Background: Epidermolysis bullosa (EB) is a condition characterized by blistering of the skin and mucous membrane after minor traumas. Four types have been described: simplex, junctional, hemidesmosal and dystrophic. The Dystrophic Epidermolysis bullosa (DEB) form results from the genetic defects of the collagen type VII alpha 1(COL7A1) gene which has been extensively studied in different populations around the world. These studies showed that the mutations affecting the Middle Eastern populations are substantially different than the rest of the world. Unfortunately, research studies of the molecular epidemiology of EB is lacking in Saudi Arabia.
Objectives: In this study, we focused on the genetic causes of the DEB form in nine affected patients of an extended family that reside in Hail area.
Patients, Materials and Methods: We compared DNA sequences of affected individuals and their unaffected relatives to identify changes that can explain the clinical presentation of the disease.
Results: Two linked homozygous mutations were discovered in COL7A1 gene in all patients: G1400Vfs*310 and c.1370C>T, P457L. Analysis of the two mutations predicted the severity of the disease.
Conclusions: The G1400Vfs*310 mutation result in truncated protein that render it unfunctiona and that it explains the observed phenotype. The second mutation is a missence mutation that may affect the predicted to be pathogenic. We are the first to implicate the two mutations in the development of DEB.
epidermolysis bullosa, kindler syndrome, transmission electron microscopy, immunohistochemical epitope mapping
There has been recent spectacular success in elucidating the molecular basis of a variety of genetic skin diseases, and the prototype of such conditions is epidermolysis bullosa (EB) which is a clinically and genetically heterogeneous group of diseases characterized by blistering of the skin and mucous membrane after minor traumas [1,2]. EB comprises four distinct major types [3,4] classified on the bases of the level of blister formation within the skin and the level of cleavage within the basement membrane zone (BMZ), as demonstrated by diagnostic transmission electron microscopy or immunohistochemical epitope mapping. These types are simplex (EBS), junctional (JEB), dystrophic (DEB) and Kindler syndrome [4].
The EBS form resulted from mutations in KRT5, KRT14, PKP1, DSP, PLEC1, TGM5, JUP, EXPH5 and DST genes [3,5]. The separation in this form with intraepidermal lesions is due to the basal keratinocytes being fragil. In contrast, the tissue separation in the JEB form is the results of mutations in LAMA3, LAMB3, LAMC2, COL17A1, ITGB4 and ITGA6 genes [3,4,6]. The defect occurs within the dermal–epidermal basement membrane. The Kindler syndrome is caused by mutations in FERMT1 gene [3,4]. Lastly, the blisters in the DEB form occur beneath the lamina densa, at the level of the anchoring fibrils (AF). In the severe cases, the morphology is characterized by being altered, reduced in number or entirely absent 7. DEB is caused by genetic defects in the COL7A1 gene encoding type VII collagen [3,7].
The variations of clinical phenotypes, can be argued, are determined by the site of the lesion and the specific nature of the underlying mutations. However, it has been observed that phenotypic variability exist between siblings. This suggests that the clinical outcome could be influenced by other factors such as genetic and/or epigenetic changes [8,9]. The DEB can be mild or severe. In the mild cases, the symptoms include a few localized blistering or nail involvement. Based on the mode of inheritance, DEB is divided into two major subtypes either dominant (DDEB) or recessive (RDEB) [7]. The latter is subdivided into additional variants distinguished by the level of expression of the collagen VII gene and the AF appearance. The severe generalized RDEB subtype (RDEBsg), also known as Hallopeau-Siemens RDEB (HS-RDEB) [7], is characterized by generalized blistering and scarring of the hands, knees, elbows and feet leading to pseudosyndactyly, and severe mucosal involvement [10-12]. In addition, these symptoms are often associated with the development of aggressive, rapidly metastasising squamous-cell carcinomas and erosions, the gastrointestinal tract, particularly the oesophagus, is affected by blistering and scarring, also corneal erosions and loss of nails [13-15]. This phenotype can be explained by the COL7A1 mutations that lead to premature termination codons (PTC). The resulting truncated protein cannot assemble into a functional AF. The failure of AF assembly may explain the negative immunohistochemical staining to type VII collagen in patients with RDEBsg.
Additional RDEB conditions, collectively designated “RDEB generalized other” (RDEB-O) [16] including the non-Hallopeau-Siemens type (non-HS RDEB) as well as the DDEB subtypes include a spectrum of less aggressive phenotypes: in these subjects, blistering and scarring can be localized or generalized but are milder and do not lead to pseudosyndactyly, and extracutaneous involvement is less frequent [15]. The RDEB-O is frequently caused by missense mutations or in-frame deletions in one or both alleles. These mutations tend to be less disruptive to the protein. The AF is reduced in number with a wisp-like morphology and the immunohistochemical staining for collagen VII is reduced [7]. In the DDEB, the majority of the reported mutations lead to glycine substitutions in the triple-helical domain of COL7A1, although splice site mutations leading to in-frame exonic skipping have also been described in a limited number of families [14].
Both dominant and recessive DEBs are caused by mutations in the type VII collagen gene, COL7A1. This gene consists of 118 exons spanning about 31.1 Kb at the chromosomal location 3p21.1 and it encodes a 9 Kb mRNA [17]. Over 700 various mutations have been, so far, reported in “The Human Gene Mutation Database” (HGMD), most are private, but small number of recurrent mutations have been described [18,19]. The mutation G2043R has been reported to be recurrent mutation in the DDEB [20] worldwide and has been reported in Hungarian, Italian, Hispanic Mexican, Scottish, Austrian, North American (Caucasian), Norwegian, Spanish and white British patients [20-25].
In RDEB few recurrent mutations have been described. The following are just some examples. The mutations R578X, 7786delG, and R2814X have been reported in COL7A1 gene in 25% of British patients with RDEB [26,27], even though the R578X mutation was first described in Italian patients [28]. The mutations 5818delC, 6573+1G→C, and E2857X are present only in Japanese patients afflicted with RDEB [12]. Whereas, mutation 425A→G, and splice site mutations leading to premature termination codon, were first described of RDEB in an Italian patient [29]. In the Italian patients with DEB, six recurrent mutations have been found: 7344G→A, 425A→G, 8441-14del21, 4783-1G→A, 497insA, and G1664A; of which the last three mutations were found in Italian patients only [30].
The molecular epidemiology of EB in the Middle East is significantly different from that found in Japan, Europe and the US [31]. None of the recurrent mutations reported for DEB in other regions were detected. In the Arab world molecular investigation of a large family from Northern Tunisia showed that the affected patients were homozygous for the c.7178delT mutation [32]. In another study, R1763X, G1483D, 4406_4415del, 1507+1G→A specific to Tunisian population; whereas the mutations R1978X, and 7344G→A, which were previously reported in other ethnic groups, were found in Tunisian patients with RDEB [33].
