Abstract
Congenital FVII deficiency is the most frequently encountered defect among the Rare Bleeding Disorders (RBD). Geographical distribution is uneven but it has been described worldwide. Factor FVII Padua (Arg304Gln) is a Type 2 variant with shows FVII activity levels which varies with the origin of the thromboplastin used in the assay system. It is low (less than 5%) using rabbit brain thromboplastins, but normal (100%) with OX-brain preparations. Thromboplastins of human origin (placenta) or human recombinant reagents yield intermediate levels (40-50% of normal). FVII antigen is normal. FVII Padua has been described in many parts of the world but it seems rare in Latin America where only a few heterozygotes cases have been described but no homozygotes. We described here the first FVII Padua case seen in Argentina and, probably, in Latin America. The proposita is a 64 year old female who had a Spanish background and no bleeding tendency. She was found to be homozygote for the Arg304Gln mutation whereas her daughter was heterozygote and also asymptomatic. The reasons for the rarity of this mutation in Argentina and in Latin America are discussed.
Key words
Factor VII, Deficiency FVII Padua, asymptomatic, diffusion, Latin America
Introduction
Congenital FVII deficiency is the most frequent defect among the Rare Bleeding Disorders. Its prevalence is estimated to be 1: 500.000 [1-3].
The first family was described in 1951 by Alexander et al. but laboratory studies had suspected that an “accelerator” of prothrombin conversion, different from FV, existed a few years before 1951 [4].
FVII deficiency is in fact one of the only two congenital coagulation disorders, that were not discovered by the description of a family with a clotting defect that did not fit the previous knowledge on blood coagulation; the other is FXIII deficiency [3].
Since 1951, several cases have been described all over the world. Most of the recent have been studied, also, by means of molecular biology techniques [1,2].
About 200 mutations, have been described, the majority missense mutations [2]. The Arg304Gln (or as listed today, the Arg364Gln) mutation has been originally described in 1978 in Padua [5,6]. Subsequently this variant was seen in many parts of the world [7-17]. A special concentration of the defect is seen in three areas, namely: Mediterranean countries, USA (African-Americans) and Japan [18].
The defect has the peculiar feature to cause a discrepant behavior in the binding to tissue factor of different origin. In other words the FVII level changes radically according with the tissue thromboplastin used in the assay system [5,19,20].
If rabbit brain thromboplastin is used, activity levels of less than 5% are obtained; if human recombinant or human placenta preparations are used the level is about 40- 50% of normal and if an OX brain thromboplastin is utilized, the level is 100% of normal [5,6]. The mutation responsible for the defect, an Arg304Gln substitution was found in 1991 [10,21,22].
This behavior is typical of FVII Padua so that a diagnosis of the defect can be made even without molecular biology techniques. There is in fact only another mutation involving the same amino acid (Arg304), that shows a similar behavior, namely FVII Nagoya (Arg304Trp) [23]. However since the Arg304Trp mutation is rare, contrary to the Arg304Gln, it can be safely assumed that a patient presenting this tissue factor dependent behavior is indeed a case of FVII Padua. FVII Padua has been rarely described at the heterozygosis or compound heterozygosis level in Latin-American, exactly in Brazil, Venezuela and Costa Rica but no homozygotes have ever been reported [14]. It has never been described in Argentina in any form, heterozygous, homozygous or compound heterozigous.
The purpose of this study is to report a case of homozygous FVII Padua from Argentina
Patients and methods
The proposita is a 64 year and female from northeastern Argentina (Chaco Province) with a Spanish background. Parents were non consanguineous. The father died after prostate carrier surgery which was referred to have been accompanied by excessive bleeding. A half-brother of the proposita (result of her father previous marriage) is reported to have frequent epistaxis but it is not known if the patient has a clotting defect.
There are no other cases of bleeding tendency in the family. The proposita’s husband died at the age of 19 because of drowning but he had a negative personal and family history for bleeding. The proposita underwent appendectomy in young age without undue bleeding; tooth extractions were also referred not to be accompanied by bleeding. Menstruations were normal and delivery by cesarean section at the age of 20 was uneventful. A diagnosis of FVII deficiency was made at the age of 60 in preparation for a colonoscopy. Because of this diagnosis, the patient underwent video colonoscopies under the protection of two aFVII concentrates (dosage unknown) without any undue bleeding or side effects. During this procedure a small polyp was removed. The control procedure was carried out 2 years later, under the protection of a total doses of 1,3 mg of aFVII concentrate. Again there was no bleeding or side effects and the procedure failed to show relapse of polyps.
