Premature closure of the foramen ovale (FO) is a rare congenital anomaly that results in a wide spectrum of hemodynamic findings. We experienced the case of a male neonate with this condition who, at birth, showed unique hemodynamics that was similar to that in hypoplastic left heart syndrome, despite having a normal sized left ventricle (LV). His systemic blood flow was dependent on patent ductus arteriosus (DA). Echocardiography on day 0 revealed retrograde flow in the ascending aorta. We could not detect flow across the interatrial septum, and diagnosed premature closure of FO. Because his LV was not hypoplastic, we considered that the systemic blood flow would be maintained by LV output after lung circulation was established. We infused prostaglandin E1 incorporated in lipid microspheres (lipo-PGE1), which confirmed the retrograde flow in the ascending aorta. We stopped the lipo-PGE1 infusion after the flow in the ascending aorta became antegrade on day 2. After DA closed, systemic blood flow was maintained by LV output, and normal circulation was established. In addition to the congenital heart anomaly, several other anomalies were noted, including a downslanting of the palpebral fissure, micrognathia, and cryptorchidism. A cytogenetic analysis revealed der(20)t(6;20) (q25.2;p13), and 6q duplication syndrome was diagnosed. The patient was discharged without any medications on day 54 despite the presence of mild pulmonary hypertension. This is probably the first report of premature closure of FO in a patient with such hemodynamics and the first one of premature closure of FO associated with 6q duplication syndrome.
premature closure of foramen ovale, 6q duplication syndrome, neonatal hemodynamics, pulmonary hypertension, hypoplastic left heart syndrome
Premature closure of the foramen ovale (FO) is a rare congenital heart anomaly. An FO diameter <2 mm and Doppler velocity >120 cm/s or an FO diameter <3 mm with a Doppler velocity measured gradient >5 mmHg have been used as criteria by various authors to diagnose this entity [1]. This congenital heart disease can result in a wide spectrum of hemodynamic findings. If FO closes during early gestation, hypoplasia of the left ventricle (LV) occurs, and if it closes during late gestation, right heart failure may occur [2-4]. 6q duplication syndrome is a rare chromosomal abnormality that is associated with congenital heart disease [5], but no reports on patients with this chromosomal abnormality and premature closure of FO are available. Here we describe a case of 6q duplication syndrome with premature closure of FO who showed unique hemodynamics.
A 29-year-old G1P0 vaginally mother delivered a male neonate at 35 weeks of gestation. She had normal intelligence and had no anomalies, but had a spontaneous abortion at 26 years of age. Her sister’s son had chromosomal abnormalities, but the details were unknown. The neonate’s birth weight was 1860 g (<10 percentile), length was 41 cm (<10 percentile), and head circumference was 30.6 cm (>50 percentile). Apgar scores were 8 and 10 at 1 and 5 min, respectively. Blood oxygen saturation (SpO2) was not >90% after birth despite oxygen administration, and he was admitted to our neonatal intensive care unit (NICU). At admission, his SpO2 was 90% with the administration of 30% oxygen. He showed no breathing difficulty and lung abnormalities were absent on the chest X-ray. However, the X-ray revealed that his right atrium was remarkably enlarged and the cardiac apex was undetectable (Figure 1a); thus, a congenital heart disease was suspected as the cause for the desaturation. Echocardiography on day 0 showed retrograde flow in the ascending aorta (Figure 1b) and the systemic blood flow was maintained through the ductus arteriosus (DA). Although we suspected hypoplastic left heart syndrome (HLHS), his LV was not hypoplastic (Figure 1c). The LV diastolic dimension was 14.2 mm, which was normal considering his body weight. We considered that HLHS was unlikely because opening of the mitral and aortic valves was not restricted. We diagnosed premature closure of FO because flow through the interatrial septum was undetectable. We considered that his hemodynamics was similar to that during the fetal period, but was unable to explain adequate LV growth. Therefore, we thought that LV output would increase after lung circulation was established, and that the systemic blood flow would be supported by LV output. We carefully monitored the hemodynamics after administering prostaglandin E1 incorporated in lipid microspheres (lipo-PGE1). On day 1, flow in the ascending aorta became bidirectional, and on day 2, retrograde flow was undetectable (Figure 1d). Because we considered that the systemic blood could be maintained by LV output because his adequate LV size, we weaned the patient off lipo-PGE1 infusion on day 2. DA closed on day 7 and his hemodynamics were stable, but mild pulmonary hypertension (PH) was detected (flow velocity of mitral valve regurgitation (MR) was 3.4 m/s: pressure gradient was 47.1 mmHg).