However, little is currently known about the molecular epidemiology of EB in Saudi Arabia patient populations. Only two studies of EB were undertaken in Saudi Arabia and both studies focused on the prevalence of EB in the eastern province of the country and the effectiveness of Phenytoin as a treatment [32,34-36]. Neither of them looked at the underlining genetic causes of the disease underlining the need for molecular genetic studies of the disease in Saudi Arabia. In this report, we have identified a large extended consanguineous Saudi family with 9 patients afflicted with the recessive form of DEB. We found a novel splice site mutation in all patients causing exon skipping and a pre-termination stop codon PTC). This study is the first genetic study of EB ever to be performed in Saudi Arabia.
Study Design
Nine Saudi patients with clinical diagnosis RDEB belonging to eight nuclear families. These families are related to each other by decent and were grouped into two branches. The two branches have a common ancestry that goes back few generations. These families reside in the city of Hail in the northern region of Saudi Arabia. The patient’s ages are between 3 months to 7 years at the onset of the study with mixed gender. They have been taken care of at the epidermolysis bullosa center at King Khalid Hospital's nursery in the city of Hail. Written informed consents was obtained from the parents for themselves and consenting for their patient and normal children to be included in this study. All Patients and their normal siblings were less than 18 years old at the time of this study. Charts of all nine patients were reviewed and data of interest were collected from the patients’ medical records. Pedigree was constructed using Progeny software. Blood samples and skin biopsies were obtained from the patients, available parents and normal siblings. Samples were sent to King Faisal Specialist Hospital and Research Center (KFSH&RC) in Riyadh for analysis. This study was approved by both centers research advisory committee. All experiments were performed as part of an approved protocol by the Institutional Review Boards of all institutions involved in the patient care and laboratory testing including sequencing as described by this section in accordance with the participating institutions guidelines and regulations that confoims to international standards.
Clinical diagnosis of DEB
The clinical features of the diseases in the 9 patients ranged from moderate to severe (Table 1). Accordingly, the patients were divided into 2 groups. Group A contained 3 patients with moderate severity and group B contained 6 patients with severe disease which presented with scars and wounds that are more extensive than the patients in group A. For the immunohistopathologic and electron microscopic studies, two biopsies were taken from each patient after the administration of local anesthetic. (1) Four millimeter (mm) punch biopsies were taken from the affected area on the right thigh by circle movement to reach the upper layer of the subcutaneous tissue. (2) Two mm punch, for normal histopathology and immunohistochemistry, from adjacent area to a new fresh bullae skin. Both biopsies were saved separately in formaldehyde/glutaraldehyde containers, fixed and sent to KFSH&RC for analysis.
Table 1. Clinical Features of the nine patients afflicted with EB
Patient number |
Family Branch |
Sex |
At the time of the study in 2014 |
Age |
Clinical features |
Severity |
Suppl. Figures |
1 |
A |
female |
12 |
- Painful erythematous fragile skin and bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia resulted as a consequence of damage to hair follicles.
- She has thinning, fall of hair and scaring alopecia of the scalp.
- She has deformed nails and some finger without nails (anonychia).
- She has Scar formations complicated in some patients with structures formations which prevent joints movements, flexion contractures of the extremities present also.
- Due to repeated erosions around fingers the wound healed with forming fusion in fingers (pseudosyndactyly) started in web space then progress till all digits eventually become totally fused and encased by atrophic scar tissue. Also she has loss of teeth.
|
Severe |
1q & 1r |
2 |
A |
male |
3 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory, hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
|
Moderate |
|
3 |
B |
male |
4 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
- He has deformed nails and some finger without nails (anonychia).
|
Moderate |
1e, 1l |
4 |
B |
male |
10 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles. He has thinning, fall of hair and scaring alopecia of the scalp.
|
Severe |
1a, 1b, 1c, 1d, 1h, 1i, 1j, 1k |
5 |
B |
female |
9 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory, hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
- She has thinning, fall of hair and scaring alopecia of the scalp.
- She has deformed nails and some finger without nails (anonychia).
- She has Scar formations complicated in some patients with structures formations which prevent joints movements, flexion contractures of the extremities present also.
- Due to repeated erosions around fingers the wound healed with forming fusion in fingers (pseudosyndactyly) started in web space then progress till all digits eventually become totally fused and encased by atrophic scar tissue. Also she has loss of teeth.
|
Severe |
1m, 1o |
6 |
A |
female |
3 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory, hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
|
Mild |
1f |
7 |
A |
female |
8 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory, hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
- She has thinning, fall of hair and scaring alopecia of the scalp.
- She has deformed nails and some finger without nails (anonychia).
- She has Scar formations complicated in some patients with structures formations which prevent joints movements, flexion contractures of the extremities present also.
- Due to repeated erosions around fingers the wound healed with forming fusion in fingers (pseudosyndactyly) started in web space then progress till all digits eventually become totally fused and encased by atrophic scar tissue. Also she has loss of teeth.
|
Severe |
|
8 |
A |
female |
5 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory, hypopigmentation and hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
|
Moderate to Severe |
1g, 1n, 1p |
9 |
A |
female |
11 |
- Painful erythematous fragile skin, bullae which become crusted erosions almost all over her body (skin and mucous membrane), but mainly over mechanical trauma area especially distal extremities. Erosions heal with post inflammatory, hypopigmentation and, hyperpigmentation. Erosions healed with atrophic scars and milia results as a consequence of damage to hair follicles.
- She has thinning, fall of hair and scaring alopecia of the scalp.
- She has deformed nails and some finger without nails (anonychia).
- She has Scar formations complicated in some patients with structures formations which prevent joints movements, flexion contractures of the extremities present also.
- Due to repeated erosions around fingers the wound healed with forming fusion in fingers (pseudosyndactyly) started in web space then progress till all digits eventually become totally fused and encased by atrophic scar tissue. Also she has loss of teeth.
|
Severe |
|
Histopathology
Sections from fixed skin biopsies were cut and mounted onto slides. H&E staining was performed for microscopic evaluations. Immunohistochemistry, was performed on selected cases to check for presence, absence, or positions of certain proteins (antigens) in situ (such as laminin, collagen type IV). To do this, 4 µm paraffin-embedded sections were cut and mounted on super frost/plus slides. Slides were placed in 60 °C for 15 min then dewaxed with xylene and hydrated. Using microwave method, antigen retrieved by heat or enzymes. Slides were then immersed in 0.5 % H2O2 followed by serum albumin to block endogenous biotin. Slides were then incubated with primary antibody followed by secondary antibody. Reaction was detected by the DAB chromogen (brown in color).