At present the patient is in good health and has no bleeding manifestations.
The daughter of the proposita is 44 year old and has never presented with any bleeding symptom. Her menstrual cycles are also reported as normal.
There are no known episodes of venous or arterial thrombosis in the patients or in the family. aPTT and PT were carried out using standard procedures. The reagent used for the aPTT was supplied by Instrumentation Laboratory, Milan, Italy. Seven reagents were used for the PT and FVII assay, namely: three rabbit brain thromboplastins (STA Neoplastin plus, Stago Laboratories, Asniers, France; PT- Fibrinogen HS Plus, Instrumentation Laboratory, Milan, Italy; Tromboplastina S, hemo_medica, Fisher Diagnostics, USA); a reagent obtained from human placenta (Thromborel S, Dade-Behring, Marburg, Germany) and two recombinant human thromboplastins (Dade-Innovin, Siemens, Marburg, Germany and Recombiplastin 2G, Instrumentation Laboratory, Milan, Italy). An ox-brain thromboplastin was also used (Thrombotest, Nygaard Laboratories, Oslo, Norway).
Factor VII clotting assay were carried out on 1:10 diluited plasma using known FVII deficient plasma as substrate and the different thromboplastins. FVII antigens level was studied by an Elisa method (Asserachrom FVII, Stago Laboratories, Asniers, France).
Dna was extracted from dried thick drops of whole blood blotted on Whatman paper. For this purpose we used the kit (QiAmp DNA minikit) supplied by QIAGEN Laboratories (Qiagen s.r.l., Milan, Italy).
Amplification of exons 1 to 8 and respective spice junctions of the FVII gene were performed using oligonucleotide primers kindly supplied by Dr. James H. (Tyler, Tx, U.S.A.) or acquired from Invitrogen (Carlsbad, Ca, U.S.A.)
Mutational analysis was performed by polymerase chain reaction (PCR) amplification using oligo 8AF (5’-GAGGTGGCAGGTGGTGGAAA-3’), 8AR (5’CGGCACAGAACATGTACTCC-3’) 8BF (5’- TGATGACCCAGGACTGCCT-3’), 8BR (5’- GGGATTTGGTGCCAGGACA-3’).
PCR was carried out in a total volume of 15 μL with 50 ng of genomic DNA, 10 mM of each primer, and 9 µl of PCR Master Mix, 2X (Promega, Madison, Wisconsin, U.S.A.). After an initial denaturation step at 95°C for 5 minutes, amplification was performed for 35 cycles (denaturation at 95°C for 1 minute, annealing at 57°C for 1 minute, and extension at 72°C for 2 minutes). PCR products were bidirectionally sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit and ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, Ca, U.S.A.).
Results
Main results of the coagulation study are summarized in Table1. Platelets and aPTT were normal. PT was variably prolonged using 6 out of the 7 thromboplastins but was fully corrected by the addition of normal plasma. On the contrary PT was normal using an OX brain thromboplastin. FVII assay was also low with the same six thromboplastins but it was normal with the OX brain preparation. FI, FII, FV and FX, the other clotting factors that could prolong the test, were normal.
Table 1. Coagulation study in the proposita. Seven differents thromboplastins were used for the evaluation of the defect. In Factor VII assays, values of less than 1% of normal have been reported as 1%.
