis--FDA review of pirfenidone and nintedanib. N Engl J Med 372: 1189-1191. [Crossref]
- Kirkham PA, Barnes PJ (2013) Oxidative stress in COPD. Chest 144: 266-273. [Crossref]
- Garber K (2012) Biochemistry: A radical treatment. Nature 489: S4-6. [Crossref]
- Sussan TE, Gajghate S, Chatterjee S, Mandke P, McCormick S, et al. (2015) Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am J Physiol Lung Cell Mol Physiol 309: L27-36. [Crossref]
- Shintani Y, Maruoka S, Gon Y, Koyama D, Yoshida A, et al. (2015) Nuclear factor erythroid 2-related factor 2(Nrf2) regulates airway epithelial barrier integrity. Allergol Int 64: S54-S63.
- Boueiz A, Damarla M, Hassoun PM (2008) Xanthine oxidoreductase in respiratory and cardiovascular disorders. Am J Physiol Lung Cell Mol Physiol 294: L830-840. [Crossref]
- Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, et al. (2011) NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol 164: 507-520. [Crossref]
- Churg A, Marshall CV, Sin DD, Bolton S, Zhou S, et al. (2012) Late intervene with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am J Respir Crit Care Med 185: 34-43.
- Wong C, Jones S (2013) Oxidative stress and macrolides in bronchiectasis-exhaling few clues. Respirology 18: 1037-1038. [Crossref]
- Hecker L, Cheng J, Thannickal VJ (2012) Targeting NOX enzymes in pulmonary fibrosis. Cell Mol Life Sci 69: 2365-2371. [Crossref]
- [Crossref] Rahman I, MacNee W (2012) Antioxidant pharmacological therapies for COPD. Curr Opin Pharmacol 12: 256-265.
- Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, et al. (2011) Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 147: 293-305. [Crossref]
- Al-Jawad FH, Al-Atabee HG, Sahib AS (2010) Usefulness of antioxidant drugs in bronchial asthma. Pak J Physiol 6: 18-21.
- Sato A, Hoshino Y, Hara T, Muro S, Nakamura H, et al. (2008) Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J Pharmacol Exp Ther 325: 380-388. [Crossref]
- Alvarado A, Arce I (2015) Molecular Biology of Chronic Obstructive Pulmonary Disease from the Bases to the Therapeutic Decision: A Review. Br J Med Medic Res 10: 1-14.
- Braman SS (2012) Refractory asthma: lessons learned from the bronchoscope. Chest 141: 575-576. [Crossref]