Electron Microscopy
Tissue from skin biopsies were fixed for at least 12 hours in 1.6% glutaraldehyde buffered to pH 7.2-7.4 with Millonig’s Phosphate buffer, washed three times for 15 minutes in Millonig’s buffer. The tissues were, then, post-fixed in 1% OsO4 buffer for at least 10 hours. Subsequently, they were washed three times for 15 minutes each in distilled water. To Dehydrate the tissues, they were subjected to a graded series of acetone at 50% and 70% concentrations two times for 15 minutes each and then at 90% acetone concentration three times for 15 minutes each. Final dehydration step was performed in 100% acetone four times for 20 minutes each. Subsequent steps involve infiltration of the tissues with acetone/resin at 1:1 ratio for at least 12 hours, followed by infiltration with resin alone for another 12 hours. The tissues were molded for electron microscopy and placed into 50ºC vacuum oven overnight. Next morning, the blocks were placed into the 70ºC oven for 48 hours to complete polymerization of the resin. To prepare the slides, semi-thin section (0.5 µm) using Ultra Microtome were prepared and stained for 1 min with Join’s stain (mix of blue stain). Transmission Electron Microscope (JEOL 1230 TEM) was used for analysis.
Polymerase Chain Reaction (PCR) and Sequencing
Genomic DNA was extracted from whole blood samples obtained from patients (and their families, when available) as described previously [37]. Briefly, Gentra Puregene Blood Extraction kit (Qiagen, Valencia, CA, USA) was used. Promoters, 118 exons, and both the 5′- and 3′- untranslated regions of the COL7A1 gene were amplified by polymerase chain reaction (PCR) using HotStarTaq DNA polymerase (Qiagen, Valencia, CA, USA). Primers were designed from within the intron regions to span the splice sites and the exon/ intron boundaries. Sequences of the primers are listed in Supplementary table 1. In addition, M13 sequences were attached to the 5′ end of each primer to allow for forward and reverse sequencing. Amplifications were performed by touchdown PCR. Extension temperature ranged from 68to55° C. PCR products were sequenced with the BigDyeR Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, California). Sequencing data were analyzed for mutation detection using SeqMan II software (DNA Star Inc., Madison, WI). Only the region were the mutation was detected in the patients were sequenced in the available parents and other siblings DNA. Mutations found were classified as either novel or reported with the help of HGMD data base. To rule out the possibility that novel mutations detected were unreported normal variations within the Saudi population, 261 DNA samples from normal individuals obtained from the blood bank of the King Faisal Specialist Hospital and Research Centre were sequenced. Sequencing data were analyzed for mutation detection using SeqMan II software (DNA Star Inc, Madison, WI). Samples with mutations were confirmed by sequencing the reverse strand using M13 Reveres primer. The effect of the mutations detected on the severity of the disease was analyzed by PolyPhn-2 and Mutation Taster. HGMD public database was used to check on the novelty of the mutation. University of California, Santa Cruz (UCSC) genomic browser was used to determine the frequency of mutations.
Supplementary Table 1. List of primers used in this study
Exon |
Forward primer |
Reverse primer |
Product size |
Promoter |
GGGACCTTGGTTTCCCTAAG |
AGCCCGAGTCCTGGTCTT |
624 |
1 |
CCCGAGATCAGGGACTTTTC |
TGGAGTTGGCTGGGTTGT |
411 |
2 |
ATCCCAAGTCCCAGTGAGG |
GGTTTCCAGGGTCTCCTCC |
422 |
3, 4 |
TGATACCCGTAACCCTCACC |
ACTCAGTCACCCACATGCTC |
539 |
5, 6 |
GAGTGACAGGTCAGCTGGG |
AGCTGGGACGTTCACCTG |
689 |
7, 8 |
GGCAGGAGGTAGGATGTCAG |
TGGGCCTTGCAGACTCAG |
551 |
9, 10 |
AGATGTGGGCTGAGGGG |
GCCTGCAAGATAACAGGGTC |
557 |
11 |
CCTTTCCTGGAACTTGGTG |
CTCCAACAGGACCAGAGCT |
448 |
11, 12 |
GTGAGACTGGTCAGTGCGG |
TGGAGGTACACTCAGACCCC |
575 |
13, 14 |
GTCCATCACTGCCATCGTC |
TCAGAGGGAAATGCTGGC |
563 |
15 |
AGGGACCAATGAGGGTATGG |
GAGAAGCAAGGGTCTGCAAG |
356 |
16, 17 |
AGACTCCCATCATCTTCCCC |
CTGGGCAATCAGGAACACAC |
586 |
18, 19 |
GACTTTCAGATCACCCCTGC |
CTCCAATCCAGAGCACAAGC |
555 |
20 |
GGTCTGAGAGGCTGGGC |
CTACCTTGCGGAGTCTGCC |
329 |
21 |
AACAGAGCCAGTGAAGTGGG |
GAGTCACTCAGAGTCGGGG |
353 |
22, 23 |
CAATAAGACAAACCGGACCC |
GGGAACAAGTTACTGAAGCGG |
565 |
24 |
TCCTGAGGCATCTGTCACAC |
CCTATTGGGTGGTCAGGAGA |
374 |
25, 26 |
CCCTGATGTGTTTCTCCAGG |
GTCCAGTGGCTGCATGATAG |
552 |
27, 28, 29 |
AGTAGGCTGATGGGGAAGGG |
