Research on drug-induced cardiomyopathy (CM) has largely focused on medically prescribed drugs due to reduced drug efficacy and complicated patient management. Investigation on other aetiologies – environmental toxins including cardiodepressant agents (ethanol), cardiostimulant agents (cocaine and amphetamine), heavy metals (cobalt, lead, mercury, arsenic, lithium or beryllium) and carbonaceous compounds (carbon monoxide or tetrachloride) have been relatively rarely. However, accumulating clinical data demonstrates that exposure to these environmental toxins has a pathogenic role mostly in populations with extreme dietary deficiency and/or long-term occupational exposure. Animal models of CM used to explore cardiotoxicity of environmental toxins have also heightened the awareness of their potential contributions to the pathogenesis of CM in the absence of any other known cause. Thus, the purpose of this paper is to review published evidence on toxic (non-medical drug) induced CM including a meta-analysis of causative toxins, pathophysiology, diagnosis and clinical management.
toxin-induced cardiomyopathy, ethanol, amphetamine, cocaine, heavy metals, carbon monoxide, carbon tetrachloride
Abbreviations
ACM: Alcoholic Cardiomyopathy; AHA: American Heart Associations; AMI: Acute Myocardial Infarction; ATP: Adenosine Triphosphate; CAD: Coronary Artery Disease; CCl4: Carbon Tetrachloride; CFCs: Chlorofluorocarbons; CM: Cardiomyopathy; CMR: Cardiac Magnetic Resonance Imaging; CNS: Central Nervous System; CO: Carbon Monoxide; COCHPA: Cocaine Associated Chest Pain; CVD: Cardiovascular; DCM: Dilated Cardiomyopathy; ECG: Electrocardiograph; Echo: Echocardiography; ESC : European Society of Cardiology; HBOT: Hyperbaric Oxygen Therapy; HF: Heart Failure; LA: Left Atrial; LV: Left Ventricular;
LVEDD: Left Ventricular End Diastolic Diameter;
LVEDV: Left Ventricular End Diastolic Volume; LVESD: Left Ventricular End Systolic Diameter;
LVESV: Left Ventricular End Systolic Volume; MI: Myocardial Infarction; ROS: Reactive Oxygen Species; RV: Right Ventricular; VAC: Volatile Organic Compound
From time immemorial, humans have consumed non-medical (recreational) drugs to lessen physical pain or to alter their mental state of consciousness. This form of drug use was limited to individuals who had attained maturity age or in very particular situations such as ceremonies. However, over time, drug use or abuse has progressively emerged as a serious social and public health problem, and in the process, generating great clinical concerns [1]. This is attributable to widespread drug abuse in many sectors of the population, the decline in the age of usage initiation and the severity of individual and health consequences of drug abuse. Originally, drug abuse was associated with the consumption of illegal drugs such as heroin. Currently, the concern has expanded to include the consumption of legalized drugs such as alcohol and tobacco, considered the gateway to the use of hard drugs such as cocaine and cannabis whose consumption is increasing [2].
On the other hand, drug-abuse prevention programs, both pharmacological and psychological treatments, have improved over the past two decades. However, prophylactic and therapeutic strategies, given the continuous change and complexity of drug use or abuse, is still insufficient [3]. Nevertheless, with increased association of drug-abuse with cardiotoxic effects and the development of cardiomyopathy (CM) including a related increase in hospitalization rates and mortality, the clinical need to understand the pathophysiology of cardiac damage including diagnosis and management has assumed some sense of urgency. The present paper discusses some basic concepts of drug-induced CM as well as reviews the definition of drugs, cardiotoxicity and CM. This paper also reviews specific drugs and toxins that cause CM, their pathophysiologic mechanisms, diagnosis and clinical management. Finally, a meta-analysis of diagnosis and treatment of toxin-induced CM is provided as well as the identification of areas of limited knowledge that may benefit from additional research.
A drug is any substance which when introduced into the living organism can modify one or more of its function [4]. Drugs may be broadly classified into medical and non-medical. Medical drugs, mostly prescription drugs, are taken to treat or to manage a specific medical condition while non-medical drugs, mostly recreational drugs, are taken to alter mental conscious state or to alleviate pain. The present paper centres on non-medical drugs that can cause CM as well as other environmental toxins capable of causing cardiotoxicity and CM. In the present context of drug-induced CM, drug use is defined as drug consumption that does not have a negative impact on health of the user while drug abuse is drug consumption associated with dependence – a set of psychological, behavioural and cognitive manifestations in which the use of the drug becomes a priority for the individual. Often, drug abuse is associated with tolerance, defined as the need to consume more of a drug to achieve the effects of previous consumption. CM on the other hand, has been defined as a progressive myocardial disease characterized by impaired systolic and/or diastolic function often accompanied by abnormalities in the cardiac conduction system in the absence of any other known or demonstrable cause [5,6]. The predominant causes of CM are genetics and cardiac or systemic diseases although the exposure to environmental toxins including prescribed and non-prescribed drugs may also cause the disease [5]. Drug-induced CM has important clinical implications including reduced efficacy of the affected prescription drugs and complications in patient management whereas CM due to widespread abuse of non-prescription drugs and chronic exposure to heavy metals have considerable health and socioeconomic effects [7-9].
Causative toxins
The predominant non-prescription drugs whose continued exposure may result in CM may be classified based on the effect on the heart as cardiodepressant (ethanol) and cardiostimulants (cocaine or amphetamines), and environmental toxins such as heavy metals (cobalt, lead, mercury, arsenic, lithium or beryllium) and carbonaceous compounds (carbon monoxide or carbon tetrachloride) [5].
Cardiodepressant:
Alcohol is one of the most commonly abused legalized non-medical drug classified as a central nervous system (CNS) depressant (also cardiodepressant). Ethanol is the active ingredient in alcohol (obtained from fermentation of sugars from different plants) responsible for myocardial damage via inducing (i) direct dose-dependent myocardial damage manifesting as acute effects on heart rhythm and left ventricular (LV) function; and (ii) chronic progressive LV dysfunction that may remain subclinical for a long time, known as alcoholic CM (ACM) [9,10]. Ethanol intoxication (plasma levels > 75 mg per 100 ml) exerts an acute cardiodepressive action by depressing myocardial contractility. The effect on the myocardium is reversible since ethanol intoxication has been demonstrated to exert a direct negative inotropic action with a reversible dose-dependent decrease in myocardial contractility [11,12]. ACM is a major cause of dilated cardiomyopathy (DCM) [13-16]. Reported incidences of ACM range between 21% and 32% of DCM in surveys conducted at referral hospitals [17] and higher (> 50%) in patient populations with a higher frequency of alcoholism [18]. A majority of alcoholics may have preclinical myocardial dysfunction based on autopsy findings of cardiomegaly and other signs of CM in alcoholics who did not exhibit overt symptoms of cardiac disease [19].
Moderate alcohol consumption has been demonstrated to produce a cardioprotective effect in both healthy individuals and those with heart diseases [20,21] creating a debate of the amount and duration required to produce a detrimental clinical effect on the heart. Moderate alcohol consumption (1-2 drinks per day or 5-25 g/day) decreases cardiovascular and all-cause mortality, and other hard clinical endpoints including CAD, ischemic strokes and amputations due to peripheral vascular diseases [22-25]. The exact duration and intensity of alcohol consumption preceding preclinical and symptomatic heart failure (HF) is not definitely known but the evidence of consumption of > 90g/day of alcohol (8 to 21 standard drinks) for an average duration of drinking of 5 years has been described [26-29]. However, it is important to note that the potential cardiac injury with chronic alcohol abuse is not beverage-specific nor quantity-specific but varies based on population studied, individual characteristics, genetic and environmental factors [30].
Pathogenesis:
The pathogenesis of ACM is complicated by varied outcomes such as cardioprotection of moderate alcohol consumption and the lack of the disease on some heavy alcohol users. Nevertheless, heavy drinking has been linked with asymptomatic and symptomatic LV systolic and/or diastolic dysfunction [30]. Environmental factors such as cobalt and arsenic, and genetic predisposition (HLA-B8, alcohol dehydrogenase alleles) have been suggested as possible triggers or abettors of aetiopathogenesis of ACM. An epidemic of ACM among Quebec beer-drinkers in the 1960s was associated with a 42% mortality rate but the high prevalence of ACM disappeared after brewers discontinued adding cobalt to stabilize the beer [31,32]. Genetic factors on the other hand, determine the rate of alcohol metabolism and thus plays an important role in determining the interaction between alcohol and its metabolites, and the heart [33-36].
Alcohol quantity and environmental and genetic factors influence the pathogenesis of ACM but the exact mechanisms underlying cardiac injury is poorly understood. Current evidence suggests the main pathogenic mechanisms of ACM may be due to the interference in carbohydrate metabolism, protein synthesis, alterations in oxidative status, and mitochondrial function, disruption of transduction signals, and induction of apoptosis [10]. However, ethanol itself is a direct noxious cardiac agent in a progressive, cumulative and dose-dependent manner, and its effect may be independent of nutritional, vitamin or mineral factors [9,10,37]. Ethanol alters the permeability of sarcoplasmic reticulum to calcium ions to reduce the efficiency by which calcium activates muscle contraction and decreases the synthesis of cardiac proteins in both contractile actin-myosin complex and in mitochondria predominantly in alcoholics with high blood pressure. A metabolite acetaldehyde and free radicals may also contribute to reduced protein synthesis [10]. Ethanol can also induce myocardial injury by increasing the expression of certain genes (pro-apoptotic protein Bax and caspase-3 enzyme activity), which can promote programmed cell death leading to myocyte loss [38-40]. It is also possible that other cell types or systems such as the sympathetic nervous system, renin-angiotensin system, cytokines and natriuretic peptides are activated and contribute to the overall myocardial injury. In addition to direct toxic effect of ethanol on the myocardium to cause apoptosis, necrosis and myocyte loss, other mechanism may influence the development of alterations in cardiac function and structure [40].
Diagnosis:
Heavy drinkers usually have lower LVEF, greater end-diastolic volumes, lower mean fractional shortening and a greater LV mass compared to healthy controls in a dose-dependent manner [10,40], which are independent of nutritional status or other habits such as tobacco smoking [41]. Echocardiographic abnormalities such as increased left atrial dimension, increased LV wall thickness and decreased fractional shortening precede the onset of clinical symptoms or physical findings [42,43]. Despite changes in LV structure and function, ACM lacks pathognomonic features that distinguishes the disease from other causes of HF, which is further complicated by the presence of other risk factors for CM [30]. History of alcoholism is a key factor for ACM diagnosis strengthened by the lack of other inciting factors such as prescribed drugs (. doxorubicin) and non-prescribed medication (cocaine) or ischemic heart disease [30]. Clinically, ACM manifests as four-chamber dilatation, low cardiac output and preserved or decreased LV wall thickness. Clinical signs of HF such as third heart sound, elevated jugular venous pressure and cardiomegaly with or without rales may be observed in decompensated states [44,45]. Concomitant liver disease due to cirrhosis may complicate diagnosis with the clinical picture is less clear. Supraventricular arrhythmias associated with heavy alcohol intake, as well as sudden cardiac death, are complications of ACM [46-48]. Cause of death in ACM patients mimic those of idiopathic DCM – progressive HF and sudden cardiac death [26]. Alcoholics with concomitant ACM and liver cirrhosis carry an ominous prognosis [49].
Management:
The ESC and AHA guidelines recommend Clinical management of ACM patients should begin with total abstinence from alcohol in addition to the standard management of HF [5]. Alcohol cessation has been associated with reversibility of depressed LV function and/or improvement of symptoms and signs of HF [50]. However, prognosis remains poor with a mortality rate of 40% to 50% within three to six years if the patient does not remain abstinent. Patients with chronic alcoholism may exhibit nutritional deficiencies (vitamins, minerals such as thiamine, selenium or zinc), which can precipitate or aggravate the development of ACM, and may require supplement thiamine and folate [5].
Cardiostimulants
Psychomotor stimulants (also cardiostimulants) alter mental function and behaviour to produce excitement and euphoria, depressed sensation of fatigue, and increased motor activity. Exposure to cardiostimulants has been associated with adverse cardiovascular events such as tachycardia, supraventricular or ventricular arrhythmias, impaired conduction, hypertensive crises, acute coronary syndrome, cardiac shock and cardiac arrest [9]. Animal and autopsy studies and individual case reports have linked exposure to stimulants to structural and functional changes in cardiomyocytes and clinical manifestations of cardiomyopathy and congestive HF [51,52]. Commonly used and abused major psychomotor stimulants are cocaine, amphetamines or methamphetamines [9].
Cocaine:
Cocaine (benzoylmethylecgonine) is a synthetic chemical derivative the coca plant leaves and a strong CNS stimulant used as a recreational drug. It is absorbed from all mucous membranes of the body, and as a result, has several consumption routes including snorting, inhaling as smoke or dissolving and injecting into a vein. Its consumption triggers psychotic behaviours while its withdrawal or suppression may result in potentially life threatening conditions including reactive depression and/or cocaine psychosis [53,54]. Cocaine is considered a sympathomimetic drug, which acts by inhibiting the reuptake of noradrenaline (norepinephrine) and dopamine sympathetic nerve terminals as well as via central pathways to release noradrenaline from adrenal medulla [55]. At high doses, cocaine can impair myocyte electrical activity and contractility by blocking fast sodium and potassium channels and inhibiting calcium entry into myocytes [56]. Cocaine has a short serum half-life of about 30 to 80 minutes with 90% metabolized and excreted in urine over a fortnight, which provides a means to retrospective diagnosis of a recent ingestion [57]. Concomitant consumption with alcohol potentiate the cardiotoxic effect of cocaine by forming cocaethylene, a more potent cardiotoxic cocaine metabolite [58]. Cannabis also potentiate the cardiotoxic effect of cocaine via increasing plasma concentrations of the drug [59].
The use of cocaine is associated with multiple cardiovascular complications including chest pain, myocardial infarction (MI), arrhythmias, aortic dissection and stroke [60]. It is a rare cause of DCM, which often manifests in the absence of coronary artery disease (CAD) or segmental infarct pattern, reported to occur in 10 out of 1278 cases of DCM at John Hopkins Hospital [61]. The underlying pathophysiologic mechanisms of cocaine CM are not fully understood as well as the amount and duration of cocaine is necessary to develop cocaine CM but four mechanisms have been proposed. (i) The promotion of intracoronary thrombus formation; (ii) sympathomimetic effects; (iii) increased calcium flux into myocardial cells; and (iv) increased oxidative stress [62].
It is well recognized that the use of cocaine promotes the formation of intracoronary thrombus. Cocaine stimulates platelet hyperaggregability and increases thromboxane production in the setting of coronary vasospasm. These physiologic effects promote acute formation of intracoronary thrombus and myocardial ischemia contributing to the high incidence of MI in cocaine users. Acute myocardial ischemic and extensive or recurrent MI may also play a role in LV dysfunction and CM in cocaine users. Since most cocaine users with severe regional or global LV dysfunction do not have a history of obstructive CAD or MI, myocardial dysfunction may be a consequence of transient ischemic insults possibly in the setting of vasospasm or spontaneous coronary thrombosis. It is also likely that non-ischemic events of cardiomyocyte damage may also contribute to CM in cocaine users [62].
Secondly, since cocaine acts as a strong sympathomimetic agent, there is involvement of its sympathomimetic effect in the development of CM. Cocaine ingestion stimulates the beta-adrenergic receptors in the myocardial tissues to increase the force of cardiac contraction and heart rate. Cocaine also stimulates alpha-adrenergic receptors in coronary and peripheral arteries, which increases coronary resistance, reduced coronary blood flow, increased blood pressure and increased myocardial wall stress [64,65]. Increased wall stress common in acute cocaine intoxication instead of reduced contractility plays an important role in acute depression of LV function [66]. Finally, an increase in calcium flux into the myocardial cells and increased oxidative stress alter cardiac electrophysiology resulting in ECG abnormalities such as the prolongation of the PR, QRS and QTc duration, atrioventricular nodal conduction and maximum sinoatrial conduction time. These ECG changes may trigger atrial extra-systole, and atrial fibrillation and tachycardia as well as transient ventricular tachycardia [64].
Clinical manifestations of cocaine CM are similar to other forms of DCM. However, it should be strongly suspected in young males (< 50 years of age) presenting with signs of adrenergic excess (defined as a sudden and significant increase in the serum levels of the catecholamine adrenaline and noradrenaline but with a less significant increase in dopamine transmission) accompanied by HF or LV dysfunction [9]. Cocaine CM has an acute onset without a long prodrome. Common ECG abnormalities include sinus tachycardia with frequent arrhythmias including atrial fibrillation and ventricular tachycardia [61]. Echocardiogram demonstrates ab increase in LV mass and LV dysfunction. Urine test for cocaine and its metabolite (benzoylecgonine) is required to obtain evidence of recent cocaine ingestion. The cornerstone of clinical management of cocaine is abstinence, which has been shown to reverse cardiac function [63]. Beyond abstinence, little evidence is available about the treatment of cocaine CM. However, current expert consensus guidelines recommend the management of cocaine CM should be similar to other forms of DCM with the exception of beta-blockers, which should be avoided initially, and benzodiazepine indicated to blunt adrenergic excess [5,6]. Beta-blockers can be added later in compliant patients who follow-up and abstains. Same to alcohol, LV function can improve significantly with abstinence from cocaine use. Unfortunately, the success of conventional HF treatment on patients with cocaine CM is complicated by high recidivism associated with a recurrence of LV dysfunction and symptomatic HF [5].
