Abstract
Purpose: To report a case of oculocutaneous hypopigmentation found in an infant with sever hypotonia, cardiomyopathy and agenesis of the corpus.
Case Report: This study involved baby a male who was delivered after a gestational period of 37 weeks. Product of healthy consanguineous Yamani parents with growth retardation, congenital bilateral cataracts, and dysmorphic facial, fair hair, and skin, nystagmus and he had left ventricle hypertrophy. Mutations in the gene epg5 have been identified as the cause of Vici syndrome.
Conclusion: Vici syndrome is a distinct clinical entity. Its main clinical manifestations included hypotonai, developmental delay, albinism, cataract, cardiomyopathy, immune deficiency, and agenesis of the corpus callosum.
Key words
Vici syndrome; agenesis of corpus callosum; EPG5
Introduction
Vici syndrome [OMIM242840] is a severe, recessively inherited congenital disorder characterized by the principal features of agenesis of the corpus callosum (ACC), cataracts, oculocutaneous hypopigmentation, cardiomyopathy, failure to thrive and variable.
It was first described in 1988, in two brothers with a malformation syndrome consisting of agenesis of the corpus callosum, cutaneous hypopigmentation, bilateral cataract, cleft lip and palate, and combined immunodeficiency. The sibs suffered from severe psychomotor retardation, seizures, recurrent severe respiratory infections, and chronic mucocutaneous candidiasis. They died of bronchopneumonia at ages 2 and 3 years [1,2].
Vici syndrome is a multisystem disorder associated with defective autophagy and suggest a fundamental role of the autophagy pathway in the immune system and the anatomical and functional formation of organs such as the brain and heart [3].
Clinical Report
This baby is the first product of healthy consanguineous Yamani Parents. He was full term, after an uneventful Pregnancy with normal vertex delivery at 36 weeks gestational age with birth weight 3.2 kg, a cranial circumference of 31 cm and an Apgar score of 8/8.
At 25 days old, he was hospitalized with history of cough, Shortness of breath, and fever for last three days. Associated with cyanosis, the patient had history of poor sucking and recurrent choking attacks with each feeding since birth, but unfortunately was neglected. No history of vomiting or skin rash and no history of abnormal movement. No family history of abortions or deaths.
In the examination he looked pale, cachectic and dysmorphic (Figure 1). (hypopegmented skin, eye, and blond hair, broad forehead, hyperbolism,long philtrum and micrognathia). His weight was 4,100 g (3–10th centile), length 53 cm (3–10th centile), and head circumference 33 cm (3rd centile). On ophthalmology examinations showed mild horizontal nystagmus, bilateral cataract and flair retina. Full of crepitation on bilaterl chest, no murmur with normal heart sound and had profound hypotonia.
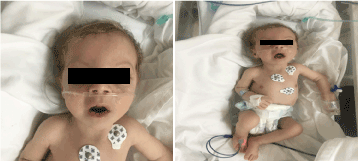
Figure 1. Clinical photo of patient at 2 months of age (a) Hypotonia, (b) blond of hair long philtrum and micrognathia
Laboratory investigation revealed microcytic and hypochromic anemia, renal profiles and coagulation were unremarkable. Alanine transaminase (ALT) and aspartate aminotransferase (AST) were elevated at 183–231 (10–45 U/L), and 193–301 (10–45 U/L), respectively. The bilirubin was normal. CK: 389 U/L and LDH: 612 U/L. The ammonia was normal 13 mmol/L. The immunologic workup revealed reduced immunoglobulins levels IgA: 0.1 g/L (0.7–4), but the IgG: 4.51 g/L (7–16), IgM: .8 g/L (0.4–2.3). Were within the normal value. Lymphocyte subsets (CD3, CD4, CD8, CD19, and CD22) counts were normal. Blood, urine and were no growth. The serological test for toxoplasma, rubella, cytomegalovirus, herpes simplex, and hepatic markers were all normal. Chest X-ray showed pneumonic infiltration and cardiomegaly. Abdominal ultrasonography was revealed bilateral hydronephrosis. Left ventricular hypertrophy was detected by echocardiographic study. Magnetic resonance imaging (MRI) of the brain confirmed agenesis of corpus callosum (Figure 2). Genetic testing was confirmed A homozygous loss of function variant of EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
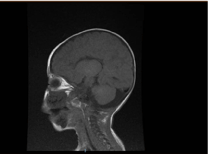
Figure 2. MRI of the brain, Sagittal T1 weighted section showing agenesis of corpus callosum
During the 2th week of admission, his condition deteriorated quickly with repeated episodes of aspiration pneumonia that required ventilation. Eventually, the patient went into cardiopulmonary arrest and died.