ATCAGTGAGACCTGTGCTGC |
566 |
30, 31 |
GGGGAATAGAGAATGGGTTGA |
TGTAAAAACCCCGAGACAGC |
355 |
32, 33, 34 |
CCTTGTTGATGGGGTGAGG |
TGCACAGGGTCTGGTGTG |
579 |
35, 36 |
TGTCTAGGGGCTTCTTCCAG |
ATCAAAGGGTCACAAGGGC |
464 |
37 |
CCCCATTTGAACCCACATA |
CTGACCTGCTGTTCTCTCC |
335 |
37, 38, 39 |
ACCCCATGACCCTCATCAC |
ACACGGGTGGGAAGACC |
549 |
40, 41, 42 |
GGCTGACTTGATGATGTCCC |
TGGAAAAGAAGAGGTCAGAGC |
689 |
43 |
GGTGAGGCTGGAGAGAAGGT |
TGAGGAGCAGGGGTAGCA |
265 |
44 |
AGTCTTACCCCTGCCAACCT |
GTGGCCCCTGAATGTAGAGA |
315 |
45, 46 |
GACCAGGAGTGGGACTTTTG |
CACTGTGGAATCACAGCCC |
393 |
47, 48, 49, 50 |
TTCCATGTGACCTCCATTCC |
GATGCAGGACGCTCGAAG |
700 |
51, 52, 53 |
CAACCTCAAGGCTTCTGGG |
CCCTCAGCCTTTCCTATCAC |
652 |
56, 57, 58 |
AGTTGAGCCTGGGGCAAAC |
CCTGAGGATAGGGGAGGAAG |
656 |
59 |
GGGTGAGTGTGATTGGTCCT |
CCACACCCTCTCACCTTTTC |
271 |
60 |
GCCCACAGGTGAGTGATG |
GGTCACCCTGTGGAAAATAGA |
253 |
61 |
TGGGGGTTTCTCAGGACA |
AATGAGGCTGGGGTCTAGG |
273 |
62 |
GCCTTAGGCTCACAGGGA |
ACCAACTCTCTCGGATGCTG |
234 |
63, 64 |
TTTTGGGGTCCTCTTAGTCC |
TTGCTACAGATCTTGGCTGTG |
290 |
65, 66, 67 |
AACCATGGAGCTACAGGCAC |
ATACGGCAGAACACAAAGGG |
617 |
68, 69, 70 |
AGCCATGATGTTTCAATGGG |
ATGGAGCCTCATGGCAAG |
514 |
71, 72 |
GCTTTCCTGAGGCCGTG |
GAAGAGAGAATGCTGGTGGC |
466 |
73 |
GAAGTGGCTCAGTGGGTTGT |
AAATGCAAATAGCGGGTGAG |
409 |
74 |
GAGTGAGGGAAGAGGGGTTG |
CCCTGCAGGAAACAAGAAAA |
227 |
75 |
AGGAGAACGTGGAGAACAGG |
CATATTTCTGGGGACCAAGC |
274 |
76, 77, 78 |
CCCTATAACCCTCTGTGATCCTG |
AGACCCCAGCCCTGCAC |
584 |
79 |
CTCTGTCTAGGGGGATGGTG |
AGACATGCACACACACAGCA |
234 |
80 |
ATTGACCCCCAAGGAAAAAG |
TCTGATGAGGGGGAGTTAGC |
270 |
81, 82, 83 |
TGACCAGTACTGCCTCAGTTTC |
GAGGAAGAGAAAGTTCAGGGC |
596 |
84, 85, 86 |
CCTAGCTCACAGGTTAGGG |
TGCCCCACTCCTCATATTTC |
554 |
87, 88, 89 |
TGGGCCTGAAATATGAGGAG |
GGTGGGTAAACTATGGGTCG |
566 |
90 |
CCGCATATTTAAGCTCTGGC |
CAAGAGGCCCCTTATGC |
370 |
91A |
GAAGGCTCAGTCCAGACACC |
AACCCCAACACGTCCACTC |
530 |
91B |
AGATCCCAGGACACACCTGA |
CTCAGACATGCGACCAAGAA |
376 |
92, 93, 94 |
GAGCGTCCCTCTGTCTGTG |
CCCCTCAAGACTGGGAAC |
742 |
95, 96, 97 |
TTCCCAGTCTTGAGGGG |
CATTGACTCAGAGGTTGGAAATC |
627 |
98 |
CACAGGACCCTCACCTCACT |
TGTACCCCAGGACTCTCTGC |
512 |
99, 100 |
CACAGGACCCTCACCTCACT |
CAGGCAGGAATCAGAGGAGT |
454 |
101, 102 |
AAGGGTGACAAGGTGAGTGTG |
CCCTCCTGCCCTCACAG |
477 |
103, 104 |
GGGCTCGTTGTATTCTAAGCC |
AGCTACCACACTGGTGGGAC |
545 |
105, 106, 107 |
GTCCCCCACCTCCACAGTAG |
CGATCAGGCCCTCCTTG |
821 |
108 |
GGAGAGATGGTGAGTGTGGG |
ATGACCCGACCATGAGCTTC |
251 |
109, 110 |
GTTGGGGTCACAGTGGAGTT |
ATCAGAGTCAGGCAGGTTGG |
393 |
111 |
ATGGGAGTTCAGGGAGGTTC |
CCAACCTGCCTGACTCTGAT |
517 |
112 |
CAGAGGCCCCGAAGGACT |
CTACCTCCCTCACCCAGCAC |
543 |
113 |
AGAGAGGGGAGCAGGTGAGT |
TACAGGGCAAGCAGTCAAGA |
537 |
114, 115 |
TCTCTACCCCTCTGCCTGTG |
GCCCTGCATTCATGGACAC |
464 |
116, 117 |
GGAAGGTGAGGACAGCTGAA |
GTCTCCTCCAGGACCCTGAC |
777 |
118 |
GTAGGGTCCCCTTCCATGTT |
CTGTACCCATGCAGAATAATACCA |
644 |
Whole Genome Genotyping Microarrays
Whole genome was genotyped with 2.7M Cytogenetic Microarray (Affymetrix, Ca, USA) which offers more than 2 million markers across the genome to detect both known and unknown broad range of chromosomal aberrations in accordance with the manufacturer’s instructions and as described previously [38]. Chromosome Software Analysis Suite (ChAS) is used to analyze the Cell files. ChAS provides options for data visualization and customized analysis in an easy to use in a graphical interface. In addition, for loss of heterozygosity analysis, Affymetrix AxiomTM gene ship was used. The result was analyzed by AutoSNPa, version 4, software.
Clinical diagnosis of EB
Nine distantly related Saudi patients were recruited for this study being treated at the Hail Epidermolysis Bullosa Center. Their sex and ages at the time of diagnosis are presented in Table 1. The patients belong to eight consanguineous nuclear families as described above. All parents of the patients and siblings are completely well without any single lesion after full examination. As described in the pedigree (Figure 1), the disease is familial and inherited in a recessive pattern. Patients were born with fragile skin, blistering and erosions mainly around trunk. Later, the bullae (Supplementary Figure 1a, and 1b) started to spread with small to moderate size that range from 1 to 3 cm in early infancy over mechanical trauma area especially around distal extremities which ruptured to erosions, Erythema. The erythema heals with Postinflammatory hypopigmentations or hyperpigmentations (Supplementary Figure 1c-1f).