Amphetamines:
Amphetamines are synthetic derivatives of phenethylamines, natural amines synthetized from phenylalanine. Adding a methyl group to amphetamines forms methamphetamine, which has a greater solubility in lipids and capacity to cross the blood brain barrier. Amphetamines are potent CNS stimulants more commonly used as a recreational drug to induce euphoria, increase alertness, intensify emotions, increase aggression, and alter self-esteem [9], and less commonly as a prescription drug to treat attention-deficit hyperactivity disorder (ADHD) and narcolepsy, a sleep disorder [67,68]. The common ingestion routes are inhaling (or smoking), swallowing (pill), snorting or injecting intravenously powder that has been dissolved in water/alcohol. It is readily absorbed from the nasal mucosa and gastrointestinal tract, and freely penetrates the blood-brain barrier [3]. Metabolism varies highly and up to 30% of the parent compound can be excreted unchanged in urine. The plasma half-life varies significantly from 5 hours to 20-30 hours depending on urine flow and pH, with drug elimination increased in acidic urine [3]. Amphetamine or its metabolites is detectable in urine for several days after ingestion but excretion can be prolonged by the administration of large doses or in the presence of alkaline urine (deliberate concomitant use of bicarbonate soda delays excretion and enhances the effect of the drug) [69]. Alcohol has been shown to potentiate the effect of amphetamines [8,9]. Ecstasy (3,4-Methylenedioxymethamphetamine) is a derivative of amphetamines and exerts a similar sympathomimetic effect, which usually begin about 20 minutes after ingestion and lasts up to six hours although the effect of larger doses can persist up to 48 hours. It is commonly ingested orally (in tablet form) at less commonly annually, and metabolized by the liver and excreted by the kidney [3].
Pharmacologically, amphetamines are substrates for the neuronal uptake transporters for catecholamine including norepinephrine, serotonin and dopamine, and cause their release from nerve terminals in the central and autonomic nervous systems [70]. In contrast to cocaine, amphetamine can inhibit the enzyme monoamine oxidase and lacks the local anaesthetic effect of inhibiting sodium channels [9]. Commonly observed cardiovascular symptoms of ingestion of amphetamines include chest pain palpitations and dyspnoea, which manifest with or without underlying CAD [9]. Cardiomyopathy has been described in 18% of individuals using metamphetamines [51], and has been suggested as a possible cause of unexplained CM in young patients due to its widespread use [52]. Ecstasy can cause MI, arrhythmias and CM [71,72]. Animal studies show repeat exposure to ecstasy or its metabolites can result in eccentric LV dilation and diastolic dysfunction, and contractile dysfunction in cardiomyocytes [71,73]. Amphetamine associated sympathetic activation results in varying degrees of tachycardia, vasoconstriction, unpredictable blood pressure effects and arrhythmias, which depends on the dose and the presence or absence of co-occurring cardiovascular disease. Hypertension is common although severe hypotension due to paradoxical central sympathetic suppression, a late state of catecholamine depletion or ventricular impairment induced by ischemia or mechanical complications) may also occur [74-76].
The pathophysiology of amphetamine-induced CM is not fully understood. However, it is suggested to be a consequence of both direct cardiotoxic effect and indirect amphetamine-induced hypertension, necrosis and ischemic [77]. Repeated exposure of amphetamine in rats show the drug may directly induce cardiomyocyte hypertrophy, myocarditis with inflammatory infiltrates and areas of necrosis, and consequently may result in eccentric LV dilation and diastolic dysfunction [73]. High doses may result in cardiac dysfunction disorder with disruption of microtubules and actin [78]. These findings suggest that potential mechanisms of amphetamine-induced CM are related to oxidative stress and catecholaminergic stimulation. In addition, recreational use of amphetamines is often marked by a repeated pattern of frequent drug application followed by a period of abstinence, which significantly alter cardiovascular and cardiovascular reflex function, and produce cardiotoxicity [79]. However, the withdrawal of amphetamine has been associated with reversibility of cardiac lesions and the administration of standard DCM therapy lead to improvement in cardiac function [80,81] (Table 1).
Table 1. Summary of cardiac effect of cardiostimulants and management options
Drug |
Cardiac effect |
Complications |
Clinical management |
Cocaine |
Inhibits re-uptake of neuronal catecholamines |
Hypertension
|
Benzodiazepines, nitrates, phentolamine, sodium nitroprusside, or hydralazine |
|
Class I sodium channel blocker, anti-arrhythmic effects |
Bradyarrhythmias (cocaine) |
Atropine (cautiously), temporary transvenous cardiac pacing |
|
|
Supraventricular tachyarrhythmias |
Observation (reverses with time), adenosine, cardioversion, beta-blocker |
Amphetamines |
Release catecholamines from nerve endings |
Ventricular arrhythmias
|
Cardioversion, lignocaine, magnesium sulphate |
|
|
Myocardial ischaemia and infarction |
Aspirin, nitrates, phentolamine, thrombolysis, or coronary intervention |
|
|
Acute heart failure |
Diuretics, inotropes, ventilation and intra-aortic balloon pumping may be necessary |
Ecstasy |
Release catecholamines from nerve endings |
Non-cardiogenic pulmonary oedema |
Ventilation |
|
|
Chronic heart failure
Endocarditis |
Conventional HF medication |
Heavy metals
Heavy metals refer to metals with relatively high densities, atomic weights or atomic numbers [82,83]. Some heavy metals such as cobalt, lead, lithium and mercury have been implicated as a cause of selective cardiotoxicity and may play an important role in the pathogenesis of various forms of CM. These heavy metals can cause structural changes in cardiomyocytes, alter myocardial contraction and the deregulation of some essential enzymes in the myocardium [84].
Cobalt:
Cobalt is a heavy metal and an important cause of reversible CM. It exists as a bivalent and trivalent molecule in circulation and produces a cytotoxic profile similar to nanoparticles, causing neurological, thyroid, and cardiological pathology [86]. Humans can be exposed to cobalt in several ways. First, cobalt is an ingredient in the manufacturing of glass, inks and pains as such occupations contact with cobalt occurs in processing plants, heavy metal industry, diamond polishing and the manufacture of ceramics [87-89]. Second, cobalt stimulates the production of red blood cells and thus used for the treatment of refractory anaemia, and by athletes, to increase red blood cells mass and consequently exercise performance [90,91]. Third cobalt is a component of high-performance, wear resistant alloys critical in the manufacture of implanted medical devices such as dental implants, coronary artery stents and metal orthopaedic prostheses [92-94]. Fourth, cobalt has been used to increase foam stability in beer [95-97]. Cobalt CM first became apparent in Quebec beer drinkers appearing as an epidemic among heavy drinkers in the mid-1960s [98,99]. The CM resembled typical DCM but was distinct from that generally seen in ACM characterized by large pericardial effusion, low output HF, purplish skin coloration and a high early mortality rate (42%). The cobalt CM disappeared after the withdrawal of cobalt from beer brewing process [31,98].
The exact pathophysiologic mechanism for developing cobalt CM is unclear but it has been suggested that cobalt may decrease myocardial contractility by a competitive antagonism with calcium [9]. Cobalt may interfere with the binding of calcium to the sarcolemma, the transport of calcium into the cardiomyocyte, and the inotropic effects of calcium [100,101]. Cobalt may also interrupt the citric acid cycle and the generation of adenosine triphosphate (ATP) by aerobic cellular respiration [102-104], inhibit the activity of respiratory chain enzymes and ATP production in mitochondria [105,106] and promote the production of reactive oxygen species [107,108]. The net result of these changes is depressed cardiac function and altered cardiac cell structure [109,110]. Experimental animal models show clinical manifestation of cobalt CM include weight loss, pericardial effusion and low voltage on ECG, modest histological changes with no inflammatory response on microscopy and ultrastructural changes including vacuolar degeneration, swollen and distorted mitochondrial, features observed in reversible experimental CM induced by alcohol and thiamine or protein deficiency [104,111-114]. The pathognomonic pathological findings in cobalt CM is the presence of dense osmophilic intra-mitochondrial particles [111,115,116].
Cobalt alone does not significantly alter mitochondrial function and cardiac contractility despite significant myocardial accumulation [117-119] indicating conditions other than exposure to cobalt potentiate or mediate the effect of cobalt. Experimental studies demonstrate minimal cardiac effect of cobalt, which became manifest in the presence of hypothyroidism or by the consumption of a low-protein, low thiamine diet [112,116]. Deficiency of thiamine and thyroxine in the presence of cobalt act at the same enzymatic sites to disrupt normal functioning of citric acid cycle [120-124]. Dietary protein may modulate the absorption of cobalt and potentiate the development of goitre [125,126] and cobalt may suppress synthesis of thyroxine [127-129] and antagonize its peripheral actions [129-131]. Due to these synergistic interactions, cobalt cardiotoxicity, dietary protein and thiamine deficiency, and hypothyroidism routinely coexist. Amino and sulfhydryl groups of amino acids has been demonstrated to provide protection against cobalt effects by combining with cobalt and preventing the chelation of cobalt with sulfhydryl groups of the myocardial tissue [99].
At present, a decrease in the medicinal utility of cobalt and the implementation of measures that were effective to reduce industrial exposure have significantly decreased the incidence of sub-acute cobalt CM. However, the classical features of cobalt CM have recently increased among patients with malfunctioning cobalt-alloy hip-prosthesis [85]. Clinically, the features of cobalt CM based on the evaluation of Quebec beer drinkers, are readily distinguishable from those of idiopathic non-ischemic CM (Table 1). Typically, patients with cobalt CM have a history of anorexia and weight loss, acute onset of HF, presence of pericardial effusion, polycythemia and thyroid abnormalities and a clinical picture dominated by peripheral hypoperfusion presenting as hypotension, lactic acidosis and cyanosis. The syndrome resembled beriberi heart disease but with the absence of therapeutic response to treatment with thiamine [90,132] (Table 2).
Table 2. Differences in cobalt CM and idiopathic non-ischemic CM
Clinical features |
Idiopathic non-ischemic CM |
Cobalt-induced CM |
Predisposing factors other than cobalt |
None |
Low-protein diet, thiamine deficiency, hypothyroidism |
Recent medical history |
Variable and non-specific |
Recent history of anorexia and weight loss |
Clinical presentation |
Typical asymptomatic or slow progressive LV dysfunction |
Rapid onset and progression of severe HF |
ECG changes |
Non-specific changes or poor R-wave progression |
Low voltage across all ECG leads, sinus tachycardia, absence of cardiac arrhythmias |
Echocardiographic changes |
Enlargement and depressed systolic function of both ventricles |
Pericardial effusion, enlargement and depressed systolic function of one or both ventricles |
Laboratory findings |
No typical findings |
Polycythemia, hypothyroidism, elevated enzymes and lactic acidosis |
Clinical course |
Typically slowly progressive over months to years |
Rapidly progressive course with cyanosis, hypotension |
Clinical outcome |
Infrequently reversible, low short-term mortality rate |
High short-term mortality rate but survivors experience complete cardiac recovery |
The diagnosis of cobalt CM requires evidence of cardiac dysfunction and reversibility of cardiac dysfunction. (i) Demonstration of bi-ventricular dilatation and systolic dysfunction in the setting of blood/tissue concentration of cobalt are elevated; and (ii) normalization of cardiac structure and function when exposure ceases and blood/tissue concentration of cobalt are comparable to those of non-exposed individuals in the absence of other interventions used in the treatment of CM. Some studies have suggested cobalt may impair diastolic filling without affecting systolic function but echocardiographic evidence of diastolic filling is a common findings in middle-aged to elderly individual and thus its non-specific to individuals exposed to cobalt relative to non-exposed controls [133-135]. Definitive treatment of cobalt CM is ceasing exposure to cobalt. In the setting of implant related causes, treatment is by surgical removal of prosthesis [136].
Lead:
Lead is a toxic heavy meatal whose excessive or chronic exposure can cause overt, clinical symptoms of cardiac and vascular damage with potential fatal consequences. It has been associated with morphological, biochemical, and functional derangements of the heart as well as perturbations in cardiac electrical and mechanical activity [137,138]. Typical routes of lead exposure are inhalation and swallowing while the main sources of lead exposure are gasoline additives, food-can soldering, lead-based paints, ceramic glazes, drinking water pipe system and folk remedies [139]. Lead exposure has been associated with cardiovascular events such as hypertension CAD, stroke, peripheral artery disease and CM but the exact role of lead in cardiovascular diseases remains incompletely understood [141,142]. The severity of lead cardiotoxicity is influenced most directly by the dose of lead and the duration of lead exposure. Other factors such as the route of exposure, age of individuals, temperature and dietary calcium intake also appear to influence the expression and severity of cardiotoxic symptoms manifested in both acute and chronic forms of lead exposure [137].
The threshold of serum levels of lead that triggers cardiac involvement and symptoms of cardiotoxicity is yet to be determined conclusively. However, environmental and occupational lead exposures that raise serum lead levels > 100 µg% and 60 µg% in adults and children respectively have been associated with transient as well as permanent cardiac and vascular lesions and functional disturbances [137]. Cardiotoxic manifestations of acute and chronic lead exposure include myocarditis [143,144], ECG abnormalities [145-147], altered heart rate [145,147], slowed ventricular systole [145,148], hypertension [149-152], and vascular degeneration [144,145] (Table 3). Clinical evidence raising suspicion for lead cardiotoxicity rely on early studies, which include histological findings of degenerative and inflammatory changes in the myocardium [143], ECG abnormalities including sinus bradycardia [143,146-148], multifocal ventricular beats [143], T-wave inversion [147], left bundle branch block [143], first degree heart block [145,146] and ectopic atrial rhythms [147]. Other cardiac changes observed in occupational lead exposure include abnormal low voltage waves, S-T interval changes, arteriosclerotic changes, intimal fatty infiltration and proliferation of perivascular connective tissue, exertional dyspnoea and abnormal cardiovascular response to exercise [145].
Table 3. Cardiotoxic manifestation of environmental lead in humans
Worker, serum lead levels (µg%) [Ref #] |
Cardiotoxic manifestations |
Lead worker (NR) [143] |
Bradycardia, LBBB, multifocal ventricular escape beats, myocarditis |
Lead workers (> 100) [146] |
AV-conduction disturbances, ectopic atrial rhythms |
Lead workers (NR) [148] |
Bradycardia, shorter ventricular ejection time, prolonged ventricular contraction time |
Adult (> 250) [149] |
Impaired beta-receptor-mediated cardiovascular function, increased heart rate at maximal physical work, decreased chronotropic responsiveness to isoproterenol, increased heart rate by 25 beats, reduced plasma renin activity |
Children (> 60) [147] |
Atrial arrhythmias abnormal PR intervals, prolonged corrected QT interval, inverter T-wave |
Children died of acute lead poisoning due to HF secondary to chronic myocarditis (NR) [144] |
Autopsy findings: myocarditis, interstitial oedema, connective tissue infiltration, compression and replacement of adjacent muscle fibres |
LBBB: Left Bundle Block Branch; HF: Heart Failure; NR: Not Reported; QTc: Corrected QT interval
Pathophysiologically, acute lead exposure affects cardiac function whereas chronic exposure affects the electrical and mechanical activity of the heart and alters myocardial function [137]. Lead may induce the generation of reactive oxygen species (ROS) and nitrogen species, and mediate the formation of free radicals, which enhances lipid peroxidation and changes in calcium and sulfhydryl homeostasis. The promotion of ROS production may trigger a cycle of oxidative stress and inflammation of the myocardial tissues [153]. In addition, the presence of inflammation may play a role in the progression of myocardial injury evident in increased expression and production of inflammatory marker in lead exposed patients [154]. Although these findings suggest a possible involvement of oxidative stress in the pathophysiology of lead cardiotoxicity, it is not clear whether these changes are a cause of oxidative damage or are a consequence of it. Oxidative stress and inflammation may be involved in raising arterial pressure via promoting endothelial dysfunction, promoting smooth muscle cells proliferation and transformation and impairing nitrogen oxide homeostasis [155-158]. Lead competes with calcium for the transport by channels and pumps involved in the movement of ion across cell membrane and between cytoplasm, endoplasmic reticulum and mitochondria contributing to the changes in cytosolic calcium ions known to regulate vascular tone and vascular smooth muscle contraction [159]. Finally, cessation of exposure or chelation therapy has been shown to reverse lead-associated cardiac changes but cases of permanent damage have also been reported [147].