Materials and methods
WES is performed on genomic DNA using the Agilent Sure Select Target Enrichment workflow to capture regions of interest from a DNA fragment library. The whole exome is sequenced on the Illumina HiSeq 2500 sequencing system with a minimum coverage of 30X of 95% of the target regions. The proband's exome DNA sequences are mapped and compared to human genome build UCSC hg19 reference sequence. Saudi Diagnostic Laboratories uses an in-house pipeline to compare the proband's sequence to the reference sequence. Assessment of coverage and quality for targeted coding exons of the known protein-coding RefSeq genes is performed. Exome analyses interrogate thousands of genetic variants in a proband using proprietary databases customized to Arab populations. Subsets of these are characterized using the American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) guidelines to classify their clinical relevance. Sanger sequencing is performed to validate all variants included in the report unless otherwise stated
Results
A homozygous loss of function variant of EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) was identified in this patient. According to ACMG guidelines (Gen in Med 17:5, 2015) it should be classified as a pathogenic variant. Homozygous Loss of Function (LOF) variants of EPG5 are a common mechanism of Vici syndrome, 242840 [3].
Discussion
Vici Syndrome, is a rare congenital multi system disorder that is inherited in an autosomal recessive pattern, it is characterized by failure to thrive, hypopigmentation, immunological defects evident by recurrent infections, neurological abnormalities of developmental delay, seizures, cataracts, cardiomyopathy and neuromuscular abnormalities; with the later only being described in a few recent case reports [3]. Vici syndrome was first reported in 1988 by Dionisi Vici, et al. [1] in the case of two brothers presenting with albinism, ACC, cataracts, cardiomyopathy, mental retardation, and immunodeficiency, and was subsequently named ‘Vici syndrome’ by del Campo, et al. [4] in 1999. Vici syndrome has been observed in both males and females, and a number of related mutations have been identified in the EPG5 gene [5].
Our patient parents have consanguinity and support autosomal recessives inheritance of Vici syndrome. Thirteen of the 31 patients, including ours, are siblings. Cases have been reported in which the parents were distant cousins [6].
Vici syndrome is due to recessive mutations in EPG5 on chromosome 18q12.3, encoding ectopic P granules protein 5 (EPG5), a key autophagy regulator in higher organisms. Autophagy is a fundamental cellular degradative pathway conserved throughout evolution with important roles in the removal of defective proteins and organelles, defence against infections and adaptation to changing metabolic demands [7].
CNS development, immune regulation and skin pigmentation in Patients with Vici syndrome implicate autophagy in a wider range of cellular processes. In the brain, autophagy has been extensively investigated as a key pathogenic mechanism in various neurodegenerative disorders [8].
Most signs and symptoms of vici syndrome are present at birth; some features like cataracts, cardiomyopathy, and immunodeficiency are not always present at birth, but are expected to evolve over the first years [9]. And most of the patients have died within the first three years of life secondary to cardiomyopathy or pneumonia [10]
2021 Copyright OAT. All rights reserv
Acknowledgements
The authors wish to thank parents of patient and to Saudi diagnostic laboratory.
References
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet 29:1-8. [Crossref]
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics 45: 83-87. [Crossref]
- Said E, Soler D, Sewry C (2012) Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A 158: 440-444. [Crossref]
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinism and agenesis of the corpus callosum with profound developmental delay: Vici syndrome, evidence for autosomal recessive inheritance. Am J Med Genet 85: 479–485. [Crossref]
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A 164A: 3170-3175. [Crossref]
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 152A: 1849-1853. [Crossref]
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: a review. Orphanet J Rare Dis 11: 21. [Crossref]
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101. [Crossref]
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Vici Syndrome. Eur J Hum Genet 22. [Crossref]
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Case Reports in Genetics 2011: 421582. [Crossref]