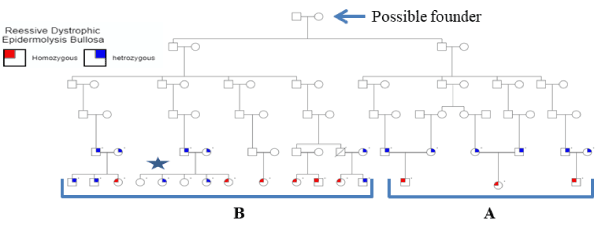
Figure 1. Print out of the pedigree of the extended family suffering from EB. Individuals with dot above their squares of circles are tested for mutations. Nine patients, marked in red, were affected and carry the two mutations described in the text. Individuals with blue mark are all heterozygous. The ones with no marks are normal. All remaining individuals were not tested either because they were not available at the time of the study or refused to participate. Diagonal line represents known death before the onset of the study. Circles represent females and squares represent males. The two branches of the extended family are marked with A and B. The sequences of the nuclear family with a star are presented in supplementary figures 3 and 4
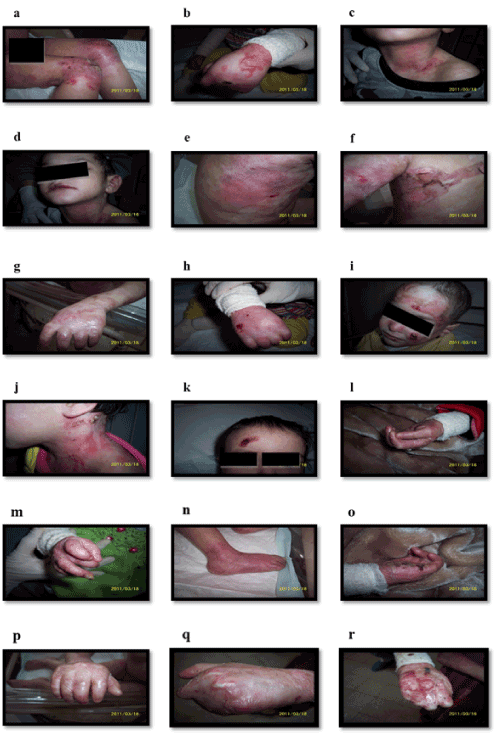
Supplementary Figure 1. Clinical features of some patients as described in Table 1. (a) Intact bullae in right thigh, (b) Bullae over dorsum of left hand, (c) Erosions in left side of neck, (d) Erosions and Erythema, (e) Erosions and Post inflammatory hypopigmentation, (f) Erosions over trunk, (g) Atrophy scar in left hand, (h) Scars and erosions, (i) Milia, (j) Crusted erosion in the neck, (k) Crusted erosions, (l) Anonychia, Loss of nail and nail dystrophy, (m) Loss of nail and nail dystrophy, (n) Loss of toe nails and erythema in right foot, (o) Nail loss and crusted erosions, (p) Fusion of fingers and milia, (q and r) Pseudosyndactyly (mitten-hand deformity) and Anonychia of both pictures
The involved areas increased gradually in size with age to encompass most of the body with mucous membrane involvements. The erosion’s inflammation damage hair follicles and result in the formation of scars and milia (1- to 4-mm white papules) (Supplementary Figure 1g-1i). Erosions frequently associate with crustations secondary to bacterial infection (Supplementary Figure 1j, and 1k). Repeated trauma causes new lesions and wounds which heal slowly with variable degree of pain.
The patient’s hair thins, and subsequently fall, Also, there were scaring of alopecia of the scalp. They have deformed nails but without nails in some fingers (Anonychia) (Supplementary Figure 1l-1o). Scar formations were complicated in some patient’s number 1, 5, 7 and 9 with flexion contractures formations which prevent joints movements.
Due to repeated erosions around fingers, the wound healed in some patients 1, 5, 7, and 9 forming fusion in fingers (pseudosyndactyly) started in web space then progress till all digits eventually become totally fused and encased by atrophic scar tissue (Supplementary Figure 1p-1r). Frequent skin examinations were performed to exclude Squamous cell carcinoma or melanoma especially in chronic erosions area. These examinations failed to detect any transformation. All patients have regular dressing with minimal trauma to skin and topical antibiotics are used regularly with each dressing. No internal organ involvement was detected. However, the cutaneous lesions had many secondary bacterial infections which sometimes complicated with septicemia. There were patients not in included in this study in the center die secondary to septicemia.
Malabsorption of iron resulted in a mixed anemia, and failure to thrive. The patients’ growth was drastically impaired due to decrease in feeding because of pain associated with oral mucosa erosions. All patients have mild to moderate hypochromic microstic anemia. These patients were prescribed vitamin and iron supplementations. They suffered from mild difficulty to swallow solid food but not soft. No difficulty in defecation or micturition. Gastrointestinal organs were not involved.
No other signs or symptoms in other areas including respiratory organs. However, ophthalmologist visits were regular to detect any erosion in the eyes. At the time of writing this manuscript, mild erosions of the eyes had been detected.
The clinical features presented here represent a recessive inherited dystrophic epidermolysis bullosa - Hallopeau-Siemens (RDEB-HS) type. These features start as being mild then progress to severe. For this reason, skin biopsies from each patient were obtained and immunoflorecnt microscopy was performed to confirm dystrophic epidermolysis bullosa diagnosis.
Histopathology
The H&E stained slide from the skin biopsy revealed a subepidermal separation (blistering), associated with mild mixed dermal inflammation and fibrosis (Figure 2a). Immunohistochemical staining for collagen type IV (Figure 2b-2c) revealed that the basement membrane (which expressed the brown-stained type IV collagen) is above the split (at the blister roof), and above that too is the epidermal keratinocytes (which expressed the brown-stained cytokeratin 14) (Figure 2d). This indicates that the separation (blistering) is at a level below the basement membrane.
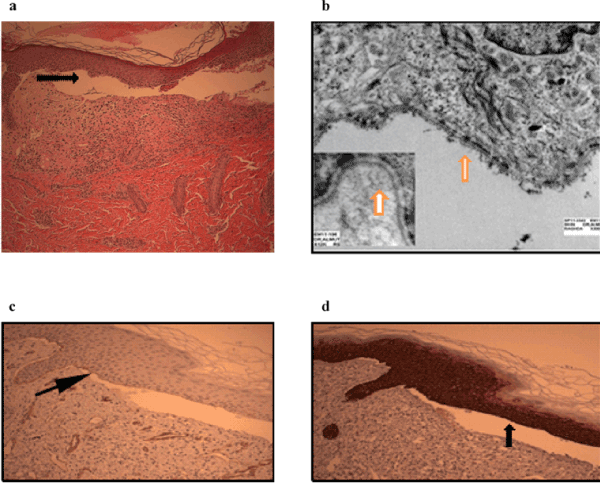
Figure 2. presents (a) H&E staining showing a subepidermal splitting (arrow), with fibrosis and mild mixed inflammation beneath it. Original magnifications at 100X. (b) Electron micrograph showing intact hemidesmosomes and basal cell ultrastructure; no preserved anchoring fibrils observed (inset). C & D. Immunohistochemical staining illustrating (arrows) that both collagens IV +ve basement membrane (c) and Cytokretain 14 +ve keratinocytes (d) at the blister roof. Pictures were taken by the authors
Electron Microscopy
On examining the skin biopsy ultrastructurally, the intermediate filaments cytoskeleton of the epidermal basal cells, as well as the basement membrane hemidesmosomes, appeared intact. However, the anchoring fibrils were absent (Figure 2b). This observation, coupled with the histopathologic and immunohistochemical pattern, was in keeping with the known clinical impression of dystrophic form of epidermolysis bullosa.
Microarray Genotyping
To rule out copy number variations of the COL7A1 gene in these patients, 2.7M Cytogenetic Microarray was used. No deletion or insertion was detected (data not shown) in all patients. Homozygosity study (Supplementary Figure 2) showed that all the patients are homozygote and that all parents were heterozygote for the region containing COL7A1 gene.