Mercury:
Mercury is a toxic environmental pollutant occurring at low-levels in water systems such as lakes, rivers, oceans as well as bio-concentration in some fatty fish species [138,160]. In 1999, Frustaci et al. [161] study on trace elements in 13 idiopathic DCM patients reported >10,000 times more mercury in the myocardium than skeletal muscle. Mean mercury concentration in the myocardium was approximately 22,000 times higher in DCM patients (178.5 µg/g) than in controls (0.008 µg/g) but there was no increase in mercury in cardiac tissue from patients with DCM due to ischemic or valvular heart disease [161]. Although mercury has been associated with a host of cardiovascular diseases, its role in the pathogenesis of CM is exacerbating viral infection by either altering early host immune response or by increasing viral replication in viral DCM [160]. Exposure to mercury potentiates encephalomyocarditis infection by reducing interferon and antibody activity or altering viral life cycle [162-164] and herpes simplex virus type 2 infection by increasing viral replication via interfering with host defence mechanisms, inhibiting cytokine production and decreasing macrophage activity [165].
The pathophysiology of mercury associated CM is disrupting the immune system homeostasis via defective autoantibody production [165-167]. Susceptible haploid genotypes may develop autoantibody response against nucleolar antigens including fibrillarin, which binds to mercury and cleavages during mercury-induced cell-death resulting in fibrillarin fragments that induce antigenic response and initiate sustained inflammation [168-170]. Mercury also alters normal immune functioning by binding with selenium to suppress the activities of selenium-dependent free radical detoxification enzymes – glutathione peroxidases and thioredoxin reductase – and thereby reduce antioxidative defences and promote free radical stress and lipid peroxidation in the human body [171-176]. Free radicals injure the retroviral genome causing mutations of previously avirulent species into virulent strains [177]. Mercury binds with selenium to form insoluble mercury selenides and reduce the bioavailability of selenium to perform its normal function as the active site moiety in various selenium dependent enzymes [178]. In contrast, reciprocal interactions are expected, high selenium levels could protect against excess mercury intoxication although at present there is little evidence from human studies to support this hypothesis [138].
Clinically, patients with mercury-associated CM may present with echocardiography, ECG and histological abnormalities. The study by Frustaci et al. [161] on trace elements in 13 idiopathic DCM reported echocardiographic evidence of increased LV end diastolic diameter (LVEDD) and significant depression of LV function but without significant valvular abnormalities [161]. On cardiac catheterization and ventriculography, mercury-associated CM patients usually reveal compromised global contractility, reduction of cardiac index and elevated LV filling pressure while coronary angiography reveals normal arteriogram. Common ECG abnormalities include frequent ventricular ectopic beats, sustained or non-sustained ventricular tachycardia. Histology of LV biopsy fragments may rule out specific myocardial diseases such as myocarditis and storage or infiltrative myocardial diseases with signs of hypertrophy with attenuation and degenerative changes of myocardial fibres. Electron microscopy may detect degenerative changes consisting of loss of contractile elements, the presence of myelin bodies and the vacuolization of mitochondria with fragmentation of mitochondria cristae [161]. Reduced exposure to mercury and the use of chelating agents, mostly oral chelators such as 2,3-dimercaptosuccinic acid (DMSA or succimer) bind to mercury and increase its excretion from the body [179].
Arsenic:
Arsenic is a metalloid found ubiquitously in nature. Key sources of human exposure to arsenic are medicinal, occupational and environmental while arsenic exposure occurs from inhalation, absorption through the skin and primarily by ingestion of contaminated water or dietary forms such as seafood [138, 180,181]. Arsenic is toxic to majority of organ systems, with the liver being the more sensitive target organ although the cardiovascular effect may be potentially fatal. Clinical studies show that exposure to arsenic induces adverse cardiovascular effects such as hypertension, diabetes mellitus, atherosclerosis, coronary heart disease, stroke and CM in a dose-dependent manner [182,183]. The extent of arsenic poisoning depends on the various factors including dose, individual susceptibility to arsenic and the age of the affected individual. Chronic arsenic exposure usually affects the vascular system causing hypertension and cardiovascular disease while acute arsenic toxicity may cause CM and hypotension [181,182].
Pathophysiologically, long-term arsenic exposure alters myocardial depolarization and may induce cardiac arrhythmias [184,185]. Acute exposure to high arsenic levels (93 mg) may cause severe hypertrophy of the ventricular wall [186]. Oxidative stress and vascular inflammation may also be involved in the pathogenetic effects of arsenic exposure. The vascular endothelium regulates the release of various mediators such as nitric oxide, angiotensin‐II, endothelin‐1, adhesion molecules and cytokines [187]. Nitric oxide is a major mediator with vasodilatory and anti-inflammatory properties and inhibits platelet adhesion and aggregation, smooth muscle cell proliferation and migration [181]. The exposure of endothelial cells to sodium arsenite induces a decline in the integrity of vascular endothelium and endothelial cytotoxicity and thereby reduces the bioavailability of nitric oxide and increasing oxidative stress [187]. Increased accumulation of arsenic in the vascular wall and increased atherosclerotic lesion formation with increased macrophage accumulation and fibrosis have been observed in arsenic-exposed mice compared to water-fed controls [188].
Clinical investigation of arsenic-induced CM depends on the evidence of arsenic intoxication in the presence of evidence of cardiac dysfunction. Reliable biological indicator of arsenic intoxication is levels of arsenic in blood, urine, hair and nails. However, arsenic metabolism from the blood occurs within a period of several hours and the lack of correlation between blood arsenic levels and that in drinking water suggests the measurement of blood arsenic levels is not a reliable indicator of long-term exposure [181]. On the other hand, since the majority of arsenic absorbed from the lungs or the gastrointestinal tract and excreted in urine within 1-2 days, measurement of urine arsenic levels is generally considered the most reliable marker of acute arsenic exposure, found to correlate well with exposure of populations living near industrial point sources of arsenic [189,190]. Chelation therapy for arsenic-induced cardiotoxicity mostly includes three drugs (i) British anti-lewisite (BAL; dimercaprol); (ii) DMPS (unithiol) and (iii) DMSA (succimer), which enhance arsenic excretion. Treatment should be initiated as rapidly as possible (within minutes to a few hours) because efficacy declines as the time interval between exposure and chelation increases [191].
Lithium:
Lithium is an alkaline metal that has long been used in medicine since the 1840s [192]. Its initial therapeutic role in the early 19th Century was the treatment for gout [193], and at present, lithium is used in the prophylaxis and treatment of depression and bipolar disorder [194,195]. It is the most effective long-term therapy for bipolar disorder, protecting against both depression and mania, and reducing the risk of suicide and short-term mortality [196]. However, its use in a population at a relatively high risk for overdose, a narrow therapeutic range and several well-characterized adverse effects limits the potential usefulness of higher doses as well as requires routine monitoring of serum concentrations to minimize the risks of toxicity [194,195]. Lithium toxicity may result from accidental or intentional overdose or due to impaired clearance [192]. Serious lithium cardiotoxicity is uncommon and generally manifests in individuals with underlying heart disease. Lithium-induced CM may result in arrhythmias including bradycardia, sinoatrial block, and first-degree atrioventricular block [192]. Same to sodium and potassium, lithium is a cation that at the cellular level competes with sodium and potassium displacing them to cause intracellular metabolic changes. Lithium excretion is almost completely through the kidney in the same manner as sodium, freely filtered with 80% reabsorption at the proximal tubules [197,198]. Sodium intake and lithium clearance have a direct relation with decreased sodium intake leading to decreased lithium clearance. Evidently, lithium intoxication is common in low salt diets, dehydration states or with a concurrent use of diuretics [197,198].
Pathophysiologically, the mechanisms underlying lithium cardiotoxicity is not fully understood. However, the monovalent nature of lithium and cations involved in cardiac electrophysiology may assist in the understanding of lithium cardiotoxicities. In cardiac electrophysiology, sodium and potassium channels and cation influxes play an important role in depolarization, cardiac contraction and repolarization within the cardiac cycle [195]. Lithium blocks cardiac sodium channels at non-toxic levels and cardiac potassium channels resulting in perturbations of the resting membrane potential by its ability to partially replace intracellular potassium [199,200]. Lithium may also affect cardiac system indirectly through its effect on the thyroid causing hyperthyroidism related CM [196]. Clinically, chronic lithium intoxication presents with non-specific features and diagnosis requires a high degree of suspicion. Common but clinically insignificant ECG changes include T-wave flattening or inversion, which is usually incidental finding and reverse with discontinuation of lithium drug, sinus bradycardia in the absence of hypotension followed by sinus bradycardia with hypotension, third degree atrioventricular block, sinus node arrest, bradycardic cardiac arrest, U waves, ST-segment depression and prolonged QT interval [200,201]. Forced diuresis (administration of large volumes of saline, loop diuretics or intravenous crystalloids) and haemodialysis for 3-6 hours or continuous hemofiltration techniques sustained for 18-44 hours enhance clearance of serum and tissue lithium [202,203].
Beryllium:
Beryllium is a metallic element naturally occurring in rocks, coal, oil, soil and volcanic dust. Main source of exposure is occupational in industries using beryllium in their products include aerospace, automotive, biomedical, defence, energy and electrical, fire prevention instruments, manufacturing, jewellery, recycling and telecommunication [204]. Beryllium may also be found in domestic consumer products including electronic devices such as television, calculators and personal computers although deliberate manipulation will result in exposure [205]. The principal route of beryllium exposure is the lung. Other incidental exposure route include hand-to-mouth dermal contact and resuspension following deposition onto clothing [205]. Exposure via the lung route is extremely toxic, which damages the mucosal lining to cause pneumonia and berylliosis (or granulomatous lung disease) – a potentially fatal and persistence ling disease that can potentate injury to other organs including the cardiovascular system [204].
The evidence of myocardial consequences of the exposure to beryllium and its compounds in humans are limited. However, severe cases of chronic beryllium disease may lead to cor pulmonale, which is hypertrophy of the right heart ventricle. In an earlier study of 17 individuals exposed to beryllium in a fluorescence lamp manufacturing plant, autopsy revealed right atrial and ventricular hypertrophy [206]. In a beryllium manufacturing plant, increased cardiac deaths have been reported that may be the result of direct beryllium toxicity to the heart or an indirect response to impaired lung function [207]. Animal studies also support the cardiotoxic effect of beryllium. Acute inhalation exposure to beryllium and its compounds caused cardiomegaly in monkeys [208]. The exposure to beryllium oxide for 15 to 40 days [209] or beryllium sulfate for 100 days [201] in dogs resulted in decreased arterial oxygen tension. Although the exact pathophysiological mechanism of beryllium cardiotoxicity remains unknown, it is postulated it is the consequence of compensatory increases in cardiac musculature due to pulmonary fibrosis cause by inhalation exposure. The decrease of arterial oxygen tension reflects the reduced ability of the lung to oxygenate blood [204].
Carbonaceous compounds
In addition to alcohol, recreation drug and heavy metals, other cytotoxic substances with the potential to cause CM are carbon compounds mosly carbon monoxide and carbon tetrachloride.
Carbon monoxide:
Carbon monoxide (CO) is an odourless, tasteless, non-irritant and colourless gas diffusing with more ease across lung alveoli than oxygen. It is a toxic by-product of incomplete combustion of carbonaceous materials. Major sources of CO exposure include fire smoke inhalation, faulty heating systems and cigarette smoking [211,212]. Domestic fire smoke is the leading domestic source of CO and a major determinant of patient survival after smoke inhalation [211,212]. The positive effects of CO that may have a clinical value for CVD include (i) vasorelaxing activity via the regulation of smooth muscle tone; (ii) anti-inflammatory effect via the regulation of the expression and release of various cytokine; and (iii) anti-apoptotic reaction that inhibits cellular apoptotic pathway in mitochondria [213]. However, cardiotoxicity limits therapeutic use of CO. Primarily, CO toxicity results from tissue hypoxia, a consequence of the binding of CO to haemoglobin. CO has a greater affinity for haemoglobin (ten times faster) relative to oxygen but disassociates very slowly decreasing oxygen carrying capacity of erythrocytes [214,215]. Haemoglobin is the haemoprotein in the blood cells responsible for transporting oxygen to the tissues, and acts as a buffer in erythrocytes contributing to the bicarbonate-carbonic acid buffering system. CO binding to haemoglobin forms carboxyhaemoglobin, which produces a leftward shift in the oxygen-dissociation curve resulting in tissue hypoxia [216].
Pathophysiologically, toxicity of CO has been linked to tissue hypoxia (ischemic injury) and direct toxic injury. High oxygen demand by myocardial tissue limits its tolerance to hypoxia and is a target organ for CO toxicity [217,218]. In addition to hypoxic injury, high sensitivity of myocardial tissues to oxygen deprivation worsened by increased oxygen demand because of increased contractility, reduced coronary blood flow reserve, and cardiomyocyte respiration inhibition, CO produces additional myocardium damage with cardiospecific mechanisms. CO binds with heme group of myoglobin with affinity of 60 times greater than that of oxygen. Carboxymyoglobin reduces mitochondrial oxygenation, impairs oxidative phosphorylation and deteriorates the energy source for the mitochondria. CO is also directly toxic for mitochondria via impairment of mitochondrial respiratory chain and decreased glutathione [219,220]. The inhibition of ATP energy sources forces the cardiomyocytes to switch to anaerobic metabolism with consequent hypoxia, lactic acidosis and apoptosis [221].
CO may also trigger transcapillary efflux enhancing leukocyte seclusion within the endothelial lining to promote oxidation of plasma low-density lipoprotein, enhance nitrosative stress because of peroxynitrite formation in the absence of nitric oxide generation and lipid peroxidation due to increased free radical production [222]. CO also exerts prothrombotic potential – triggering arterial and venous thrombosis, the development of mild and moderate LV structure abnormalities and myocardial fibrosis [223-230]. Endothelium dysfunction amplifies coronary vasospasm and increases cardiac function and contractility to induce a hyperadrenergic state, which increases the risk of arrhythmias [231]. Ischemic damage to the myocardial tissues may be amplified by peripheral circulation failure and hypotension because of impaired cardiac function. Cardiac decompensation deteriorates tissue oxygenation, which is considered the leading cause of death in severely CO-intoxicated patients [232].
Clinical features of CO-induced CM include increased heart rate and reduced systolic, diastolic blood pressure. Cardiovascular manifestations included dyspnoea (58%), palpitations (96%), chest pains (50%) and ECG changes (96%). Common ECG abnormalities included tachycardia, prolonged QT interval and ST segment changes with inverted T waves presented with premature ventricular contractions [233]. The primary target of clinical management of CO toxicity and CM is the alleviation of CO-induced hypoxia using hyperbaric oxygen therapy (HBOT) as the chosen treatment. HBOT comprises exposing the patient to 100% oxygen at pressures ranging from 2 to 2.8 atmospheres. According to Henry’s Las, blood exposed to pure oxygen at 38 degrees centigrade and 1 atmosphere will dissolve approximately 2.3 ml oxygen in 100ml and twice as much if the pressure is doubled [234]. High pressure are useful to increase the amount of dissolved oxygen into the blood to displace CO from haemoglobin within erythrocytes and re-oxygenate hypoxic tissues [235]. Although prognostication of CO-induced CM is not well defined, patients with pre-existing heart diseases such as atherosclerotic disease are at an elevated risk of morbidity after CO intoxication. Depressed coronary circulation limits the ability to compensate for increased coronary flow during myocardial hypoxia requiring increased myocardial oxygen demand, which increases time onset to angina [212].
Carbon tetrachloride:
Carbon tetrachloride (CCl4) is a clear non-inflammable heavy liquid that evaporates readily producing a sweet characteristic odour similar to chloroform. It is a manufactured chemical, which does not occur naturally in the environment. Historically, CCl4 was used to produce chlorofluorocarbons (CFCs) used as heat transfers agents in refrigerating equipment and aerosol propellants [237. It is also an ingredient in many industrial fluids and metal degreasers as well as found in in domestic cleaning supplies, spot removers for carpets and fire extinguishers but most of these uses have been discontinued [238,239]. CCl4 is absorbed through inhalation, ingestion and dermal absorption, distributed throughout the body with the highest concentration in the liver, brain, kidney, muscle, fat and blood, and excreted primarily in exhaled air. CCl4 is mostly hepatotoxic that also causes cardiac disorders [240-243]. CCl4 high dose causes acute cellular necrosis, oxidative stress and inflammation resulting in acute tissue injury and apoptotic organ failure [244,245].
The pathophysiological mechanism of CCl4 cardiotoxicity of CCl4 is not fully known but it is thought to result from increased free radical production, which plays a role in tissue degenerative process. The cardiotoxicity of CCl4 has a biphasic mechanism. The first phase involves the production of free radicals (CCl3 and CCl3OO) through the metabolism of NADPH – cytochrome P450 system, which induces lipid peroxidation [236]. Multiple cytochrome P450 enzymes have been found in cardiac tissues, including CYP 2E family of isozymes. The second phase involves the activation of tissue macrophages accompanied by the production of inflammatory and profibrogenic mediators [245]. Rodents exposed to CCl4 demonstrate increased markers of inflammation, cardiomyocyte injury (troponin, CK-MB) and 0oxidatuve stress in the heart [236,242]. Case reports have described cardiomegaly, congestive HF and cardiac fibrosis following exposure to CCl4 [236].