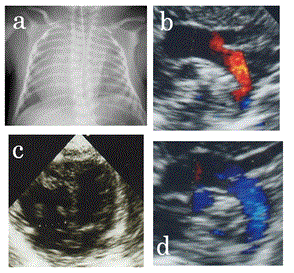
Figure 1. a. Chest X-ray on day 1. The right atrium is dilated and the cardiac apex is undetectable. b, c. Echocardiogram on day 1. b. Aortic view: aortic arch flow is retrograde. c. Four-chamber view: left ventricle is not hypoplastic. d. Echocardiography on day 2 showing antegrade aortic arch flow.
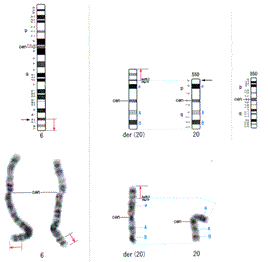
Several other anomalies were noted, including skull dehiscence, downslanted and wide palpebral fissures, a wide nose, low-set and malformed ears, high-arched palate, microstomia, thin lips, micrognathia, low hairline, micropenis, and cryptorchidism (Figure 2). Thus, we suspected a chromosomal abnormality. A cytogenetic analysis revealed a chromosomal abnormality of der(20)t(6;20) (q25.2;p13) (Figure 3), and we diagnosed 6q duplication syndrome. Because neonatal jaundice was prolonged until day 14, phototherapy was administered four times. However, he had no difficulty taking oral feeds and his body weight gain was acceptable. The patient had no cardiac problems except mild PH when he was discharged on day 54. In addition, the pressure gradient calculated from MR flow velocity was about 40 mmHg, and he did not require medications for PH.
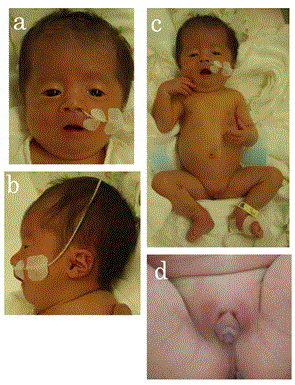
Figure 2. a, b. The patient’s face on day 32. Note the downslanted and wide palpebral fissures, the wide nose, low-set and malformed ears, microstomia, thin lips, micrognathia, and a low hairline. c. The patient’s full-length photograph. d. The patient’s external genitalia: note the micropenis and cryptorchidism.
t(6;20) (q25.2;p13) observed in the patient. Black arrows show the cutoff point.</p>
<h2 id=)
Discussion
Premature closure of FO can lead to a wide spectrum of hemodynamic findings, including findings similar to those of HLHS. LV showed adequate growth in the present case, suggesting that FO closed later in gestation. Fortunately, right heart failure was not apparent, suggesting that he was born soon after FO closed. On day 0, his hemodynamics was similar to those seen during the fetal period, thus being similar to those of HLHS. However, LV had grown adequately during the fetal period, and LV output maintained the systemic circulation after pulmonary circulation was established. In fact, normal hemodynamics was observed after pulmonary circulation was established. To the best of our knowledge, no reports are available on cases of premature closure of FO with such a clinical course. We wondered why it took so long to establish pulmonary circulation, thus his LV could not enough functioned by day 2. This may have been the cause for the slow decrease in pulmonary vascular resistance after birth. Our patient had a chromosomal abnormality, and the decrease in pulmonary vascular resistance is known to be delayed in some patients with chromosomal abnormalities such as trisomy 21. Premature closure of FO is sometimes associated with PH, but a previous study reported that PH with premature closure of FO tends to show a faster decrease in pulmonary vascular resistance than neonatal PH resulting from other causes such as respiratory distress syndrome or meconium aspiration syndrome [1]. However, PH was prolonged in our patient and mild PH was present on day 54 when he was discharged. This observation also supports the hypothesis that the PH was caused by premature closure of FO associated with the chromosomal abnormality. These observations support our hypothesis that the persistent high pulmonary vascular resistance in this patient was due to LV functional failure. To the best of our knowledge, no reports have been published on such hemodynamics seen in patients with premature closure of FO.