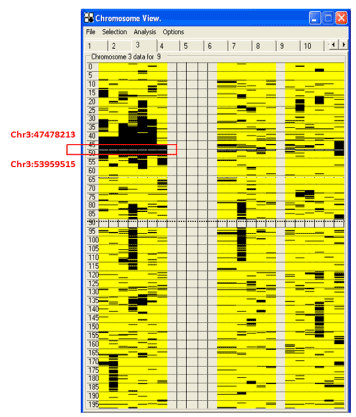
Supplementary Figure 2. shows the output of homozygosity mapping of the AutoSNPa software. The homozygous region (left side) shared by all 9 patients, but not with other normal family members, is boxed in red and the codon locations are indicated. The right side represents the homozygous regions of the parents and the normal siblings
Sequencing
The sequencing of the PCR products of the COL7A1 DNA obtained from all patients revealed two homozygous mutations in all 9 patients. The most significant mutation is a G deletion mutation at position 4199 (c.4199delG) which resulted in the change of the second amino acid of exon 37 at position 1400 from Glycine to Valine and a frame shift leading to a stop codon after 310 amino acids (G1400Vfs*310; Figure 3a). All parents and some siblings (see pedigree) were heterozygous (Supplementary Figure 3) shows the sequencing of the largest nuclear family from the extended family. To assess the frequency of this mutation in Saudi population, 261 DNA samples from healthy Saudi individuals were sequenced. No homozygous change for this mutation was found). Only 2 samples were heterozygous for this mutation (2/261= 0.00766). To check whether this mutation has been reported previously, HGMD date base did not list this change in COL7A1 gene as part of its comprehensive list of published mutations. Moreover, Mutation Taster website was used to analyze the mutation farther (Figure 3b). This variant was not found in the 1000 genome and ExAC databases. It appears that the mutation is novel and restricted to one extended family presented here residing in the northern province of Saudi Arabia.
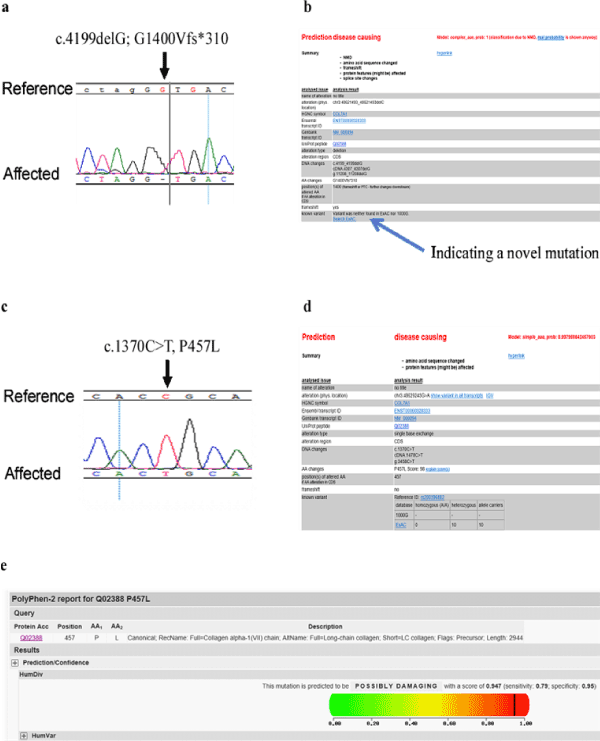
Figure 3. Print out of the sequences generated by Seq-Man software which displays the mutations detected in patients. (a) Sequence of the first mutation in all patients showing the deletion of G as compared to reference sequence. (b) is a MutationTtaster output showing that the mutation is novel and damaging. (c) Sequence of the second mutation found in all patients. Print screen of MutationTaster (d) and Polyphen 2 (e) outputs showing that this mutation is damaging
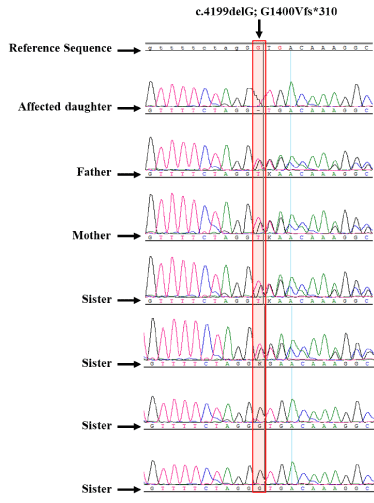
Supplementary Figure 3. showing the chromatograms of a representative family sequencing, marked with a star in figure 1. The sex and relationship of all family members are shown. Two father, mother and two of the sisters are heterozygous while the other two are normal. The mutation is highlighted with a pink box
The other variance was also detected as homozygous missense mutation in all 9 patient DNA sequences (c.1370C>T, P457L) in exon 11 (Figure 3c). We employed PolyPhen-2 (Figure 3e) and Mutation Taster (Figure 3d) website for the prediction of whether it has a high probability of causing disease. Both website predicted it to be disease causing with high probability. All family members follow Mendelian inheritance (Supplementary Figure 4) as in the first mutation. Mutation Taster indicated that this mutation is known (rs200396882). UCSC genomic browser web site was used to analyze the rs200396882 variant (Supplementary Figure 5). The frequency of it is extremely low (0.008%) supporting the prediction that it cause disease. The DNA around this mutation was sequenced in the 261 normal control samples. Three heterozygous and 2 homozygous samples were found. It is not clear whether these 2 homozygous samples are from normal individual. It may be that the two samples came from affected individuals.
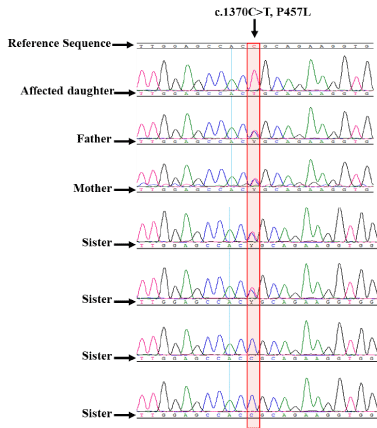
Supplementary Figure 4. showing the chromatograms of the second mutation. The missense mutation is shown for the same family in the supplementary figure 3. The patient is homozygous for the mutation. The father, mother and two of the sisters are heterozygous while the other two sisters are normal. The mutation is highlighted with a pink box
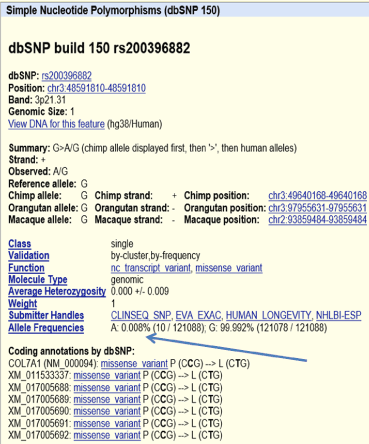
Supplementary Figure 5. presents the UCSC genomic browser output describing the second mutation characteristics. What is important here is the frequency of the change is 0.008% (pointed to it by an arrow)
In this report, we have identified a Saudi specific mutation in the COL7A1 gene that result in the development of EB disease very early in life. The extended family we studied here resides in Hail city in the northern province of Saudi Arabia. There are no molecular studies reported on cases of EB in Saudi Arabia. Only two studies mentioned in the introduction of this report described EB disease in the eastern region of the country [32,34-36]. We are the only group to our knowledge to study the molecular changes of people affected by the disease in Saudi Arabia. Our study is restricted to one extended family in one region of the country. It is not clear whether the cases in the eastern region are related to our case family studied here.