Clinical management of CCL4 cardiotoxicity is challenging because of the lack of proven therapies. However, HBOT has been found to play a therapeutic role in both animal and human CCl4 intoxication. HBOT appears to inhibit the mixed function oxidase system responsible for conversion of CCl4 to hepatotoxic free radicals [246-249]. HBOT should be considered for potentially severe CCl4 exposure prior to the onset of liver function abnormalities because of a delicate balance between oxidative processes that are therapeutic and those that mediate hepatotoxicity [250]. Recently, Al-Rasheed et al. [236] demonstrated the efficacy of silymarin in combination with chlorogenic acid and melatonin in ameliorating the toxicity of CCl4 induced cardiac damage, which may support the use of this combination as an effective drug in the treatment of cardiomyopathy induced by toxic agents.
Meta-analysis of clinical evaluation
An electronic search in PubMed was performed using a combination of the following search terms: environmental toxins (ethanol, cocaine, amphetamines, cobalt, lead, mercury, arsenic, lithium OR beryllium) AND cardiomyopathy OR cardiotoxicity AND serum biomarkers, electrocardiography, echocardiography OR cardiac magnetic resonance from inception to May 2019. Studies excluded were case series, case reports and review articles. In total, the initial electronic search retrieved 427 articles. A hierarchical review of titles, abstracts and full paper yielded 17 relevant studies investigating diagnosis and/or management of toxin-induced CM. Studies enrolling asymptomatic and/or symptomatic patients exposed to environmental toxins were included in this meta-analysis. Supplementing the electronic search, a manual search of reference lists from the included studies were screened for more relevant studies, which yielded an additional four studies bringing the total to 21 studies published between 1959 and 2017 [31,251-270]. Table 4 provides a summary of the main characteristics of interest in the 21 included studies.
Table 4. Summary of characteristics of the included studies
Author |
Year |
No. |
Age |
Males |
Patient selection |
Test |
Study findings |
Evans [251] |
1959 |
20 |
NR |
NR |
Symptomatic chronic alcohol use |
ECG |
ECG abnormalities are common in alcoholic CM and may assist in early diagnosis |
Priest [252] |
1966 |
37 |
NR |
NR |
Patients hospitalized for alcoholic abuse |
ECG |
Reversible ECG abnormalities are common in alcoholics mostly T-wave changes |
Attar [253] |
2017 |
100 |
35 |
98 |
Daily alcohol intake > 80 g/day for > 5 years |
ECG/Echo |
ECG and echo changes are common in alcoholics prior to symptomatic cardiac disorders but reversible during early stages |
Brickner [254] |
1991 |
30 |
35 |
30 |
Chronic cocaine users in inpatient drug rehabilitation program |
ECG/Echo |
Cocaine use is associated with increased LV mass index and wall thickness, and LV hypertrophy |
Gitter [255] |
1991 |
101 |
NR |
NR |
Cocaine-related chest pain on admission |
ECG, serum marker |
Abnormal ECG is common in cocaine use patients with chest pain, mostly ST-T changes |
Zimmerman [256] |
1991 |
48 |
29 |
34 |
Cocaine users with chest pain |
ECG |
Significant cases of MI in cocaine users and persistence of ECG abnormalities |
Hollander [257] |
1994 |
246 |
33 |
176 |
Cocaine associated chest pain |
ECG |
MI is common among cocaine users. ECG is common but non-specific |
Hollander [258] |
1995 |
130 |
38 |
NR |
Cocaine associated chest pain |
ECG |
Low mortality rate for patients hospitalized for cocaine use with acute onset of complications |
Weber [259] |
2000 |
250 |
33 |
192 |
Cocaine associated chest pain |
ECG |
Incidence of AMI is high in among cocaine users presenting with chest pain |
Weber [260] |
2003 |
344 |
38 |
198 |
Cocaine associated chest pain |
ECG |
Cocaine associated chest pain with no evidence of ischemia or CVD complications have a low risk of death or MI 30 days post discharge |
Kozor [261] |
2014 |
20 |
37 |
19 |
Self-reported regular cocaine users |
CMR |
Cocaine users have increased aortic stiffness and systolic BP associated with greater LV mass |
Maceira [262] |
2014 |
94 |
37 |
81 |
Asymptomatic chronic cocaine users |
ECG,CMR, blood test |
CMR is able to detects CVD in a majority of asymptomatic cocaine users |
Wijetunga [263] |
2003 |
21 |
41 |
19 |
Methamphetamine users with CM |
Echo |
Methamphetamine use appears to produce CM in some users |
Haning [264] |
2007 |
158 |
36 |
98 |
Methamphetamine dependents |
ECG |
Common ECG changes predominantly prolonged QTc increasing risk for ventricular arrhythmias |
Voskoboinik [265] |
2015 |
20 |
35 |
14 |
Patients diagnosed with MAC |
ECG, Echo, CMR |
MAC patients with reverse takotsubo pattern and lesser ventricular dilatation are more likely to achieve early recovery of ventricular function |
Kueh [266] |
2016 |
30 |
40 |
25 |
Amphetamine-associated CM and evidence of HF |
Echo |
Amphetamine associated CM is predominant in young men marked with severe LV dilation and dysfunction persisting despite treatment |
Bazmi [267] |
2017 |
230 |
34 |
196 |
Acute amphetamine users with positive urine test |
ECG, Echo |
ECG and echo common in a majority of patients but cardiac complications were common |
Kesteloot [31] |
1968 |
16 |
NR |
NR |
Chronic Quebec beer drinkers |
ECG, RVG, chest x-ray |
Associates carbon toxicity in chronic alcoholics to dietary deficiency of sulfhydryl-group containing amino acids and proteins |
Horowitz [268] |
1988 |
30 |
41 |
21 |
Cemented tungsten carbide workers |
ECG, RVG, chest x-ray |
There is no clear evidence of overt systolic LV dysfunction despite prolonged occupational exposure although duration of exposure has a correlation with LV dysfunction |
Yoon [269] |
2014 |
626 |
42 |
11 |
Admitted in ED with acute CO poisoning |
ECG |
CO induced CM was rare in all patients but with favorable prognosis, myocardial stunning due to catecholamine surge most likely plays a central role in the development of CO-induced CM |
Cha [270] |
2016 |
43 |
60 |
27 |
Patients hospitalized for CO poisoning |
ECG, Echo |
CO poisoning and myocardial injury experience CM induces reversible global ventricular dysfunction and Takotsubo-like pattern |
AMI: Acute Myocardial Infarction; BP: Blood pressure; CM: Cardiomyopathy; CMR: Cardiac Magnetic Resonance Imaging; CO: Carbon Monoxide; CVD: Cardiovascular; ECG: Electrocardiograph; Echo: Echocardiograph; HF: Heart Failure; LV: Left Ventricular; MAC: Methamphetamine-Induced Cardiomyopathy; NR: Not Reported; RVG: Radionuclide Ventriculography
Findings
In all, the 21 studies included in the present meta-analysis enrolled 2,594 patients with either asymptomatic or symptomatic cardiac dysfunction after the exposure to toxins known to cause CM [31,251-270]. In seventeen (17) studies, the enrolled patients were predominantly male (74%) [253,254,256-270] and youthful (mean age=37.8, range=29-60). Study designs varied: thirteen (13) were prospective cohort studies [31,251-254,257,260-262,264,266,268,270], seven (7) were retrospective cohort studies [255,256,258,259,263,265,263] and one was a cross-sectional study [267]. Diagnostic tests common in all the 21 studies were electrocardiography and/or echocardiography, and less commonly serum cardiac biomarkers [255,262]. On the other hand, diagnostic use of cardiac magnetic resonance (CMR) imaging was rare, only used by three studies [261,262,265] and in two earlier studies, radionuclide ventriculography and chest x-ray [31,268]. Various toxins causing CM were investigated: three studies investigated ethanol (alcohol) [251-253], nine cocaine [254-262], five amphetamines [263-267], two cobalt [31,268], while the remaining two investigated carbon monoxide [269,270].
In patients with toxin-induced CM, despite the heterogeneity in causative substances (ethanol, cocaine, CO, amphetamines or cobalt), ECG abnormalities are common presentations. In the present pooled analysis of 14 studies [251-253,255-260,262,264,267-269], 1,045 out of 1,807 patients presented with ECG abnormalities translating into an event rate (incidence) of 59.3% (event rate: 0.593; 95% CI: 0.517 to 0.665; p = 0.017) (Figure 1). The most common ECG abnormality was ST-T changes reported in 11 studies [31,253,255-257,260,262,264,268-270] occurring in 350 out of 1199 patients and event rate of 27.2% (0.272; 95% CI: 0.214 to 0.339; p = 0.000) (Figure 2). The second common ECG abnormality was sinus tachycardia (defined as heart rate > 100 beats per minute) affecting 166 out of 712 patients translating into 26.3% event rate (0.263; 95% CI: 0.229 to 0.300) (Figure 3).
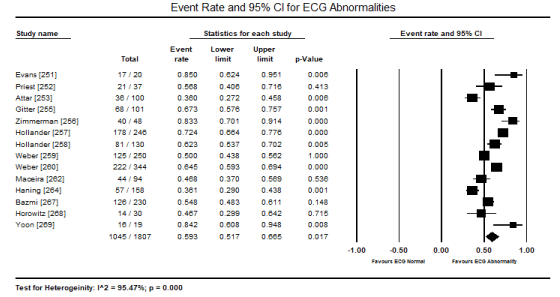
Figure 1. Forest plot for event rate for ECG abnormalities
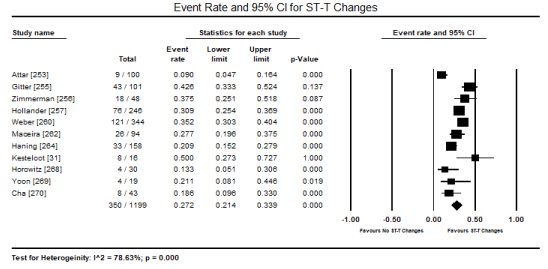
Figure 2. Forest plot for event rate for ECG ST-T changes
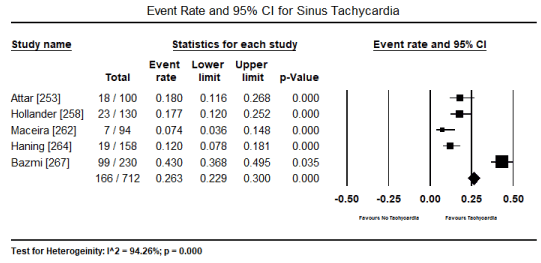
Figure 3. Forest plot for event rate for sinus tachycardia
In patients with severe or persistent ECG abnormalities referred for hospitalization, additional echocardiographic evaluations were performed to assess pathological changes in cardiac function and structure. The present findings show that echocardiographic changes are common in patients with toxic-induced CM. Pooled analysis of raw data (number of affected patients) from three studies [253,267,269] revealed 106 out of 244 patients had evidence of cardiac dysfunction, which translated into an event rate of 48.2% (0.482; 95% CI: 0.185 to 0.793; p = value 0.921) (Figure 4). However, the high heterogeneity across the studies could be due to variability in patient selection, which included both asymptomatic and symptomatic patients with insufficient data to compare echocardiographic changes between the two patients groups. Similarly, the event rate of LV dysfunction was 35.4% in five studies [265-267,269,270] (event rate 0.354; 95% CI: 0.087 to 0.760; p = 0.502) (Figure 5). Pooled analysis from five studies [253,262,263,265,266] showed LVEDD (> 45 mm) was common in 57 out of 265 patients (event rate 0.324; 95% CI: 0.098 to 0.679; p = 0.332) (Figure 6). Additional analysis of LVEDD (mean values) from three studies [254,266,267], revealed weighted mean LVEDD (mm) was significantly high (weighted mean 52.1 mm; 95% CI: 9.072 to 58.002; p = 0.000) (Figure 7). In two studies [255,269] that enrolled symptomatic CM patients, cardiac biomarkers (troponin or creatinine) showed 80.7% had elevated cardiac biomarkers although they were non-specific to toxin-induced CM.
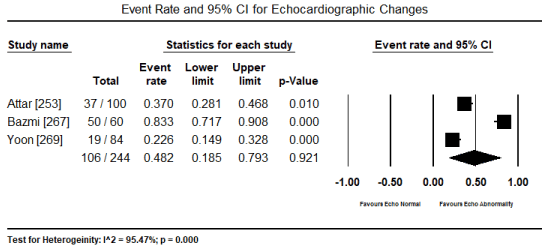
Figure 4. Forest plot for event rate for echocardiography changes
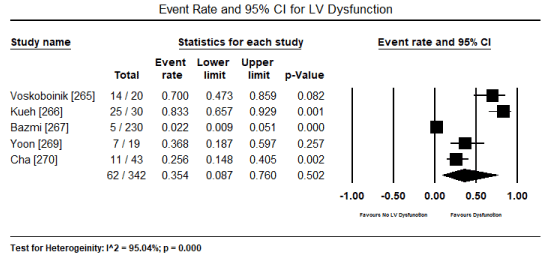
Figure 5. Forest plot for event rate for LV dysfunction
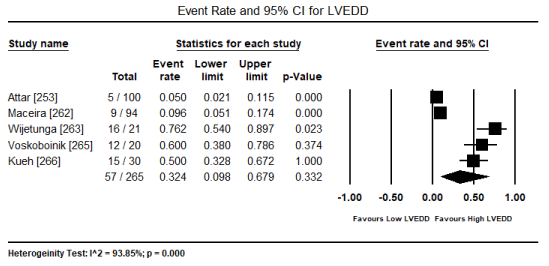
Figure 6. Forest plot for event rate for LVEDD
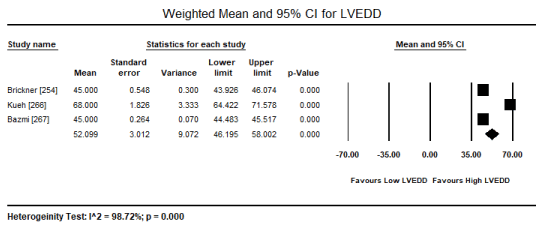
Figure 7. Forest plot for weighted mean LVEDD
Data on prognostication and management of toxin-induced CM was insufficient (provided in different formats) to support meta-analysis. However, individual studies indicated cardiac dysfunction (ventricular changes) and ECG abnormalities prior to symptomatic cardiac dysfunction or during the early stages of CM are largely transient and may reverse during normal hospital duration [252,253]. Consequently, ECG and echocardiographic changes may be potential markers for ongoing cardiotoxic effects prior to the development of symptomatic cardiac disorders [253]. Patients with reversible takotsubo-like pattern of LV dysfunction have favorable prognosis with high possibility of achieving early recovery of ventricular function [265,270]. In contrast, chest pain complaints in the setting of ischemia or CVD complications may suggest an increased risk of death [260]. The transient nature of toxin-induced CM supports pooled analysis of five studies [251,257,266,267,270] that reported 21 deaths out of 569 patients translating into a low event rate of 5.2% (0.052; 95% CI: 0.020 to 0.126; p = 0.000) (Figure 8). Furthermore, pooled analysis of two studies [257,258] indicate only 3.5% (13 out of 376) of the patients developed HF. The withdrawal of exposure to causative toxins combined with HF medication for patients with symptomatic cardiac dysfunction have proved to be effective in improving cardiac function and resolving symptoms [252,253,257,258].
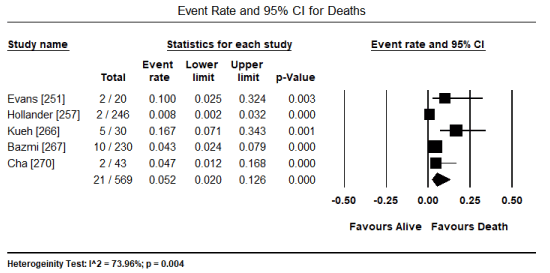
Figure 8. Forest plot for event rate for deaths
Effective management of HF remains problematic partly due to heterogeneous aetiology and variable symptomatology [271]. Cardiomyopathy is an important cause of HF but which has received relatively little research attention. A disproportionate number of studies investigating CM have focused on idiopathic (genetic) and ischemic aetiologies, and less commonly on secondary aetiologies including environmental toxins mostly alcohol, cardiostimulants, heavy metals and carbonaceous compounds. The present meta-analysis reveal that changes in ECG, echocardiography and cardiac biomarkers may precede the development of myocardial impairment and may be useful indicators for the development of CM. ECG abnormalities were common in 59.3% of the included patients, echocardiography changes in 48.2% of the included patients, and cardiac biomarkers were elevated in 81% of symptomatic patients.