6q duplication syndrome is a rare chromosomal abnormality, and many cases result from an unbalanced segregation of familial reciprocal translocation [6]. A maternal cousin of this patient had chromosomal abnormalities, and the patient’s mother had a spontaneous abortion. These observations support the speculation that the mother had a reciprocal chromosomal translocation. Many cases with this syndrome have congenital heart disease [5]. However, this is probably the first report of a case of 6q duplication syndrome associated with premature closure of FO. The patient’s clinical features, such as downslanted and wide palpebral fissures, a wide nose, low-set and malformed ears, a high-arched palate, microstomia, thin lips, micrognathia, low hairline, micropenis, cryptorchidism, and low birth weight, are in line with the diagnosis of this syndrome [5]. However, prolonged neonatal jaundice has not been reported previously in patients with this syndrome. Most patients with this syndrome die early in life [5], and the prognosis depends on the degree of organ dysfunction. Several adult cases with this syndrome have been reported [7-9]. Our patient had mild PH at discharge, and we consider that control of PH was very important in his survival.
In conclusion, we experienced a case of premature closure of FO complicated with 6q duplication syndrome in a patient who showed unique hemodynamics at birth. This is probably the first report of premature closure of FO with such hemodynamics and the first case premature closure of the FO complicated with 6q duplication syndrome. Control of PH is very important factor affecting the prognosis in such cases.
- Gupta U, Abdulla R, Bokowski J (2011) Benign outcome of pulmonary hypertension in neonates with a restrictive patent foramen ovale versus result for neonates with an unrestrictive patent foramen ovale. Pediatr Cardiol 32: 972-976.
- Schall SA, Dalldorf FG (1984) Premature closure of the foramen ovale and hypoplasia of the left heart. Int J Cardiol 5: 103-107. [Crossref]
- Bharati S, Patel AG, Varga P, Husain AN, Lev M (1991) In utero echocardiographic diagnosis of premature closure of the foramen ovale with mitral regurgitation and large left atrium. Am Heart J 122: 597-600. [Crossref]
- Coulson CC, Kuller JA (1994) Nonimmune hydrops fetalis secondary to premature closure of the foramen ovale. Am J Perinatol 11: 439-440.
- Seel C, Hager HD, Jauch A, Tariverdian G, Zschocke J (2005) Survival up to age 10 years in a patient with partial duplication 6q: case report and review of the literature. Clin Dysmorphol 14: 51-54. [Crossref]
- Villa O, del Campo M, Salido M, Gener B, Astier L (2007) Small supernumerary marker chromosome causing partial trisomy 6p in a child with craniosynostosis. Am J Genet Part A 143A: 1108-1113.
- D'Emma C, Crippa L, Delozier C, Michail E, Graber P (1982) [Familial observation of partial trisomy 6, and probable partial monosomy 18q by parental translocation]. J Genet Hum 30: 39-50. [Crossref]
- Smith A, Jauch A, Slater H, Robson L, Sandanam T (1999) Syndromal obesity due to paternal duplication 6(q24.3-q27). Am J Med Genet 84: 125-131. [Crossref]
- Namihira T, Hattori N, Shiroma S, Miyazato Y (2004) Autosomal recessive juvenile Parkinson’s disease with partial trisomy of chromosome 6q syndrome: a case report. Psychiatry Clin Neurosci 58:672-673.