Two mutations were found in all 9 patients c.1370C>T, P457L in exon 11 and c.4199delG, G1400Vfs*310 in exon 37. The first mutation is a known SNP (rs200396882) with very low frequency (0.008%). It is not clear whether this frequency includes cases of normal but homozygous for this variant. We used two predictive web based programs (MutationTaster and Polyphen-2) to predict whether the SNP can be disruptive. As shown in Figure 3c, the mutation is damaging with high probability.
This mutation is located in one of the nine Fibronectin III-like domains (FNIII) located just downstream of the N-terminus region in the amino-terminal non-collagenous (NC-1) domain which consist of the first 28 amino acid [17,39,40]. The 9 domains (numbered FNIII-1 to FNIII-9; counting from the N-terminus region) have homology to adhesive proteins and they are thought of as attachment or anchoring points to the basement membrane structures. Each of the 9 domains contains repeat sequences. The P457L mutation is within the third (FNIII-3) domain which is expected to weaken or even eliminate at least one of the 9 anchoring points. In all, the location of the mutation and the prediction of PolyPhen2 and Mutation Taster pointed to the high likely hood that this mutation to be responsible for at least some of the phenotypes observed.
The second mutation, p. G1400Vfs*310 is in exon 37 just 9 exons after the NC-1 domain within the triple helix that is part of the central collagenous domain (the next 84 exons to the NC-1 domain) [17,39,40] domain. This domain is characterized by Gly-X-Y repeat sequences. The p. G1400Vfs*310 substitute Glycine with Valine disrupting one of the Gly-X-Y repeat. The glycine substitution in the triple-Helical region has been shown to cause a spectrum of dystrophic EB phenotypes [41]. More importantly, the mutation would result in a premature termination resulting in a truncated protein removing most of the triple helix domain and the subsequent C-terminal (NC-2) domain. Most of the mutations reported consist of nonsense or small insertion or deletion mutations. This type of mutations leads to premature termination codon (PTC). In general PTC generates low levels of mRNA transcripts, presumably due to nonsense-mediated decay [42,43]. One would expect that nonsense-mediated decay would lead to the loss of the entire protein or at least reduced amount. Unfortunately, we could not determine the nonsense mediated decay or the level of expression of COLVIIA protein because none of the patients were willing or able to provide any more skin biopsies at the time we discovered the mutation.
This study was funded by an internal fund of KFSH&RC and by student grant from King Abdulaziz City for Science and Technology (KACST).
BA performed the experiments described here except the histopathology and the electron microscopy and wrote the initial draft of the manuscript as part of her Master of Science degree. FA is the attending physician who cared for the patients and performed the histopathology. AA performed the electron microscopy. EA is the academic supervisor of BA who helped in writing this manuscript. AH is the principal scientist who supervised the day to day laboratory experiments, edited the manuscript, and submitted it for publication.
Parents concented for themselves and for their patients and their normal siblings who participated in the study since they are less than 18 years old at the time of the study. All experiments were performed as part of an approved protocol by Institutional Review Boards of all institutions involved in the patient care and laboratory testing including sequencing as described by the method section in accordance with the participating institutions guidelines and regulations that confoims to international standards. Cocents were obtained from parents to use their photograph and identifying information for this publication.
All data referred to in this manuscript is available and will be supplied upon request.
None of the authors has any conflict of interest as the funding came from participating institutions they work for.
View Supplementary Data
- Fine JD (2010) Inherited epidermolysis bullosa. Orphanet J Rare Dis 5:12. [Crossref]
- Tabor A, Pergolizzi JV Jr, Marti G, Harmon J, Cohen B, et al. (2017) Raising Awareness Among Healthcare Providers about Epidermolysis Bullosa and Advancing Toward a Cure. J Clin Aesthet Dermatol 10: 36-48. [Crossref]
- Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW et al. (2014) Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 70: 1103-1126. [Crossref]
- Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, et al. (2008) The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 58: 931-950. [Crossref]
- EV F (1999) The molecular biology of epidermolysis bullosa simplex. In: Fine J-D, Bauer EA, McGuire J, Moshell A, National Epidermolysis Bullosa R, eds. Epidermolysis bullosa : clinical, epidemiologic, and laboratory advances, and the findings of the National Epidermolysis Bullosa Registry. Baltimore: Johns Hopkins University Press.
- Varki R, Sadowski S, Pfendner E, Uitto J (2006) Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet 43: 641-652. [Crossref]
- Varki R, Sadowski S, Uitto J, Pfendner E (2007) Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet 44: 181-192. [Crossref]
- Bodemer C, Tchen SI, Ghomrasseni S, Séguier S, Gaultier F, et al. (2003) Skin expression of metalloproteinases and tissue inhibitor of metalloproteinases in sibling patients with recessive dystrophic epidermolysis and intrafamilial phenotypic variation. J Invest Dermatol 121: 273-279. [Crossref]
- Titeux M, Pendaries V, Tonasso L, Decha A, Bodemer C, et al. (2008) A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum Mutat 29: 267-276. [Crossref]
- Ishiko A, Masunaga T, Ota T, Nishikawa T (2004) Does the position of the premature termination codon in COL7A1 correlate with the clinical severity in recessive dystrophic epidermolysis bullosa? Exp Dermatol 13: 229-233. [Crossref]
- Tamai K, Ishida-Yamamoto A, Matsuo S, et al. (1997) Compound heterozygosity for a nonsense mutation and a splice site mutation in the type VII collagen gene (COL7A1) in recessive dystrophic epidermolysis bullosa. Lab Invest 76: 209-217. [Crossref]
- Tamai K, Murai T, Mayama M, Kon A, Nomura K, et al. (1999) Recurrent COL7A1 mutations in Japanese patients with dystrophic epidermolysis bullosa: positional effects of premature termination codon mutations on clinical severity. Japanese Collaborative Study Group on Epidermolysis Bullosa. J Invest Dermatol 112: 991-993. [Crossref]
- Cianfarani F, Zambruno G, Castiglia D, Odorisio T (2017) Pathomechanisms of Altered Wound Healing in Recessive Dystrophic Epidermolysis Bullosa. Am J Pathol 187:1445-1453. [Crossref]
- Dang N, Murrell DF (2008) Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol 17: 553-568. [Crossref]
- Horn HM, Tidman MJ (2002) The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol 146: 267-274. [Crossref]
- Pfendner EG, Lucky AW (1993) Dystrophic Epidermolysis Bullosa. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(R). Seattle (WA): University of Washington, Seattle.