Electrocardiogram
The present meta-analysis finds ECG changes are common in patients exposed to cardiotoxic agents with or without symptoms of cardiac dysfunction. In support of these findings, consensus guidelines recommend the use of ECG in the initial testing and routine monitoring of electrical perturbations in patients suspected/diagnosed with toxin-induced CM [5]. Electrophysiological alterations is a principal manifestation of cardiotoxicity and underscores the use of ECG as a preclinical test for electrical aberrations in patients suspected with toxin-induced CM [272]. Several cogent reasons exist that justify ECG monitoring in patients suffering from CM. (i) Three (3) important manifestations of cardiac injury are alterations in electrical properties; alterations in contractility; and death. Alterations in electrical properties precede the last two and thus ECG monitoring is obligatory. (ii) ECG provides information on the general health of the patient. (iii) Pre-test ECG can identify certain subclinical cardiac abnormalities to warrant additional diagnostic tests. (iv) Some cardiotoxin substances may produce ECG changes without producing detectable myocardial changes [273].
Typical ECG changes associated with toxin-induced CM include perturbations in the heart rate in terms of electrical impulse generation disorders or impulse conduction pathology [253]. For patients suspected with toxin-induced CM, ECG recordings should include heart rate, heart rhythm, QT interval (> 0.55 milliseconds is considered prolonged), supraventricular and ventricular arrhythmias, atrioventricular/ventricular cardiac blocks, T-waves and myocardial ischemia (ST segment and T-wave changes) [274]. Sinus tachycardia and arrhythmias such as ventricular and atrial fibrillation are common rhythm disorders. Conduction disturbance is dose-dependent and increases with increased exposure to toxins sometimes leading to HF and even sudden cardiac death. In CM associated with chronic ethanol intoxication, ectopic are a common feature mostly atrial premature contractions and ventricular premature contractions [253]. To maximize the diagnostic effectiveness of ECG test, attention should be given to recording schedule, lead systems and proper interpretation [264,267,269]. In a prospective study of patients admitted for alcohol abuse [252], ECG abnormalities preceded the development of myocardial dysfunction. ECG abnormalities were reversible suggesting they may not be attributable to myocardial damage but to direct toxic effect of the alcohol itself or the breakdown of the enzymatic process [252].
Despite its prognostic and diagnostic value, ECG changes are both non-specific and non-sensitive for accurate diagnosis of toxin-induced CM. The Cocaine Associated Chest Pain (COCHPA) Study Group [257] evaluating 242 patients with cocaine-induced CM found poor diagnostic sensitivity (89.9%) and positive predictive value (17.9%). Amin et al. [275] confirmed the same poor predictive value in the use of ECG in diagnosing AMI. The two studies demonstrate that although ECG has utility in the diagnosis of CM, poor sensitivity and positive predictive value makes it unreliable in accurately distinguishing benign CM from actual ischemia. Normal variant of ECG patterns such as early repolarization in younger patients may lead to misinterpretation further complicating the accuracy of ECG test in the diagnosis of CM [276,277]. Although non-specific, the present findings suggest ECG test is important to raise clinical suspicion for toxin cardiotoxicity in the absence of any other known cause in patients exposed to toxins. Abnormal ECG findings may suggest early stages of cardiotoxicity to warrant additional diagnostic tests to confirm CM [253]. Since toxin-induced CM is largely reversible if detected in early stages, ECG may be a useful initial test because of widespread availability, non-invasive nature and ease of use.
Cardiac imaging
For patients admitted to the intensive care unit due to tachycardia of very abnormal ECG changes, additional non-invasive imaging should be performed to confirm the diagnosis of toxin-induced CM. It has been demonstrated that cardiotoxicity and/or toxin-induced CM leads to marked changes in cardiac morphology and/or functionality. Clinically relevant variables measured by echocardiography include LV ejection fraction (LVEF), regional wall motion abnormalities, aortic root diameter, and cardiac wall size (RVEDV, LVESV, LVEDV, LVEDP, LVEDD and LVESD). In the present meta-analysis, echocardiography was the preferred non-invasive modality in assessing cardiac dysfunction used in six studies [253,254,263,265-267,270] whereas the use of CMR was rare, appearing only in three studies [261,262,265] with one study using both echocardiography and CMR imaging [265]. Typical echocardiographic changes in patients with long-term exposure to toxins include increases in LV mass, LV dimensions and LV volumes, and septal and LV wall thickness, and left atrial (LA) dimensions [253,262,263,265-267]. The common cut off points indicating impaired cardiac function or structure mentioned on two studies are LVEDD > 55 or 65 mm; LA > 42 mm; RV > 19 mm; intraventricular septum thickness ≥ 11 mm and global hypokinesia [263,265]. Disease progression can be slowed or stopped by the withdrawal of the exposure to toxins, which may also improve cardiac function and relieve cardiac symptoms during the early stages of the disease evaluated by ECG and echocardiogram [265].
Although the use of echocardiography is widespread, CMR is the current gold standard technique for the assessment of ventricular dimensions and function, with high accuracy, reproducibility and reference values for both LV and RV [278,279]. CMR can provide a comprehensive evaluation of the cardiovascular system as well as has a unique capability for tissue characterization, which has been applied for the assessment of various CM [280-282]. In cocaine-induced CM, CMR imaging compared to controls showed increased LVESV (30±8 vs 26±5 mL/m2; p < 0.01), increased LV mass index (80±13 vs 69±9 g/m2; p < 0.05) and increased RVESV (36±9 vs 28±4) with significantly depressed LVEF (59±5 vs 68±4) and RVEF (56±5 vs 65±5). Myocardial late gadolinium enhancement (LGE) may indicate myocardial damage (262). The findings showed that in patients with cocaine-induced CM, CMR reveals RV involvement in about 25% of the patients and can detect up to 71% in asymptomatic cocaine users [262]. Despite the high detection rate, CMR is not widely available for routine clinical practice, which explains the use of echocardiography in a majority of the studies as well as in clinical settings.
Cardiac biomarkers
Cardiac biomarkers may provide supplementary evidence of cardiac dysfunction. Patients with CM due to cardiostimulant use (amphetamine or cocaine) exhibit elevated levels of cardiac biomarkers. In patients with amphetamine abuse and CM, Bazmi et al. [267] found very high levels of CK-MB (35.9±2.3 U/mL) and troponin I (0.6±0.2 ng/mL) indicating cardiac damage. In a study of cocaine users, Gitter et al. [255] reported elevated levels of total creatine kinase (> 200U/L) in 43% of the patients rising to 47% at some time during the course of hospitalization. Yoon et al. [269] enrolled patients with CO-induced CM in which all (100%) had elevated levels of cardiac biomarkers (creatine kinase-MB > 5 µg/L; troponin T hs > 0,014 ng/ml or troponin I > 0.04 ng/ml) who underwent additional ECG tests to evaluate cardiac injury. Despite the use of cardiac biomarkers in detecting subclinical changes, they are non-specific to toxin-induced CM. The specificity of creatine kinase MB and myoglobin in the diagnosis CM in patients exposed to cocaine is decreased but the specificity of troponin is not affected. Patients with cocaine-induced CM with elevated troponin concentration have a high incidence of underlying coronary disease [267]. Although troponin elevation is highly specific for cardiomyocyte injury in any clinical scenario, it does not indicate the underlying mechanism of injury, which may include both toxin and non-toxin causes such as plaque rapture, coronary vasoconstriction leading to decreased oxygen delivery, demand ischemia or pre-existing atherosclerosis, acute inflammation, direct myocardial toxicity or a combination of all these factors [267].
Prognostication
Despite differences in the primary toxin causing CM across the included studies, a consensus of a dose-dependent (duration or cumulative quantity) cardiotoxic effect emerged. The longer the duration of toxin exposure, the greater the severity of cardiotoxicity and cardiac dysfunction. Attar et al. [253] found, in comparison to the duration of exposure of alcohol (5 to 8 years vs. > 8 years), ECG abnormalities was 21.8% vs. 42.67% and echocardiographic abnormalities was 21.87% vs. 44.11% respectively. Further, the study reported chronic alcohol intoxication induces cardiotoxicity irrespective of malnutrition [255]. Cumulative or dose-dependent effect of cocaine-induced cardiotoxicity > 6 years was associated with high blood pressure (110/70 mm Hg) while > 12 months was associated with increased ventricular wall thickness (LV hypertrophy) [254]. LV hypertrophy in cocaine users provides a substrate facilitating the development of CVD events including myocardial ischemia and arrhythmias. Further, LV hypertrophy has been associated with decreased coronary flow reserve, decreased sub-endocardial perfusion and increased myocardial oxygen consumption, factors which favour the development of myocardial ischemia in the setting of cocaine-induced coronary artery vasoconstriction and/or thrombus [254].
Concomitant disorders also indicate poor prognosis. The incidence of MI in cocaine users presenting to the emergency department with chest pains suggest possible chronic myocardial changes as causes of ischemia. Cocaine associated chest pain with no evidence of ischemia or CVD complications over a 9 to 12 hour period have a lower risk of death of MI 30 days post discharge [260]. Alcohol and other illicit drug use potentiate cardiotoxic effect of cocaine due to the effect of alcohol on systolic blood pressure and the effect of smoking on vascular stiffness [261]. Despite duration of exposure and concomitant use of other cardiotoxic agents, generally, toxin-induced CM has a favourable prognosis. In the COCHPA study [257], none of the 246 patients with cocaine associated chest pain had sustained ventricular arrhythmia on arrival and congestive HF developed only in 2% of the patients. Amin et al. [275] reported only one case of congestive HF and one case of non-sustained ventricular tachycardia among 22 patients with AMI due to cocaine use. In patients on amphetamine intoxication, reverse takotsubo pattern and lesser ventricular dilatation have potential for early recovery of ventricular function while those with evidence of myocardial fibrosis and ventricular enlargement have reduced probability of recovery [265].
The withdrawal of exposure to toxins reverses cardiac dysfunction and relieve symptoms. In follow-up echocardiography in 19 out of 20 patients with amphetamine-induced CM at a median of 38 days after initial diagnosis, 6 out of 19 patients had normalized LVEF (≥ 50%) within 6 weeks of diagnosis. LVEF improved significantly from 18.3% to 61.5% on follow-up [265]. Clinical features associated with favourable prognosis (early recovery) include the lack of echocardiographic evidence of chronicity including smaller LV cavity and LV size at diagnosis, shorter duration of drug-use, elevated cardiac enzymes and reversible takotsubo pattern [265]. Severity of cardiac dysfunction – severe LV dilation (LVEDD = 6.8±1.0 cm) and severe LV dysfunction (LVEF 22±8%) is refractory to optimal HF medical therapy since LV size remained significantly dilated with minimally improved in LV function [266]. In CO-induced CM, patients with global dysfunction have a more rapid normalization of cardiac function compared to takotsubo-like dysfunction [270]. Yoon et al. [269] also found patients with CO-induced CM normalized their LV function and abnormalities on wall motion with no reported incidence of death.
Implications
The present findings reveal that electrical and morphofunctional cardiac changes associated with toxin-induced CM are largely transient and potentially reversible at the subclinical stage of the disease. The withdrawal of exposure to the causative toxin halts the pathological process but in some cases, the earlier pathological changes could go unheeded for some years and the resulting CM will no longer subside after such abstinence. It is important therefore that early myocardial damage from exposure to toxins is detected to improve the possibility of reversal of myocardia dysfunction. Initial and routine ECG and echocardiographic tests are thus important to detect subclinical electrical and morphofunctional changes, especially in populations exposed to toxins and presenting with CM with no known cause.
Environmental toxics, mostly ethanol, cardiostimulant drugs, heavy metals and carbonaceous compounds have rarely been considered in the pathogenesis of non-ischemic DCM yet clinical observations suggest the exposure to these toxins may play an important pathogenic role in the development of DCM mostly in populations with extreme dietary deficiency and occupational exposure. Recreational use of cardiodepressant (alcohol) and cardiostimulants (cocaine and amphetamine), occupational or dietary exposure to heavy metals (cobalt, lead, mercury, arsenic, lithium or beryllium) or carbonaceous compounds (carbon monoxide and carbon tetrachloride) are the common causes of toxin-induced CM. Pathophysiology varies based on the causative toxin but it may involve direct toxic effect on the myocardium causing apoptosis, necrosis and myocyte loss or indirectly through alterations in cardiac function and structure. Alcohol, smoking and deficiency of some dietary nutrients may potentiate cardiotoxicity of toxins. The basis of the diagnosis of toxin-induced CM is demonstrable evidence of toxins in the blood or tissue. Elevated cardiac biomarkers and ECG abnormalities although non-specific raise the clinical suspicion for toxin-induced CM. Non-invasive imaging using echocardiography or cardiac magnetic resonance evaluate cardiac structure and function to detect abnormalities and confirm diagnosis in the absence of any other known cause. The duration of exposure and the extent of cardiac dysfunction are important prognostic factors while the withdrawal of exposure to toxins and HF medication may improve cardiac function and relieve symptoms. Cardiac dysfunction is reversible if detected early supporting the use of initial and routine monitoring by ECG and echocardiography. For CM due to carbon compounds (monoxide or tetrachloride), hyperbaric oxygen therapy improves blood and tissue oxygenation, and chelation therapy for CM due to heavy metals reverses cardiotoxic effects and relieves symptoms.
- Daley DC (2013) Family and social aspects of substance use disorders and treatment. J Food Drug Anal 21: S73-S76. [Crossref]
- Gupta A, Mandala A, Wee Y (2017) Drug-Induced Cardiomyopathy: An Institutional Experience. Heart, Lung and Circulation 26: S132-S133.
- Ghuran A, Nolan J (2000) Recreational drug misuse: issues for the cardiologist. Heart 83: 627-633. [Crossref]
- Zinberg NE, Harding WM, Apsler R (1978) What is drug abuse? Journal of Drug Issues 8: 9-35.
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113: 1807-1816. [Crossref]
- Iqubal A, Haque SE, Sharma S, Ansari MA, Khan V, Iqubal MK (2018) Clinical updates on drug-induced cardiotoxicity. Int J Pharm Sci Res 9: 16-26.
- Figueredo VM (2011) Chemical cardiomyopathies: the negative effects of medications and non-prescribed drugs on the heart. Am J Med 124: 480-488. [Crossref]
- Klimas J (2012) Drug-induced cardiomyopathies. In Cardiomyopathies - From Basic Research to Clinical Management 2012 Feb 15.