Test |
Proposita |
Normal value |
Comments |
Platelets count |
390 |
150-350 x109/L |
Sysmex XT 2000i |
aPTT |
26 sec. |
26-36 sec. |
APTT-SP (Instrumentation Laboratory) |
PT (rabbit brain) |
55.8 sec |
14-15 sec. |
PT-Fibrinogen HSPlus(Instrument laboratory) |
PT (rabbit brain) |
35.9 sec. |
12-14 sec. |
STA Neoplastin Plus (Stago Laboratory) |
PT (rabbit brain) |
83.6 sec. |
14-15 sec. |
Tromboplastina S (Hemo Medica) |
PT (human placenta) |
17.3 sec. |
13-15 sec. |
Thromborel (Dade- Behring) |
PT (human recombinant) |
13.7 sec. |
10-12 sec. |
Recombiplastin 2G (Instrumentation Laboratory) |
PT (human recombinant) |
13.1 sec. |
10-12 sec. |
Innovin Dade (Siemens) |
Ox-brain thromboplastin |
40 sec. |
38-42 sec. |
Thrombotest (Nyagaard Laboratories) |
PT + Normal plasma |
16 sec. |
14-16 sec. |
PT-Fibrinogen HSPlus(Instrument laboratory) |
FVII assays: |
Rabbit brain |
1% |
80-120 |
PT-Fibrinogen HSPlus(Instrument laboratory) |
Rabbit brain |
1% |
80-120 |
STA Neoplastin Plus (Stago Laboratory) |
Rabbit brain |
1% |
80-120 |
Tromboplastina S (Hemo Medica) |
Human placenta |
29% |
80-120 |
Thromborel (Dade Behring) |
OX brain |
96% |
80-120 |
Thrombotest (Nyagaard Laboratories) |
Human recombinant |
53% |
80-120 |
Recombiplastin 2G (Instrumentation Laboratory) |
Human recombinant |
39% |
80-120 |
Dade Innovin (Siemens) |
FI, FII, FV, FVIII, FIX, FX, FXI, FXII, FXIII |
|
80-120 |
|
Factor VII Antigen |
105% |
80-120 |
Asserachrom FVII (Stago Laboratories) |
The level of FVII varied between 0.6% to about 96% using the tissue thromboplastins of different origin. It was low with the three rabbit brain thromboplastins (RBT) whereas it was about 40-50% of normal using the human placenta thromboplastin or the two human recombinant thromboplastins and it was 96% of normal using the OX brain preparation. All other clotting factors were within normal limits. Factor VII antigen was 105% of normal.
The daughter had a normal aPTT and a slightly prolonged or borderline PT. FVII level was 37-52% with RBTs and 63-74 of normal with the thromboplastin of human placenta or with the human recombinant ones. Genetic analysis showed that the proposita was homozygote for an Arg304Gln mutation in exon 8 whereas the daughter was heterozygote for the same mutation (Figure 1). The study of the other exons failed to show any other abnormality.
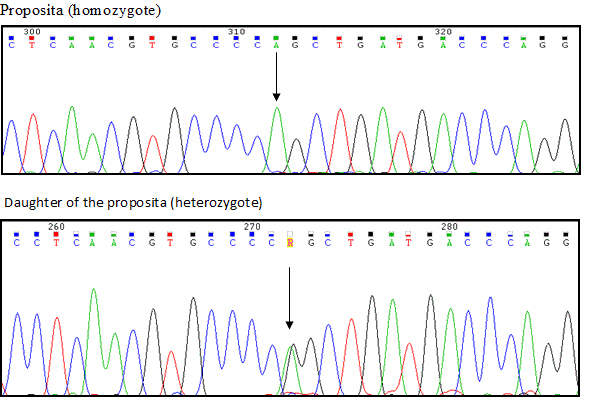
Figure 1. DNA sequencing pattern in the proposita (top) and in her daughter (bottom). The proposita is homozygote for Arg304Gln whereas the daughter is heterozygote for the some mutation. No other mutation are present in the two propositas.
Discussion
The Arg304Gln mutation, together with the Ala294Val represent the most frequent mutations in FVII deficiency [2]. It is interesting to note that, despite the fact that the two mutations are located in the catalytic domain controlled by exons 8, they are different. In fact the Ala249Val mutations is a Type 1 defect (low activity and low antigen) whereas the Arg304Gln (FVII Padua) mutation is a Type 2 defect (low activity, normal antigen) [2,5,6].
Factor VII Padua was first reported in Italy in 1978 on the basis of peculiar clotting tests which showed a discrepancy in the FVII level according to the thromboplastin used in the assay system. Factor VII was low 4-5% of normal using rabbit brain and lung thromboplastin but it was normal when an OX-brain thromboplastin was used. Thromboplastins of human origin (placenta) or human recombinant yielded levels of about 30-50% of normal [5,6]. The identity of behavior between human placenta thromboplastin and human recombinant preparation had been established in 1993 [17].
In 1991 it was demonstrated that the defect was due to in Arg304Gln mutation in exon 8 [10,21].