- Christiano AM, Hoffman GG, Chung-Honet LC, et al. (1994) Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics 21: 169-179. [Crossref]
- Kon A, Pulkkinen L, Ishida-Yamamoto A, Hashimoto I, Uitto J (1998) Novel COL7A1 mutations in dystrophic forms of epidermolysis bullosa. J Invest Dermatol 111: 534-537. [Crossref]
- Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T (2004) Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic-specific and worldwide recurrent mutations. Arch Dermatol Res 295: 442-447. [Crossref]
- Mellerio JE, Salas-Alanis JC, Talamantes ML, Horn H, Tidman MJ, et al. (1998) A recurrent glycine substitution mutation, G2043R, in the type VII collagen gene (COL7A1) in dominant dystrophic epidermolysis bullosa. Br J Dermatol 139: 730-737. [Crossref]
- Christiano AM, Morricone A, Paradisi M, Angelo C, Mazzanti C, et al. (1995) A glycine-to-arginine substitution in the triple-helical domain of type VII collagen in a family with dominant dystrophic epidermolysis bullosa. J Invest Dermatol 104: 438-440. [Crossref]
- Cserhalmi-Friedman PB, Karpati S, Horvath A, Christiano AM (1997) Identification of the glycine-to-arginine substitution G2043R in type VII collagen in a family with dominant dystrophic epidermolysis bullosa from Hungary. Exp Dermatol 6: 303-307. [Crossref]
- Rouan F, Pulkkinen L, Jonkman MF, Bauer JW, Cserhalmi-Friedman PB, et al. (1998) Novel and de novo glycine substitution mutations in the type VII collagen gene (COL7A1) in dystrophic epidermolysis bullosa: implications for genetic counseling. J Invest Dermatol 111: 1210-1213. [Crossref]
- Wessagowit V, Ashton GH, Mohammedi R, Salas-Alanis JC, Denyer JE, et al. (2001) Three cases of de novo dominant dystrophic epidermolysis bullosa associated with the mutation G2043R in COL7A1. Clin Exp Dermatol 26: 97-99. [Crossref]
- Winberg JO, Hammami-Hauasli N, Nilssen O, Anton-Lamprecht I, Naylor SL, et al. (1997) Modulation of disease severity of dystrophic epidermolysis bullosa by a splice site mutation in combination with a missense mutation in the COL7A1 gene. Hum Mol Genet 6: 1125-1135. [Crossref]
- Mellerio JE, Dunnill MG, Allison W, Ashton GH, Christiano AM, et al. (1997) Recurrent mutations in the type VII collagen gene (COL7A1) in patients with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 109: 246-249. [Crossref]
- Mohammedi R, Mellerio JE, Ashton GH, Eady RA, McGrath JA (1999) A recurrent COL7A1 mutation, R2814X, in British patients with recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol 24: 37-39. [Crossref]
- Dunnill MG, Richards AJ, Milana G, Mollica F, Eady RA, et al. (1994) A novel homozygous point mutation in the collagen VII gene (COL7A1) in two cousins with recessive dystrophic epidermolysis bullosa. Hum Mol Genet 3: 1693-1694. [Crossref]
- Gardella R, Zoppi N, Ferraboli S, Marini D, Tadini G, et al. (1999) Three homozygous PTC mutations in the collagen type VII gene of patients affected by recessive dystrophic epidermolysis bullosa: analysis of transcript levels in dermal fibroblasts. Hum Mutat 13: 439-452. [Crossref]
- Gardella R, Castiglia D, Posteraro P, Bernardini S, Zoppi N, et al. (2002) Genotype-phenotype correlation in italian patients with dystrophic epidermolysis bullosa. J Invest Dermatol 119: 1456-1462. [Crossref]
- Abu Sa'd J, Indelman M, Pfendner E, Falik-Zaccai TC, Mizrachi-Koren M, et al. (2006) Molecular epidemiology of hereditary epidermolysis bullosa in a Middle Eastern population. J Invest Dermatol 126: 777-781. [Crossref]
- Ouragini H, Cherif F, Kassar S, Floriddia G, Pascucci M, et al. (2009) Dystrophic epidermolysis bullosa phenotypes in a large consanguineous Tunisian family. J Dermatol Sci 54: 114-120. [Crossref]
- Ouragini H, Cherif F, Brick SA, Nouira S, Floriddia G, et al. (2010) Mutational survey of recessive dystrophic epidermolysis bullosa in Tunisian families unveils a spectrum of private, ethnic specific and world-wide recurrent mutations. J Dermatol Sci 57: 144-146. [Crossref]
- Abahussein AA, al-Zayir AA, Mostafa WZ, Okoro AN (1992) Recessive dystrophic epidermolysis bullosa treated with phenytoin. Int J Dermatol 31: 730-732. [Crossref]
- Abahussein AA, al-Zayir AA, Mostafa WZ, Okoro AN (1993) Epidermolysis bullosa in the eastern province of Saudi Arabia. Int J Dermatol 32: 579-581. [Crossref]
- Abahussein AA, al-Zayir AA, Mostafa WZ, Okoro AN (1993) Inherited epidermolysis bullosa. Int J Dermatol 32: 561-568. [Crossref]
- Al-Saud BK, Al-Sum Z, Alassiri H, Al-Ghonaium A, Al-Muhsen S, et al. (2013) Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J Clin Immunol 33: 1325-1335. [Crossref]
- Alsum Z, Hawwari A, Alsmadi O, Al-Hissi S, Borrero E, et al. (2013) Clinical, immunological and molecular characterization of DOCK8 and DOCK8-like deficient patients: single center experience of twenty-five patients. J Clin Immunol 33: 55-67. [Crossref]
- Christiano AM, Greenspan DS, Lee S, Uitto J (1994) Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. The Journal of biological chemistry 269: 20256-20262. [Crossref]
- Pulkkinen L, Uitto J (1999) Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biol 18: 29-42. [Crossref]
- Christiano AM, McGrath JA, Tan KC, Uitto J (1996) Glycine substitutions in the triple-helical region of type VII collagen result in a spectrum of dystrophic epidermolysis bullosa phenotypes and patterns of inheritance. Am J Hum Genet 58: 671-681. [Crossref]
- Cui Y, Hagan KW, Zhang S, Peltz SW (1995) Identification and characterization of genes that are required for the accelerated degradation of mRNAs containing a premature translational termination codon. Genes Dev 9: 423-436. [Crossref]
- Christiano AM, Amano S, Eichenfield LF, Burgeson RE, Uitto J (1997) Premature termination codon mutations in the type VII collagen gene in recessive dystrophic epidermolysis bullosa result in nonsense-mediated mRNA decay and absence of functional protein. J Invest Dermatol 109: 390-394. [Crossref]