- Urbano‐Marquez A, Fernandez‐Sola J (2004) Effects of alcohol on skeletal and cardiac muscle. Muscle Nerve 30: 689-707. [Crossref]
- Knochel JP (1983) Cardiovascular effects of alcohol. Ann Intern Med 9: 849-854. [Crossref]
- Guarnieri T, Lakatta EG (1990) Mechanism of myocardial contractile depression by clinical concentrations of ethanol. A study in ferret papillary muscles. J Clin Invest 85: 1462-1467. [Crossref]
- Yokohama, Japan, 2-8 July 2000. Abstracts. Alcohol Clin Exp Res 24: 159A-229A. [Crossref]
- Gavazzi A, De Maria R, Parolini M, Porcu M (2000) Italian multicentre cardiomyopathy study group: Alcohol abuse and dilated cardiomyopathy in men. Am J Cardiol 85: 1114-1118. [Crossref]
- Skotzko CE, Vrinceanu A, Krueger L, Freudenberger R. Alcohol use and congestive heart failure: incidence, importance, and approaches to improved history taking. Heart Fail Rev 14: 51-55. [Crossref]
- Piano MR (2000) Alcoholic cardiomyopathy: incidence, clinical characteristics, and pathophysiology. Chest 121: 1638-1650. [Crossref]
- Regan TJ (1990) Alcohol and the cardiovascular system. Jama 264: 377-381. [Crossref]
- Segel LD, Klausner SC, Gnadt JT, Amsterdam EA (1984) Alcohol and the heart. Med Clin North Am 68: 147-161. [Crossref]
- Davidson DM (1989) Cardiovascular effects of alcohol. West J Med 151: 430-439. [Crossref]
- Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA et al. (2009). Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcohol Clin Exp Res 33: 206-219. [Crossref]
- Hines LM, Rimm EB (2001) Moderate alcohol consumption and coronary heart disease: a review. Postgrad Med J 77: 747-752. [Crossref]
- Beulens JW, Algra A, Soedamah-Muthu SS, Visseren FL, Grobbee DE et al. (2010) Alcohol consumption and risk of recurrent cardiovascular events and mortality in patients with clinically manifest vascular disease and diabetes mellitus: the Second Manifestations of ARTerial (SMART) disease study. Atherosclerosis 212: 281-286. [Crossref]
- Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath Jr CW, Doll R. Alcohol consumption and mortality among middle-aged and elderly US adults. N Eng J Med 337: 1705-1714. [Crossref]
- Bos S, Grobbee DE, Boer JM, Verschuren WM, Beulens JW (2010) Alcohol consumption and risk of cardiovascular disease among hypertensive women. Eur J Cardiovasc Prev Rehabil 17: 119-126. [Crossref]
- Costanzo S, Di Castelnuovo A, Donati MB, Iacoviello L, de Gaetano G. Alcohol consumption and mortality in patients with cardiovascular disease: a meta-analysis. J Am Coll Cardiol 55: 1339-1347. [Crossref]
- Fauchier L, Babuty D, Poret P, Casset-Senon D, Autret ML et al. (2000) Comparison of long-term outcome of alcoholic and idiopathic dilated cardiomyopathy. Eur Heart J 21: 306-314. [Crossref]
- Kupari M, Koskinen P, Suokas A, Ventilä M (1990) Left ventricular filling impairment in asymptomatic chronic alcoholics. Am J Cardiol 66: 1473-1477. [Crossref]
- Lazarevic AM, Nakatani S, Nešković AN, Marinković J, Yasumura Y et al. (2000) Early changes in left ventricular function in chronic asymptomatic alcoholics: relation to the duration of heavy drinking. J Am Coll Cardiol 35: 1599-1606. [Crossref]
- Kupari M, Koskinen P, Suokas A (1991) Left ventricular size, mass and function in relation to the duration and quantity of heavy drinking in alcoholics. Am J Cardiol 67: 274-279. [Crossref]
- George A, Figueredo VM (2011) Alcoholic cardiomyopathy: a review. J Card Fail 17: 844-849. [Crossref]
- Kesteloot H, Roelandt J, Willems J, Claes JH, Joossens JV (1968) An enquiry into the role of cobalt in the heart disease of chronic beer drinkers. Circulation 37: 854-864. [Crossref]
- Weber KT (1998) A Quebec quencher. Cardiovasc Res 40: 423-425. [Crossref]
- Djoussé L, Gaziano JM (2008) Alcohol consumption and heart failure: a systematic review. Curr Atheroscler Rep 10: 117-120. [Crossref]
- Hines LM, Stampfer MJ, Ma J, Gaziano JM, Ridker PM et al. (2001) Genetic variation in alcohol dehydrogenase and the beneficial effect of moderate alcohol consumption on myocardial infarction. N Eng J Med 344: 549-555. [Crossref]
- Fernández-Solà J, Nicolás JM, Oriola J, Sacanella E, Estruch R et al. (2002) Angiotensin-converting enzyme gene polymorphism is associated with vulnerability to alcoholic cardiomyopathy. Ann Intern Med 137:3 21-26. [Crossref]
- Estruch R, Fernández‐Solá J, Sacanella E, Paré C, Rubin E et al. (1995) Relationship between cardiomyopathy and liver disease in chronic alcoholism. Hepatology 22: 532-538. [Crossref]
- Bing RJ (1978) Cardiac metabolsim: its contributions to alcoholic heart disease and myocardial failure. Circulation 58: 965-970. [Crossref]
- Zakhari S (1997) Alcohol and the cardiovascular system: molecular mechanisms for beneficial and harmful action. Alcohol Health Res World 21: 21-30. [Crossref]
- Chen DB, Wang L, Wang PH (2000) Insulin-like growth factor I retards apoptotic signaling induced by ethanol in cardiomyocytes. Life Sci 67: 1683-1693. [Crossref]
- Urbano-Márquez A, Estruch R, Fernández-Solá J, Nicolás JM, Pare JC et al. (1995) The greater risk of alcoholic cardiomyopathy and myopathy in women compared with men. Jama 274: 149-154. [Crossref]
- Dancy M, Leech G, Bland JM, Gaitonde MK, Maxwell JD (1985). Preclinical left ventricular abnormalities in alcoholics are independent of nutritional status, cirrhosis, and cigarette smoking. Lancet 325: 1122-1125. [Crossref]
- Mathews Jr EC, Gardin JM, Henry WL, Del Negro AA, Fletcher RD et al. (1981) Echocardiographic abnormalities in chronic alcoholics with and without overt congestive heart failure. Am J Cardiol 47: 570-578. [Crossref]
- Iacovoni A, De Maria R, Gavazzi A (2010) Alcoholic cardiomyopathy. J Cardiovasc Med 11: 884-892. [Crossref]
- Laonigro I, Correale M, Di Biase M, Altomare E (2009) Alcohol abuse and heart failure. Eur J Heart Fail 11: 453-462. [Crossref]
- De Keulenaer GW, Brutsaert DL (1999) Dilated cardiomyopathy: changing pathophysiological concepts and mechanisms of dysfunction. J Card Surg 14: 64-74. [Crossref]
- George A, Figueredo VM (2010) Alcohol and arrhythmias: a comprehensive reviewJ Cardiovasc Med 11: 221-228. [Crossref]
- Greenspon AJ, Schaal SF (1983) The holiday heart: electrophysiologic studies of alcohol effects in alcoholics. Ann Intern Med 98: 135-139. [Crossref]
- Koskinen P, Kupari M (1992) Alcohol and cardiac arrhythmias. BMJ 304: 1394-1395. [Crossref]
- Henriksen JH, Møller S (2009) Cardiac and systemic haemodynamic complications of liver cirrhosis. Scand Cardiovasc J 43: 218-225. [Crossref]
- Pavan D, Nicolosi GL, Lestuzzi C, Burelli C, Zardo F et al. (1987) Normalization of variables of left ventricular function in patients with alcoholic cardiomyopathy after cessation of excessive alcohol intake: an echocardiographic study. Eur Heart J 8: 535-540. [Crossref]
- Wijetunga M, Seto T, Lindsay J, Schatz I (2003) Crystal methamphetamine‐associated cardiomyopathy: tip of the iceberg? J Toxicol Clin Toxicol. 41: 981-986. [Crossref]
- Yeo KK, Wijetunga M, Ito H, Efird JT, Tay K et al. (2007) The association of methamphetamine use and cardiomyopathy in young patients. Am J Med 120: 165-171. [Crossref]
- Brady KT, Lydiard RB, Malcolm R, Ballenger JC (1991) Cocaine-induced psychosis. J Clin Psychiatry 52: 509-512. [Crossref]
- Wetli CV, Fishbain DA (1985) Cocaine-induced psychosis and sudden death in recreational cocaine users. J Forensic Sci 30: 873-880. [Crossref]
- Chiueh CC, Kopin IJ (1978) Centrally mediated release by cocaine of endogenous epinephrine and norepinephrine from the sympathoadrenal medullary system of unanesthetized rats. J Pharmacol Exp Ther 205: 148-154. [Crossref]
- Mouhaffel AH, Madu EC, Satmary WA, Fraker Jr TD (1995) Cardiovascular complications of cocaine. Chest 107: 1426-1435. [Crossref]
- Burke WM, Ravi NV (1990) Urinary excretion of cocaine. Ann Intern Med 112: 548-549. [Crossref]
- Henning RJ, Wilson LD, Glauser JM (1994) Cocaine plus ethanol is more cardiotoxic than cocaine or ethanol alone. Crit Care Med 22: 1896-1906. [Crossref]
- Lukas SE, Sholar M, Kouri E, Fukuzako H, Mendelson JH (1994) Marihuana smoking increases plasma cocaine levels and subjective reports of euphoria in male volunteers. Pharmacol Biochem Behav 48: 715-721. [Crossref]
- Maraj S, Figueredo VM, Lynn MD (2010) Cocaine and the heart. Clin Cardiol 33: 264-269. [Crossref]
- Felker GM, Hu W, Hare JM, Hruban RH, Baughman KL et al. (1999) The spectrum of dilated cardiomyopathy. The Johns Hopkins experience with 1,278 patients. Medicine 78: 270-283. [Crossref]
- Awtry EH, Philippides GJ (2010) Alcoholic and cocaine-associated cardiomyopathies. Prog Cardiovasc Dis 52: 289-299. [Crossref]
- Henzlova MJ, Smith SH, Prchal VM, Helmcke FR (1991) Apparent reversibility of cocaine-induced congestive cardiomyopathy. Am Heart J 122: 577-579. [Crossref]
- Kloner RA, Hale S, Alker K, Rezkalla S (1992) The effects of acute and chronic cocaine use on the heart. Circulation 85: 407-419. [Crossref]
- Mehta PM, Grainger TA, Lust RM, Movahed A, Terry J et al. (1995) Effect of cocaine on left ventricular function: relation to increased wall stress and persistence after treatment. Circulation 91: 3002-3009. [Crossref]
- Isabelle M, Vergeade A, Moritz F, Dautréaux B, Henry JP et al. (2007) NADPH oxidase inhibition prevents cocaine-induced up-regulation of xanthine oxidoreductase and cardiac dysfunction. J Mol Cell Cardiol 42: 326-332. [Crossref]
- Torres AR, Whitney J, Gonzalez-Heydrich J (2008) Attention-deficit/hyperactivity disorder in pediatric patients with epilepsy: review of pharmacological treatment. Epilepsy Behav 12: 217-233. [Crossref]
- Billiard M (2008) Narcolepsy: current treatment options and future approaches. Neuropsychiatr Dis Treat 4: 557-566. [Crossref]
- Reidenberg MM (1976) Some Extraneuronal Interactions of Drugs of Abuse: AN Overview. Ann N Y Acad Sci 281: 1-10. [Crossref]
- Turnipseed SD, Richards JR, Kirk JD, Diercks DB, Amsterdam EA (2003) Frequency of acute coronary syndrome in patients presenting to the emergency department with chest pain after methamphetamine use. J Emerg Med 24: 369-373. [Crossref]
- Shenouda SK, Varner KJ, Carvalho F, Lucchesi PA (2009) Metabolites of MDMA induce oxidative stress and contractile dysfunction in adult rat left ventricular myocytes. Cardiovasc Toxicol 9: 30-38. [Crossref]
- Mizia-Stec K, Gąsior Z, Wojnicz R, Haberka M, Mielczarek M et al. (2008) Severe dilated cardiomyopathy as a consequence of Ecstasy intake. Cardiovasc Pathol 17: 250-253. [Crossref]
- Shenouda SK, Lord KC, McIlwain E, Lucchesi PA, Varner KJ (2008) Ecstasy produces left ventricular dysfunction and oxidative stress in rats. Cardiovasc Res 79: 662-670. [Crossref]
- Mouhaffel AH, Madu EC, Satmary WA, Fraker Jr TD (1995) Cardiovascular complications of cocaine. Chest 107: 1426-1435. [Crossref]
- Henry JA, Jeffreys KJ, Dawling S (1992) Toxicity and deaths from 3, 4-methylenedioxymethamphetamine ("ecstasy"). Lancet 340: 384-387. [Crossref]
- Smit AA, Wieling W, Voogel AJ, Koster RW, Van Zwieten PA (1996) Orthostatic hypotension due to suppression of vasomotor outflow after amphetamine intoxication. Mayo Clin Proc 71: 1067-1070. [Crossref]
- Albertson TE, Derlet RW, Van Hoozen BE (1999) Methamphetamine and the expanding complications of amphetamines. West J Med 170: 214-219. [Crossref]
- Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ (2000) Role of quinones in toxicology. Chem Res Toxicol 13: 135-160. [Crossref]
- Badon LA, Hicks A, Lord K, Ogden BA, Meleg-Smith S et al. (2002) Changes in cardiovascular responsiveness and cardiotoxicity elicited during binge administration of Ecstasy. J Pharmacol Exp Ther 302: 898-907. [Crossref]
- Islam MN, Kuroki H, Hongcheng B, Ogura Y, Kawaguchi N et al. (1995) Cardiac lesions and their reversibility after long-term administration of methamphetamine. Forensic Sci Int 75: 29-43. [Crossref]
- Lopez JE, Yeo K, Caputo G, Buonocore M, Schaefer S (2009) Recovery of methamphetamine associated cardiomyopathy predicted by late gadolinium enhanced cardiovascular magnetic resonance. J Cardiovasc Magn Reson11: 46. [Crossref]
- Srivastava S, Goyal P (2010) Novel biomaterials: decontamination of toxic metals from wastewater. Springer Science & Business Media.
- Brathwaite RL, Rabone SD (1985) Heavy metal sulphide deposits and geochemical surveys for heavy metals in New Zealand. J Roy Soc New Zeal 15: 363-370.
- Oyinloye BE, Ajiboye BO, Ojo OA, Nwozo SO, Kappo AP (2016) Cardioprotective and antioxidant influence of aqueous extracts from Sesamum indicum seeds on oxidative stress induced by cadmium in wistar rats. Pharmacogn Mag 12: S170-S174. [Crossref]
- Packer M (2016) Cobalt cardiomyopathy: a critical reappraisal in light of a recent resurgence. Circ Heart Fail 9: e003604. [Crossref]
- Tilney R, Burg MR, Sammut MA (2016) Cobalt cardiomyopathy secondary to hip arthroplasty: an increasingly prevalent problem. Case Rep Cardiol 2017: 5434571. [Crossref]
- Barceloux DG (1999) Cobalt. J Toxicol Clin Toxicol 37: 201-206. [Crossref]
- Lauwerys R, Lison D (1994) Health risks associated with cobalt exposure: an overview. Sci Total Environ 150: 1-6. [Crossref]
- Christensen JM (1995) Human exposure to toxic metals: factors influencing interpretation of biomonitoring results. Sci Total Environ 166: 89-135. [Crossref]
- Lippi G, Franchini M, Guidi GC (2006) Blood doping by cobalt. Should we measure cobalt in athletes? J Occup Med Toxicol 1:18. [Crossref]
- Duckham JM, Lee HA (1976) The treatment of refractory anaemia of chronic renal failure with cobalt chloride. Q J Med 45: 277-294. [Crossref]
- Cobb AG, Schmalzreid TP (2006) The clinical significance of metal ion release from cobalt-chromium metal-on-metal hip joint arthroplasty. Proc Inst Mech Eng H 220:385-398. [Crossref]
- Hanawa T (2009) Materials for metallic stents. J Artif Organs 12: 73-79. [Crossref]
- Roach M (2007) Base metal alloys used for dental restorations and implants. Dent Clin North Am 51:603-627. [Crossref]
- Alexander CS (1972) Cobalt-beer cardiomyopathy. A clinical and pathologic study of twenty-eight cases. Am J Med 53: 395-417. [Crossref]
- Morin Y, Daniel P (1967) Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J 97: 926-928. [Crossref]
- Kesteloot H, Roelandt J, Willems J, Claes JH, Joossens JV (1968) An enquiry into the role of cobalt in the heart disease of chronic beer drinkers. Circulation 37: 854-864. [Crossref]
- Rona G, Chappel CI (1973) Pathogenesis and pathology of cobalt cardiomyopathy. Recent Adv Stud Cardiac Struct Metab 2: 407-422. [Crossref]
- Hashimoto H, Yanagisawa T, Taira N (1989) Differential antagonism of the negative inotropic effect of gentamicin by calcium ions, Bay K 8644 and isoprenaline in canine ventricular muscle: comparison with cobalt ions. Br J Pharmacol 96: 906-912. [Crossref]
- Harrow JA, Das PK, Dhalla NS (1978) Influence of some divalent cations on heart sarcolemmal bound enzymes and calcium binding. Biochem Pharmacol 27: 2605-2609. [Crossref]
- Kleinfeld M, Stein E (1968) Action of divalent cations on membrane potentials and contractility in rat atrium. Am J Physiol 215: 593-599. [Crossref]
- Dingle JT, Heath JC, Webb M, Daniel M (1962) The biological action of cobalt and other metals. II. The mechanism of the respiratory inhibition produced by cobalt in mammalian tissues. Biochim Biophys Acta 65: 34-46. [Crossref]
- Wiberg GS (1968) The effect of cobalt ions on energy metabolism in the rat. Can J Biochem 46: 549-554. [Crossref]
- Grice HC, Goodman T, Munro IC, Wiberg GS, Morrison AB (1969) Myocardial toxicity of cobalt in the rat. Ann N Y Acad Sci 156: 189-194. [Crossref]
- Hervouet E, Pecina P, Demont J, Vojtísková A, Simonnet H, Houstek J et al. (2006) Inhibition of cytochrome c oxidase subunit 4 precursor processing by the hypoxia mimic cobalt chloride. Biochem Biophys Res Commun 344: 1086-1093. [Crossref]
- Clyne N, Hofman-Bang C, Haga Y, Hatori N, Marklund SL et al. (2001) Chronic cobalt exposure affects antioxidants and ATP production in rat myocardium. Scand J Clin Lab Invest 61: 609-614. [Crossref]
- Lewis CP, Demedts M, Nemery B (1991) Indices of oxidative stress in hamster lung following exposure to cobalt(II) ions: in vivo and in vitro studies. Am J Respir Cell Mol Biol 5: 163-169. [Crossref]
- Speijers GJ, Krajnc EI, Berkvens JM, van Logten MJ (1982) Acute oral toxicity of inorganic cobalt compounds in rats. Food Chem Toxicol 20: 311-314. [Crossref]
- Huy ND, Morin PJ, Mohiuddin SM, Morin Y (1973) Acute effects of cobalt on cardiac metabolism and mechanical performance. Can J Physiol Pharmacol 51: 46-51. [Crossref]
- Haga Y, Clyne N, Hatori N, Hoffman-Bang C, Pehrsson SK, Ryden L (1996) Impaired myocardial function following chronic cobalt exposure in an isolated rat heart model. Trace Elem Electrolytes 13: 69-74.