After the first cases, several other patients were described in many parts of the world. A least three main areas of the world show a concentration of the defect, namely the Mediterranean area, the USA (Afro- Americans) and Japan [13,16,18,22].
Since these three areas have nothing in common it is plausible to conclude for a multifounder effect. This indicates that Arg304 is a hot spot for mutations. The description of an Arg304Trp mutation (FVII Nagoya) is in agreement with the hot-spot assumption [23].
The substitution of Arg304 with tryptophan instead of glutamine (FVII Nagoya) does not change the peculiar clotting pattern [23].
Contrary to FVII Padua, FVII Nagoya appears rare, having been described so far only in two patients in Japan [23]. Since the clotting pattern is the same regardless of the mutated glutamine or tryptophan it seems that the Arg304 plays an important role in binding to tissue factor. This is in agreement with the observation that mutation at Pro303Thr is not followed by the appearance of the typical thromboplastin dependent coagulation pattern [24].
These is another area of FVII strictly involved in binding to tissue factor, namely the first EGF domain controlled by exon 4 [25-29].
The exact relation between these two area of FVII involved in binding to tissue factor has not been fully clarified yet [25,26]. It seems that the EGF area (exon 4) mainly binds the protease to its receptor whereas interaction with at least one additional protease domain residue (exon 8) is needed for the full development of the catalytic activity of the bound protease [27].
This apparently is the first homozygote patient with this mutation seen not only in Argentina but also in Latin-America.
A few heterozygotes and a compound heterozygote (combined with the Gly365Cys mutation) have been reported from Brazil, Venezuela and Costa Rica [14,29,30]. These are the only cases listed in FVII mutation Data Bases.
Whether this is due to genetic reasons or to a defective diagnosis remains to be proven. The latter hypothesis seems more likely since this rarity is strange in view of the fact that several cases have been described in European countries which were in the past the source of emigrants to Argentina (mainly Spain, Italy and Israel).
The proposita refers a Spanish background. This is not surprising since occasional patients with a clotting pattern compatible with a FVII Padua diagnosis have been described in Spain [31]. However there is no molecular biology demonstration of the defect. The intense genetic crossing that occurred in the past among the people of the Mediterranean countries and in Argentina herself may fully justify the finding.
There are a few other cases of FVII deficiencies seen in Argentina but they lack molecular biology studies [32-35]. The clotting data available for some of these patients seem to exclude that they were cases of FVII Padua [35]. Due to the paucity or even absence of bleeding symptoms shown by patients with FVII Padua, the administration of aFVII concentrates in the proposita was unjustified. Thrombotic events both arterial and venous have been reported in these patients after replacement therapy [36,37]. Furthermore, when dealing with patients with congenital FVII deficiency, clinical manifestations have always to be kept in mind besides the results of the clotting tests.
A final consideration is indicated. There is today a wide discrepancy between the number of RBD reported in Latin American and a population of about 600 million. Such discrepancy should be eliminated much to the benefit of patients who suffer for a bleeding tendency without knowing the reason for it. Hemophilia A or B and VW Disease patients are probably more easily observed, diagnosed and treated. However even RBD, taken altogether, represent an important segment of the bleeding patients.
We hope this study will stimulate in Argentina and Latin American countries at large, the search for rare bleeding disorders as it has been recently proposed for FX deficiency [38].
2021 Copyright OAT. All rights reserv
Acknowledgment
This study was supported in part by the “Associazione Emofilia ed altre coagulopatie delle tre Venezie”.
References
- Greenberg DL, Davie EW (2006) The blood coagulation factors: their complementary DNA, genes and expression. In: Colman RW, Hirsh J, Marder VJ, Clower AW, George JN, et al. (Eds) Hemostasis and thrombosis Lippincott Williams and Wilkins, Philadelphia p. 21.
- Perry DJ (2002) Factor VII deficiency. Br J Haematol 118: 689-700.