- Sandusky GE, Henk WG, Roberts ED (1981) Histochemistry and ultrastructure of the heart in experimental cobalt cardiomyopathy in the dog. Toxicol Appl Pharmacol 61: 89-98. [Crossref]
- Sandusky GE, Crawford MP, Roberts ED (1981) Experimental cobalt cardiomyopathy in the dog: a model for cardiomyopathy in dogs and man. Toxicol Appl Pharmacol 60: 263-278. [Crossref]
- Alexander CS (1967) Electron microscopic observations in alcoholic heart disease. Br Heart J 29: 200-206. [Crossref]
- Suzuki T (1967) Electron microscopic study on myocardial lesions in thiamine deficient Tohoku J Exp Med 91: 249-259. [Crossref]
- Lin JH, Duffy JL (1970) Cobalt-induced myocardial lesions in rats. Lab Invest 23: 158-162. [Crossref]
- Rona G (1971) Experimental aspects of cobalt cardiomyopathy. Br Heart J 33: 171-174. [Crossref]
- Endoh H, Kaneko T, Nakamura H, Doi K, Takahashi E (2000) Improved cardiac contractile functions in hypoxia-reoxygenation in rats treated with low concentration Co(2+). Am J Physiol Heart Circ Physiol 279: H2713–H2719. [Crossref]
- Pery-Man N, Houeto P, Coirault C, Suard I, Perennec J et al. (1996) Hydroxocobalamin vs cobalt toxicity on rat cardiac and diaphragmatic muscles. Intensive Care Med 22: 108-115. [Crossref]
- Clyne N, Persson B, Havu N, Hultman E, Lins LE et al. (1990) The intracellular distribution of cobalt in exposed and unexposed rat myocardium. Scand J Clin Lab Invest 50: 605-609. [Crossref]
- Heggtveit HA, Grice HC, Wiberg GS (1970) Cobalt cardiomyopathy. Experimental basis for the human lesion. Pathol Microbiol (Basel) 35: 110-113. [Crossref]
- Macho L, Palkovic M (1963) The influence of endocrine glands on carbohydrate VII. The effect of thyroxine on citric and pyruvic acid levels in blood during the pyruvate tolerance test. Arch Int Physiol Biochim 71: 758-761.
- Shulman GI, Ladenson PW, Wolfe MH, Ridgway EC, Wolfe RR (1985) Substrate cycling between gluconeogenesis and glycolysis in euthyroid, hypothyroid, and hyperthyroid J Clin Invest 76: 757-764. [Crossref]
- Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM et al. (2001) Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest 108: 733-737. [Crossref]
- Hyyti OM, Ning XH, Buroker NE, Ge M, Portman MA (2006) Thyroid hormone controls myocardial substrate metabolism through nuclear receptor-mediated and rapid posttranscriptional mechanisms. Am J Physiol Endocrinol Metab 290: E372-E379. [Crossref]
- Griffith WH, Paveck PL, Mulford DJ (1942) Relation of sulphur amino acids to toxicity of cobalt and nickel in rats. J Nutr 23: 603-612.
- Taylor DM (1962) The absorption of cobalt from the gastrointestinal tract of the rat. Phys Med Biol 6 :445-451. [Crossref]
- Shtenberg AI, Kusevitskii IA, Abolyn EE (1964) Effect on thyroid gland of cobalt in low-protein diets with varying iodine contents. Fed Proc Transl Suppl 23: 763-766. [Crossref]
- Kriss JP, Carnes WH, Gross RT (1955) Hypothyroidism and thyroid hyperplasia in patients treated with cobalt. J Am Med Assoc 157: 117-121. [Crossref]
- Carnes WH, Greenspan FS, Kriss JP, Lew W (1956) Alterations in chick thyroid function induced by cobalt. Endocrinology 59: 555-564. [Crossref]
- Smith TJ, Drummond GS, Kappas A (1987) Cobalt-protoporphyrin suppresses thyroid and testicular hormone concentrations in rat serum: a novel action of this synthetic heme analogue. Pharmacology 34: 9-16. [Crossref]
- Schleisner P (1957) Cobalt polycythaemia and thyroid hormone. Acta Med Scand 157: 33-38 [Crossref]
- Sullivan JF, Egan JD, George RP (1969) A distinctive myocardiopathy occurring in Omaha, Nebraska: clinical aspects. Ann N Y Acad Sci 156: 526-543. [Crossref]
- Abhayaratna WP, Marwick TH, Smith WT, Becker NG (2006) Characteristic of left ventricular diastolic dysfunction in the community: an echocardiographic survey. Heart 92: 1259-1264. [Crossref]
- Redfield MM, Jacobsen SJ, Burnett JC, Jr, Mahoney DW, Bailey KR et al. (2003) Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. Jama 289: 194-202. [Crossref]
- Sung RY, Huang GY, Shing MK, Oppenheimer SJ, Li CK et al. (1997) Echocardiographic evaluation of cardiac function in paediatric oncology patients treated with or without anthracycline. Int J Cardiol 60: 239-248. [Crossref]
- Bonilla HM, Bhimaraj A (2018) a case of cobalt cardiomyopathy. J AM Coll Cardial 71: A2386.
- Kopp SJ, Barron JT, Tow JP (1988) Cardiovascular actions of lead and relationship to hypertension: a review. Environ Health Perspect 78: 91-99. [Crossref]
- Alissa EM, Ferns GA (2011) Heavy metal poisoning and cardiovascular disease. J Toxicol [Crossref]
- Markowitz M (2000) Lead poisoning. Pediatr Rev 21: 327-335. [Crossref]
- Staessen JA, Bulpitt CJ, Fagard R, Lauwerys RR, Roels H et al. (1994) Hypertension caused by low-level lead exposure: myth or fact? J Cardiovasc Risk 1: 87-97. [Crossref]
- Schwartz J (1995) Lead, blood pressure, and cardiovascular disease in men. Arch Environ Health 50: 31-37. [Crossref]
- Navas-Acien A, Guallar E, Silbergeld EK, Rothenberg SJ (2007) Lead exposure and cardiovascular disease: a systematic review. Environ Health Perspect 115: 472-482. [Crossref]
- Read JL, Williams JP (1952) Lead myocarditis: Report of a case. Am Heart J 44: 797-802. [Crossref]
- Kline TS (1960) Myocardial changes in lead poisoning. AMA J Dis Child 99: 48-54. [Crossref]
- Stofen D (1974) Environmental lead and the heart. J Mol Cell Cardiol 6: 285-290. [Crossref]
- Myerson RM, Eisenhauer JH (1963) Atrioventricular conduction defects in lead poisoning. Am J Cardiol 11: 409-412. [Crossref]
- Silver W, Rodriguez-Torres R (1968) Electrocardiographic studies in children with lead poisoning. Pediatrics 41: 1124-1127. [Crossref]
- Dimitrova M (1972) Modifications de la fonction contractile du myocarde dans le saturnisme chronique. Mal. Prof. Med. Trav. Secllr. Soc 33: 383-387.
- Bertel O, Bühler FR, Ott J (1978) Lead-induced hypertension: blunted beta-adrenoceptor-mediated functions. Br Med J 1: 551. [Crossref]
- Kirkby H, Gyntelberg F (1985) Blood pressure and other cardiovascular risk factors of long-term exposure to lead. Scand J Work Environ Health 11 :15-19. [Crossref]
- Boscolo P, Galli G, Iannaccone A, Martino F, Porcelli G, Troncone L (1981) Plasma renin activity and urinary kallikrein excretion in lead-exposed workers as related to hypertension and nephropathy. Life Sci 28: 175-184. [Crossref]
- Jhaveri RC, Lavorgna L, Dube SK, Glass L, Khan F, Evans HE (1979) Relationship of blood pressure to blood lead concentrations in small children. Pediatrics 63: 674-676. [Crossref]
- Gonick HC, Ding Y, Bondy SC, Ni Z, Vaziri ND (1997) Lead-induced hypertension: interplay of nitric oxide and reactive oxygen species. Hypertension 30: 1487-1492. [Crossref]
- Mishra KP, Singh VK, Rani R, Yadav VS, Chandran V et al. (2003) Effect of lead exposure on the immune response of some occupationally exposed individuals. Toxicology 188: 251-259. [Crossref]
- Vaziri ND, Ding Y, Ni Z, Gonick HC (1997) Altered nitric oxide metabolism and increased oxygen free radical activity in lead-induced hypertension: effect of lazaroid therapy. Kidney Int 52: 1042-1046. [Crossref]
- Kaji T, Suzuki M, Yamamoto C, Mishima A, Sakamoto M, Kozuka H (1995) Severe damage of cultured vascular endothelial cell monolayer after simultaneous exposure to cadmium and lead. Arch Environ Contam Toxicol 28: 168-172. [Crossref]
- Fujiwara Y, Kaji T, Yamamoto C, Kozuka H (1995) Stimulatory effect of lead on the proliferation of cultured vascular smooth-muscle cells. Toxicology 98: 105-110. [Crossref]
- Ding Y, Vaziri ND, Gonick HC (1998) Lead-Induced Hypertension: II. Response to Sequential Infusions of L-Arginine, Superoxide Dismutase, and Nitroprusside. Environ Res 76: 107-113. [Crossref]
- Goldstein GW (1993) Evidence that lead acts as a calcium substitute in second messenger metabolism. Neurotoxicology 14: 97-101. [Crossref]
- Cooper LT, Rader V, Ralston NV (2007) The roles of selenium and mercury in the pathogenesis of viral cardiomyopathy. Congest Heart Fail 13: 193-199. [Crossref]
- Frustaci A, Magnavita N, Chimenti C, Caldarulo M, Sabbioni E et al. (1999) Marked elevation of myocardial trace elements in idiopathic dilated cardiomyopathy compared with secondary cardiac dysfunction. J Am Coll Cardiol 33: 1578-1583. [Crossref]
- Gainer JH (1977) Effects of heavy metals and of deficiency of zinc on mortality rates in mice infected with encephalomyocarditis virus. Am J Vet Res 38: 869-872. [Crossref]
- Toyama I, Kolmer JA (1918) The influence of arsphenamine and mercuric chloride upon complement and antibody production. J Immunol 3: 301-316.
- Webb JL (1966). Mercurials. In: Webb JL, ed. Enzyme and Metabolic Inhibitors. New York, NY: Academic Press, pp.729-985.
- Christensen MM, Ellermann-Eriksen S, Rungby J, et al. (1966) Influence of mercuric chloride on resistance to generalized infection with herpes simplex virus type 2 in mice. Toxicology 114: 57-66. [Crossref]
- Johansson U, Sander B, Hultman P (1997) Effects of the murine genotype on T cell activation and cytokine production in murine mercury-induced autoimmunity. J Autoimmun 10: 347-355. [Crossref]
- Johansson U, Hansson-Georgiadis H, Hultman P (1998) The genotype determines the B cell response in mercury-treated mice. Int Arch Allergy Immunol 116: 295-305. [Crossref]
- Yang JM, Baserga SJ, Turley SJ, et al. (2001) Fibrillarin and other snoRNP proteins are targets of autoantibodies in xenobiotic-induced autoimmunity. Clin Immunol 101: 38-50. [Crossref]
- Pollard KM, Pearson DL, Bluthner M, et al. (2000) Proteolytic cleavage of a self-antigen following xenobiotic-induced cell death produces a fragment with novel immunogenic properties. J Immunol 165: 2263-2270. [Crossref]
- Kubicka-Muranyi M, Kremer J, Rottmann N, et al. (1996) Murine systemic autoimmune disease induced by mercuric chloride: T helper cells reacting to self-proteins. Int Arch Allergy Immunol 109: 11-20. [Crossref]
- South PK, Morris VC, Levander OA, et al. (2001) Mortality in mice infected with an amyocarditic coxsackievirus and given a subacute dose of mercuric chloride. J Toxicol Environ Health A 63: 511-523. [Crossref]
- Blum H, Rollinghoff M, Gessner A (1996) Expression and co-cytokine function of murine thioredoxin/adult T cell leukaemia-derived factor (ADF). Cytokine 8: 6-13. [Crossref]
- Schenk H, Vogt M, Droge W, et al. (1996) Thioredoxin as a potent costimulus of cytokine expression. J Immunol 156: 765-771. [Crossref]
- Yamauchi A, Masutani H, Tagaya Y, et al. (1992) Lymphocyte transformation and thiol compounds; the role of ADF/thioredoxin as an endogenous reducing agent. Mol Immunol 29: 263-270. [Crossref]
- Biguet C, Wakasugi N, Mishal Z, et al. (1994) Thioredoxin increases the proliferation of human B-cell lines through a protein kinase C-dependent mechanism. J Biol Chem 269: 28865-28870. [Crossref]
- Bertini R, Howard OM, Dong HF, et al. (1999) Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med 189: 1783-1789. [Crossref]
- Beck MA, Levander OA, Handy J (2003) Selenium deficiency and viral infection. J Nutr 133: 1463S-1467S. [Crossref]
- Moller-Madsen B (1994) Localization of mercury in CNS of the rat. An autometallographic study. Pharmacol Toxicol 75: 1-41. [Crossref]
- Forman J, Moline J, Cernichiari E, et al. (2000) A cluster of pediatric metallic mercury exposure cases treated with meso-2,3-dimercaptosuccinic acid (DMSA). Environ Health Perspect 108: 575-577. [Crossref]
- Ratnaike RN (2003) Acute and chronic arsenic toxicity. Postgrad Med J 79: 391-396. [Crossref]
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J et al. (2011) Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol 31: 95-107. [Crossref]
- Chiou HY, Huang WI, Su CL, Chang SF, Hsu YH et al. (1997) Dose-response relationship between prevalence of cerebrovascular disease and ingested inorganic arsenic. Stroke 28: 1717-1723. [Crossref]
- Tseng CH, Chong CK, Tseng CP, Hsueh YM, Chiou HY et al. (2003) Long-term arsenic exposure and ischemic heart disease in arseniasis-hyperendemic villages in Taiwan. Toxicol Lett 137: 15-21. [Crossref]
- Cullen NM, Wolf LR, St Clair D (1995) Pediatric arsenic ingestion. Am J Emerg Med 13: 432-435. [Crossref]
- Mumford JL, Wu K, Xia Y, Kwok R, Yang Z et al. (2007) Chronic arsenic exposure and cardiac repolarization abnormalities with QT interval prolongation in a population-based study. Environ Health Perspect 115: 690-694. [Crossref]
- Quatrehomme G, Ricq O, Lapalus PH, Jacomet Y, Ollier A (1992) Acute arsenic intoxication: forensic and toxicologic aspects (an observation). J Forensic Sci 37: 1163-1171. [Crossref]
- Balakumar P, Kaur J (2009) Arsenic exposure and cardiovascular disorders: an overview. Cardiovasc Toxicol 9: 169-176. [Crossref]
- Simeonova PP, Hulderman T, Harki D, Luster MI (2003) Arsenic exposure accelerates atherogenesis in apolipoprotein E (-/-) mice. Environ Health Perspect 111: 1744-1748. [Crossref]
- Polissar L, Lowry-Coble K, Kalman DA, Hughes JP, van Belle G et al. (1990) Pathways of human exposure to arsenic in a community surrounding a copper smelter. Environ Res 53: 29-47. [Crossref]
- Milham Jr S, Strong T (1974) Human arsenic exposure in relation to a copper smelter. Environ Res 7: 176-182.
- Kosnett MJ (2013) The role of chelation in the treatment of arsenic and mercury poisoning. J Med Toxicol 9: 347-354. [Crossref]
- Serinken M, Karcioglu O, Korkmaz A (2009) Rarely seen cardiotoxicity of lithium overdose: complete heart block. Int J Cardiol 132: 276-278. [Crossref]
- Waring WS (2007) Delayed cardiotoxicity in chronic lithium poisoning: discrepancy between serum lithium concentrations and clinical status. Basic Clin Pharmacol Toxicol 100: 353-355. [Crossref]
- Waring WS (2006) Management of lithium toxicity. Toxicol Rev 25: 221-330. [Crossref]
- Corriveau S, Gardhouse A, Soth M, Ainsworth C (2016) Lithium Toxicity. Canadian Journal of General Internal Medicine 8: 68-71.
- McKnight RF, Adida M, Budge K, Stockton S, Goodwin GM et al. (2012) Lithium toxicity profile: a systematic review and meta-analysis. Lancet 379: 721-728. [Crossref]
- Gray J, (ed.) (2007) e-Therapeutics. Ottawa (ON): Canadian Pharmacists Association. Retrieved from http://www.etherapeutics.ca.normedproxy.lakeheadu.ca. Accessed May 12, 2012.
- Sadock B J, Sadock VA. (2007) Synopsis of Psychiatry, 10th edition. Philadelphia (PA): Lippincott Williams and Wilkins.
- Tilkian A, Schroeder J, Kao J, Hultgren H (1976) The cardiovascular effects of lithium in man: a review of literature. Am J Med 61: 665-670. [Crossref]
- Brady H, Horgan J (1988) Lithium and the heart: unanswered questions. Chest 93: 166-169. [Crossref]
- Offerman S, Alsop J, Lee J, Holmes J (2010) Hospitalized lithium overdose cases reported to the California Poison Control System. Clin Toxicol 48: 443-448. [Crossref]
- Jaeger A, Sauder P, Kopferschmitt J, Tritsch L, Flesch F (1993) When should dialysis be performed in lithium poisoning? A kinetic study in 14 cases of lithium poisoning. J Toxicol Clin Toxicol 31: 429-437. [Crossref]
- Van Bommel EF, Kalmeijer MD, Ponssen HH (2000) Treatment of life-threatening lithium toxicity with high-volume continuous venovenous hemofiltration. Am J Nephrol 20: 408-411. [Crossref]
- Cooper RG, Harrison AP (2009) The uses and adverse effects of beryllium on health. Indian J Occup Environ Med 13: 65-76. [Crossref]
- Deubner DC, Lowney YW, Paustenbach DJ, Wamerdam J (2001) Contribution of incidental exposure pathways to total beryllium exposures. Appl Occup Environ Hyg. 16: 568-578. [Crossref]
- Hardy HL, Tabershaw IR (1946) Delayed chemical pneumonitis occurring in workers exposed to beryllium compounds. J Ind Hyg Toxicol 28: 197-211. [Crossref]
- Ward E, Okun A, Ruder A, Fingerhut M, Steenland K (1992) A mortality study of workers at seven beryllium processing plants. Am J Ind Med 22: 885-904. [Crossref]
- Schepers GW (1964) Biological action of beryllium reaction of the monkey to inhaled aerosols. Ind Med Surg 33: 1-16. [Crossref]
- Hall RH, Scott JK, Laskin S, Stroud CA, Stokinger HE (1950) Acute Toxieity of Inhaled Beryllium. III. Observations correlating Toxieity with the Physieochemical Properties of Beryllium Oxide Dust. Indust. Hyg Occupational Med.2: 25-48. [Crossref]
- Stokinger HE, Sprague GF, Hall RH, Ashenburg NJ, Scott JK et al. (1950) Acute Inhalation Toxicity of Beryllium. I. Four Definitive Studies of Beryllium Sulfate at Exposure Concentrations of 100, 50, 10 and 1 Mg. per Cubic Meter. Indust. Hyg. & Occupational Med 1: 379-397. [Crossref]
- Cobb N, Etzel RA (1991) Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. Jama 266: 659-663. [Crossref]
- Zevin S, Saunders S, Gourlay SG, Jacob P, Benowitz NL (2001) Cardiovascular effects of carbon monoxide and cigarette smoking. J Am Coll Cardiol 38: 1633-1638. [Crossref]
- Kim HH, Choi S (2018) Therapeutic aspects of carbon monoxide in cardiovascular disease. Int J Mol Sci 19: 2381. [Crossref]
- Lee FY, Chen WK, Lin CL, Kao CH (2015) Carbon monoxide poisoning and subsequent cardiovascular disease risk: a nationwide population-based cohort study. Medicine 94: [Crossref]
- Huang CC, Chung MH, Weng SF, Chien CC, Lin SJ et al. (2014) Long-term prognosis of patients with carbon monoxide poisoning: a nationwide cohort study. PLoS One 9: e105503. [Crossref]
- Roughton FJ (1970) The equilibrium of carbon monoxide with human haemoglobin in whole blood. Ann N Y Acad Sci 174: 177-188. [Crossref]
- Cramlet SH, Erickson HH, Gorman HA (1975) Ventricular function following acute carbon monoxide exposure. J Appl Physiol 39: 482-486. [Crossref]
- Silver DA, Cross M, Fox B, Paxton RM (1996) Computed tomography of the brain in acute carbon monoxide poisoning. Clin Radiol 51: 480-483. [Crossref]
- Alonso JR, Cardellach F, Lopez S, Casademont J, Miró O (2003) Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol Toxicol 93: 142-146. [Crossref]
- Taskiran D, Nesil T, Alkan K (2007) Mitochondrial oxidative stress in female and male rat brain after ex vivo carbon monoxide treatment. Hum Exp Toxicol.26: 645-651. [Crossref]
- Thom SR, Fisher D, Xu YA, Notarfrancesco K, Ischiropoulos H (2000) Adaptive responses and apoptosis in endothelial cells exposed to carbon monoxide. Proc Natl Acad Sci U S A 97: 1305-1310. [Crossref]
- Lippi G, Rastelli G, Meschi T, Borghi L, Cervellin G (2012) Pathophysiology, clinics, diagnosis and treatment of heart involvement in carbon monoxide poisoning. Clin Biochem 45: 1278-1285. [Crossref]
- Franchini M, Mannucci PM (2007) Short‐term effects of air pollution on cardiovascular diseases: outcomes and mechanisms. J Thromb Haemost 5: 2169-2174. [Crossref]
- Baccarelli A, Martinelli I, Zanobetti A, Grillo P, Hou LF et al. (2008) Exposure to particulate air pollution and risk of deep vein thrombosis. Arch Intern Med 168:920-927. [Crossref]
- Lippi G, Favaloro EJ, Franchini M, Guidi GC (2008) Air pollution and coagulation testing: a new source of biological variability? Thromb Res 123: 50-54. [Crossref]
- Baccarelli A, Martinelli I, Pegoraro V, Melly S, Grillo P et al. (2009) Living near major traffic roads and risk of deep vein thrombosis. Circulation 119: 3118–3124. [Crossref]
- Franchini M, Mannucci PM (2009) Particulate air pollution and cardiovascular risk: short-term and long-term effects. Semin Thromb Hemost 35:665-670. Crossref[]
- Lippi G, Franchini M, Montagnana M, Filippozzi L, Favaloro EJ et al. (2010) Relationship between 24-h air pollution, emergency department admission and diagnosis of acute coronary syndrome. J Thromb Thrombolysis 29: 381-386. [Crossref]
- Franchini M, Mannucci PM (2011) Thrombogenicity and cardiovascular effects of ambient air pollution. Blood 118: 2405-2412. [Crossref]
- Franchini M, Mannucci PM (2012) Air pollution and cardiovascular disease. Thromb Res 129: 230-234. [Crossref]
- Andre L, Boissière J, Reboul C, Perrier R, Zalvidea S et al. (2010) Carbon monoxide pollution promotes cardiac remodeling and ventricular arrhythmia in healthy rats. Am J Respir Crit Care Med 181: 587-595. [Crossref]
- Anderson RF, Allensworth DC, DeGroot WJ (1967) Myocardial toxicity from carbon monoxide poisoning. Ann Intern Med 67: 1172-1182. [Crossref]
- Ismail MM, El-Ghamry H, Shaker OG, Fawzi MM, Ibrahim SF (2013). Some biomarkers in carbon monoxide-induced cardiotoxicity. J Environ Anal Toxicol 3: 2161-0525. [Crossref]
- End E, Long CW (1942) Oxygen under Pressure in Carbon Monoxide Poisoning. I. Effect on Dogs and Guinea Pigs. Journal of Industrial Hygiene and Toxicology 24: 302-306.
- Smith G, Sharp GR (1960) Treatment of carbon monoxide poisoning with oxygen under pressure. Lancet 276: 905-906
- Al-Rasheed NM, Al-Rasheed NM, Faddah LM, Mohamed AM, Mohammad RA et al. (2014) Potential impact of silymarin in combination with chlorogenic acid and/or melatonin in combating cardiomyopathy induced by carbon tetrachloride. Saudi journal of biological sciences 21: 265-274. [Crossref]
- Holbrook H. 1991. Carbon tetrachloride. In: Kroschwitz JI, Howe-Grant M, editors. Kirk-Othmer encyclopedia of chemical technology. Vol 5. 4th ed. New York, NY: John Wiley & Sons. pp. 1062–72
- Doherty RE (2000) A history of the production and use of carbon tetrachloride, tetrachloroethylene, trichloroethylene and 1, 1, 1-trichloroethane in the United States: part 1—historical background; carbon tetrachloride and tetrachloroethylene. Environmental Forensics 1: 69-81.
- Odabasi M (2008) Halogenated volatile organic compounds from the use of chlorine-bleach-containing household products. Environ Sci Technol 42: 1445-1451. [Crossref]
- Xiao J, Liong EC, Ching YP, Chang RC, So KF et al. (2012) Lycium barbarum polysaccharides protect mice liver from carbon tetrachloride-induced oxidative stress and necroinflammation. J Ethnopharmacol 39: 462-470. [Crossref]
- Ozturk F, Ucar M, Ozturk IC, Vardi N, Batcioglu K (2003) Carbon tetrachloride-induced nephrotoxicity and protective effect of betaine in Sprague-Dawley rats. Urology 62: 353-356. [Crossref]
- Jayakumar T, Sakthivel M, Thomas PA, Geraldine P (2008) Pleurotus ostreatus, an oyster mushroom, decreases the oxidative stress induced by carbon tetrachloride in rat kidneys, heart and brain. Chem Biol Interact 176: 108-120. [Crossref]
- Botsoglou NA, Taitzoglou IA, Botsoglou E, Zervos I, Kokoli A et al. (2009) Effect of long‐term dietary administration of oregano and rosemary on the antioxidant status of rat serum, liver, kidney and heart after carbon tetrachloride‐induced oxidative stress. J Sci Food Agr 89: 1397-1406.
- Karakus E, Karadeniz A, Simsek N, Can I, Kara A et al. (2011) Protective effect of Panax ginseng against serum biochemical changes and apoptosis in liver of rats treated with carbon tetrachloride (CCl4). J Hazardous Mater 15; 195:208-213. [Crossref]
- Shi H, Dong L, Jiang J, Zhao J, Zhao G et al. (2013) Chlorogenic acid reduces liver inflammation and fibrosis through inhibition of toll-like receptor 4 signaling pathway. Toxicology 303: 107-114. [Crossref]
- Larcan A, Laprevote-Heully MC, Lambert H, Guine JM, Galoisy P (1973) Intoxication by ingestion of a massive dose of carbon tetrachloride. Recovery in probable relation to early hyperbaric oxygen therapy. Eur J Toxicol Hyg Environ 6: 286-289. [Crossref]
- Truss CD, Killenberg PG (1982) Treatment of carbon tetrachloride poisoning with hyperbaric oxygen. Gastroenterology 82: 767-769. [Crossref]
- Burk RF, Reiter R, Lane JM (1986). Hyperbaric oxygen protection against carbon tetrachloride hepatotoxicity in the rat: Association with altered metabolism. Gastroenterology 90: 812-818. [Crossref]
- Burkhart KK, Hall AH, Gerace R, Rumack BH (1991) Hyperbaric oxygen treatment for carbon tetrachloride poisoning. Drug Saf 6: 332-338. [Crossref]
- Thom SR (2006) Antidotes in depth: hyperbaric oxygen. In Flomenbaum NE, et al., (Eds) Goldfrank's toxicologic emergencies, 8th Ed. New York, NY: McGraw-Hill
- Evans W (1959) The electrocardiogram of alcoholic cardiomyopathy. Br Heart J. 21: 445-456. [Crossref]
- Priest RG, Binns JK, Kitchin AH (1966) Electrocardiogram in alcoholism and accompanying physical disease. Br Med J 1: 1453. [Crossref]
- Attar HD, Aboobaker A, Saleem M, Irfan HB, Aman SD. Electrocardiographic and Echocardiographic Abnormalities in Chronic Alcoholics. Al Ameen J Med Sci 10: 10-15.
- Brickner ME, Willard JE, Eichhorn EJ, Black J, Grayburn PA (1991) Left ventricular hypertrophy associated with chronic cocaine abuse. Circulation 84: 1130-1135. [Crossref]
- Gitter MJ, Goldsmith SR, Dunbar DN, Sharkey SW (1991) Cocaine and chest pain: clinical features and outcome of patients hospitalized to rule out myocardial infarction. Ann Intern Med 115: 277-282. [Crossref]
- Zimmerman JL, Dellinger RP, Majid PA (1991) Cocaine-associated chest pain. Ann Emerg Med 20: 611-615. [Crossref]
- Hollander JE, Hoffman RS, Gennis P, Fairweather P, DiSano MJ et al. (1994) Prospective multicenter evaluation of cocaine‐associated chest pain. Acad Emerg Med 1: 330-339. [Crossref]
- Hollander JE, Hoffman RS, Burstein JL, Shih RD, Thode HC et al. (1995) Cocaine-associated myocardial infarction: mortality and complications. Arch Intern Med 155: 1081-1086. [Crossref]
- Weber JE, Chudnofsky CR, Boczar M, Boyer EW, Wilkerson MD et al. (2000) Cocaine-associated chest pain: how common is myocardial infarction? Acad Emerg Med 7: 873-877. [Crossref]
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE (2003) Validation of a brief observation period for patients with cocaine-associated chest pain. N Eng J Med 348: 510-517. [Crossref]
- Kozor R, Grieve SM, Buchholz S, Kaye S, Darke S et al. (2014) Regular cocaine use is associated with increased systolic blood pressure, aortic stiffness and left ventricular mass in young otherwise healthy individuals. PLoS One 9: e89710. [Crossref]
- Maceira AM, Ripoll C, Cosin-Sales J, Igual B, Gavilan M et al. (2014) Long-term effects of cocaine on the heart assessed by cardiovascular magnetic resonance at 3T. J Cardiovasc Magn Reson 16: 26. [Crossref]
- Wijetunga M, Seto T, Lindsay J, Schatz I (2003) Crystal methamphetamine‐associated cardiomyopathy: tip of the iceberg? J Toxicol Clin Toxicol 41: 981-986. [Crossref]
- Haning W, Goebert D (2007) Electrocardiographic abnormalities in methamphetamine abusers. Addiction 102: 70-75. [Crossref]
- Voskoboinik A, Ihle J, Kaye D (2015) Amphetamine-induced cardiomyopathy: Patterns and predictors of recovery. Heart Lung Circ 24: S195.
- Kueh SH, Gabriel RS, Lund M, Sutton T, Bradley J et al. (2016) Clinical characteristics and outcomes of patients with amphetamine-associated cardiomyopathy in South Auckland, New Zealand. Heart Lung Circ 25: 1087-1093. [Crossref]
- Bazmi E, Mousavi F, Giahchin L, Mokhtari T, Behnoush B (2017) Cardiovascular complications of acute amphetamine abuse: cross-sectional study. Sultan Qaboos Univ Med J 17: e31-e37. [Crossref]
- Horowitz SF, Fischbein A, Matza D, Rizzo JN, Stern A et al. (1988) Evaluation of right and left ventricular function in hard metal workers. Br J Ind Med 45:742-746. [Crossref]
- Yoon JH, Lee JS, Jung YS, Min YG, Park JS et al. (2014) Carbon Monoxide Induced Cardiomyopathy: Epidemiology, Clinical Characteristics, and Prognosis. Circ J 78: 1437–1444. [Crossref]
- Cha YS, Kim H, Hwang SO, Kim JY, Kim YK et al. (2016) Incidence and patterns of cardiomyopathy in carbon monoxide-poisoned patients with myocardial injury. Clin Toxicol 54: 481-487. [Crossref]
- Savarese G, Lund LH (2017) Global public health burden of heart failure. Card Fail Rev 3: 7-11. [Crossref]
- Finkel JB, Marhefka GD (2011) Rethinking cocaine-associated chest pain and acute coronary syndromes. In Mayo Clinic Proceedings. 86: 1198-1207. [Crossref]
- Bloom S (1981) Reversible and irreversible injury: Calcium as a major determinant. Cardiac Toxicology 1: 179-199.
- Simonson E, Cady Jr LD, Woodbury M (1962) The normal QT interval. Am Heart J 63: 747-753. [Crossref]
- Amin M, Gabelman G, Karpel J, Buttrick P (1990) Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol 66: 1434-1437. [Crossref]
- Frishman WH (1998) Carvedilol. N Engl J Med 339: 1759-1765. [Crossref]
- Basu S, Senior R, Raval U, van der Does R, Bruckner T, Lahiri A (1997) Beneficial effects of intravenous and oral carvedilol treatment in acute myocardial infarction: a placebo-controlled, randomized trial. Circulation 96: 183-191. [Crossref]
- Maceira AM, Prasad SK, Khan M, Pennell DJ (2006) Normalized left ventricular systolic and diastolic function by steady state free precession cardiovascular magnetic resonance. J Cardiovasc Magn Reson 8: 417-426. [Crossref]
- Maceira AM, Prasad SK, Khan M, Pennell DJ (2006) Reference right ventricular systolic and diastolic function normalized to age, gender and body surface area from steady-state free precession cardiovascular magnetic resonance. Eur Heart J 27: 2879-2888. [Crossref]
- Mahrholdt H, Wagner A, Judd RM, Sechtem U, Kim RJ (2005) Delayed enhancement cardiovascular magnetic resonance assessment of non-ischaemic cardiomyopathies. Eur Heart J 26: 1461-1474. [Crossref]
- Maceira AM, Joshi J, Prasad SK, Moon JCC, Perugini E et al. (2005) Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 111: 195-202. [Crossref]
- Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED et al. (2006) Cardiovascular magnetic resonance, fibrosis and prognosis in dilated cardiomyopathy. J Am Coll Cardiol 48: 1977-1985. [Crossref]