- Girolami A, Cosi E, Santarossa C, Ferrari S, Luigia Randi M (2015) The Story of Serum Prothrombin Conversion Accelerator, Proconvertin, Stable Factor, Cothromboplastin, Prothrombin Accelerator or Autoprothrombin I, and Their Subsequent Merging into Factor VII. Semin Thromb Hemost 41: 366-373. [Crossref]
- Alexander B, Goldstein R, Landwehr G, Cook Cd (1951) Congenital SPCA deficiency: a hitherto unrecognized coagulation defect with hemorrhage rectified by serum and serum fractions. J Clin Invest 30: 596-608. [Crossref]
- Girolami A, Fabris F, Dal Bo Zanon R, Ghiotto G, Burul A (1978) Factor VII Padua: a congenital coagulation disorder due to an abnormal factor VII with a peculiar activation pattern. J Lab Clin Med 91: 387-395.
- Girolami A, Patrassi G, Cappellato G, Casonato A (1979) Another Family with the Factor VII Padua Clotting Defect. Acta Haematol 62: 4–11.
- Bernardi F, Liney DL, Patracchini P, Gemmati D, Legnani C, et al.(1994) Molecular defects in CRM+ factor VII deficiencies: modelling of missense mutations in the catalytic domain of FVII. Br J Haematol 86: 610-8. [Crossref]
- Elmahmoudi H, Ben-Lakhal F, Elborji W, Jlizi A, Zahra K, et al. (2012) Identification of genetic defects underlying FVII deficiency in 10 patients belonging to eight unrelated families of the North provinces from Tunisia. Diagn Pathol 7: 92-96. [Crossref]
- Giansily-Blaizot M, Schved JF (2005) Potential predictors of bleeding risk in inherited factorVII deficiency. Clinical, biological and molecular criteria. Thromb Haemost 94: 901-906.
- O'Brien DP, Gale KM, Anderson JS, McVey JH, Miller GJ, et al. (1991) Purification and characterization of factor VII 304-Gln: a variant molecule with reduced activity isolated from a clinically unaffected male. Blood 78: 132-140.
- Sekiya A, Morishita E, Maruyama K, Asakura H, Nakao S, et al. (2012) Significant decrease in factor VII activity by tissue thromboplastin derived from rabbit brain in a patient with congenital factor VII deficiency (FVII Padua). Rinsho Ketsueki 53: 357-360.
- Shurafa MS, Kumar A, Fair DS, James HL (1993) The molecular defect in factor VII Detroit is due to substitution of Arg FASEB J 7: 115.
- Triplett DA, Brandt JT, Batard MA, Dixon JL, Fair DS (1985) Hereditary factor VII deficiency: heterogeneity defined by combined functional and immunochemical analysis. Blood 66: 1284-1287. [Crossref]
- Herrmann FH, Wulff K, Auerswald G, Schulman S, Astermark J, et al. (2009) Greifswald Factor FVII Deficiency Study Group. Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia 15: 267-280. [Crossref]
- Horellou MH, Chalendard J, Juin F, Conard J, Gouault-Heilmann M, et al. (1993) Source of tissue factor leading to discrepant plasma FVII levels in eight FVII-deficient patients of African origin. J Thromb Heamost 5: 1-2.
- Kirkel D, Lin TW, Fu SW, Dlott JS, Sahud MA, et al. (2010) Asymptomatic factor VII deficiency: gene analysis and structure-function relationships. Blood Coagul. Fibrinolysis 21: 91-94. [Crossref]
- Girolami A, Sartori MT, Steffan A, Fadin MA (1993) Recombinant thromboplastin is slightly more sensitive to factor VII Padua than standard thromboplastins of human origin. Blood Coagulation & Fibrinolysis 4: 497-498.
- Girolami A, Berti de Marinis G, Bonamigo E, Allemand E (2011) Worldwide diffusion of FVII Arg304Gln coagulation defect (FVII Padua). Eur J Haematol. 86: 135–139.
- Girolami A, Scarparo P, Bonamigo E, Treleani M, Lombardi AM (2012) Homozygous FVII deficiencies with different reactivity towards tissue thromboplastins of different origin. Hematology 17: 350-354.
- Girolami A, Berti de Marinis G, Bonamigo E, Sartori R, Vettore S (2010) Ox brain versus rabbit brain thromboplastin assays are the best tool for a preliminary diagnosis of the Arg304Gln factor VII defect (FVII Padua). Acta Haematol 124: 229-234. [Crossref]
- James HL, Kumar A, Girolami A, Hubbard J, Fair DS (1991) Variant coagulation factors X and VII with point mutations in highly conserved motif in the substrate binding pocket. Comparative molecular modelling. Thromb Haemost 65: 932
- James HL, Girolami A, Hubbard JG, Kumar A, Fair DS. (1993) The dysfunction of coagulation factor VII Padua results from substitution of arginine-304 by glutamine. Biochim Biophys Acta 1172: 301-305.
- Matsushita T, Kojima T, Emi N, Takahashi I, Saito H (1994) Impaired human tissue factor-mediated activity in blood clotting factor VIINagoya (Arg304-->Trp). Evidence that a region in the catalytic domain of factor VII is important for the association with tissue factor. J Biol Chem 269: 7355-7363. [Crossref]
- Peyvandi F, Jenkins PV, Mannucci PM, Billio A, Zeinali S, et al. (2000) Molecular characterisation and three-dimensional structural analysis of mutations in 21 unrelated families with inherited factor VII deficiency. Thromb Haemost 84: 250-257.
- Chaing S, Clarke B, Sridhara S, Chu K, Friedman P, et al. (1994) Severe factor VII deficiency caused by mutations abolishing the cleavage site for activation and altering binding to tissue factor. Blood 83: 3524-3535. [Crossref]
- Clarke BJ, Ofosu FA, Sridhara S, Bona RD, Rickles FR, et al. (1992) The first epidermal growth factor domain of human coagulation factor VII is essential for binding with tissue factor. FEBS Lett 298: 206-210.
- Takamiya O, Takeuchi S (1998) Dysfunctional factor-VII variant (FVII Tondabayashi) with R79Q: determination of mutated site with monoclonal anti-human factor-vii antibody (B101/ B1). Clin Chem 44: 1993-1995.
- O'Brien DP, Kemball-Cook G, Hutchinson AM, Martin DM, Johnson DJ, et al. (1995) Surface plasmon resonance studies of the interaction between factor VII and tissue factor. Demonstration of defective tissue factor binding in a variant FVII molecule (FVII-R79Q). Biochemistry 33: 14162-14169.
- Dickinson CD, Kelly CR, Ruf W.(1996) Identification of surface residues mediating tissue factor binding and catalytic function of the serine protease factor VIIa. Proc Natl Acad Sci USA 93: 14379-14384. [Crossref]
- Rodrigues DN, Siqueira LH, Galizoni AM, Arruda VR, Annichino-Bizzacchi JM (2003) Prevalence of factor VII deficiency and molecular characterization of the F7 gene in Brazilian patients. Blood Coagul Fibrinolysis 14: 289-292.
- Pardo A, Oteyza JP, Blanco L, Correa MA, Cesar J, et al. (1987) Study of different factor VII deficiency variants in nine families from Spain. Haemostasis 17: 268-272.
- Saavedra H, Olerto D, Serrano J, Miranda Soto R, Alvarado M, et al. (2013) Deficit de FVII hereditario. Hematologia 17: 176.
- Ramos A, Frogioni M, Mónaco S, Picón A, Alonso L, et al. (2011) Déficit aislado de factor VII en población infantil, con antecedentes personales y/o familiares de sangrado ó TP alterado Acta Bioquim. Clin- Latinoam.
- Nakkache M, Zelarayan K, Duarte P, Riveros D (2008) Deficiencia congénita de factor VII: serie de casos. XX Congreso del Grupo Cooperativo Latinoamericano de Hemostasia y Trombosis, XV Congreso del Grupo Cooperativo Argentino de Hemostasia y Trombosis. Buenos Aires, Argentina.
- Bergna, LJ, Dours, MT, Gonzalez de Mondini N, Martinez Canaveri AA (1981) Deficiencia congenita de factor VII. Presentacion de 4 casos. Medicina 41: 242-248.
- Girolami A, Bertozzi I, Rigoni I, Muzzolon R, Vettore S (2011) Congenital FVII deficiency and thrombotic events after replacement therapy. J Thromb Thrombol 32: 362-367. [Crossref]
- O'Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM (2006) Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 295: 293-298. [Crossref]
- Girolami A, Molina MA, Lopez Galletti ML, Ferrari S, Sambado L, el at. (2016) A family with factor X deficiency from Argentina: a compound heterozygosis because of the combination of a new mutation (Gln138Arg) with an already known one (Glu350Lys). Blood Coagul Fibrinolysis. [Crossref]