Objective: We examined the effects of tofacitinib on vascular remodeling-including intraluminal myofibroblast proliferation-in a murine model of allergic vasculitis with eosinophil infiltration.
Methods: We exposed C57BL/6 mice to ovalbumin (OVA) and aluminum and exposed the positive controls to aerosolized OVA daily for 7 days. We administered tofacitinib (0.1 g/kg) intraperitoneally to the other group of mice in parallel with daily exposure to aerosolized OVA for 7 days. On days 3 and 7, we performed bronchoalveolar lavage (BAL) and excised the lungs for pathological analysis. We determined histological findings of vasculitis and measured concentrations of IL-4, IL-5, IL-6, IL-13, and TGF-β in the BAL fluid (BALF). We then performed a semi-quantitative analysis of pathological changes in the pulmonary arteries according to the severity of vasculitis.
Results: In mice treated with tofacitinib, the number of eosinophils in the BALF was reduced significantly when compared with that from control mice treated with just OVA. In the tofacitinib-treated group, TGF-β concentration in the BALF was significantly decreased, pathological scores were significantly reduced, and the number of intraluminal myofibroblasts in the pulmonary arteries was lower than in the control group.
Conclusion: Tofacitinib reduced eosinophil infiltration and decreased IL-4, IL-5, IL-6, IL-13, and TGF-β in this murine model of allergic vasculitis with eosinophil infiltration. Our data suggest that tofacitinib suppresses pulmonary vascular remodeling through JAK inhibition.
tofacitinib, JAK, allergic vasculitis, IL-6, myofibroblast
IL-4: Interleukin-4; IL-5: Interleukin-5; IL-13: Interleukin-13; TGF β: Transforming growth factor- β; TNF α: Tumor Necrosis Factor- α
The vasculitides are heterogeneous conditions characterized by their ability to cause various type of vessel inflammation with or without necrosis. The antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV) are three separate diseases with necrotizing vasculitis – granulomatosis with polyangiitis (GPA; formerly known as Wegener’s granulomatosis), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA; previously known as Churg-Strauss syndrome). The factors characterizing EGPA are bronchial asthma, eosinophilic pulmonary infiltration, peripheral blood eosinophilia, and systemic necrotizing vasculitis of small- and medium-sized vessels with or without granulomas [1-3]. The vasculitis that accompanies EGPA causes serious damage to the skin, nerves, digestive canal, lungs, and other organs.
Systemic glucocorticoids are the first line of treatment for EGPA, and most patients rely on continuous glucocorticoid therapy [4].
Treatment of EGPA with Mepolizumab, an anti-interleukin-5 monoclonal antibody, has been associated with significantly high remission rates compared with those in the placebo-treated group [5]. However, these rates fell short of the required 53% remission. Although not conclusive, the results of this study suggested that mepolizumab was an effective suppressor of Th2 cytokines (IL-4, IL5, and IL-13).
The etiology underlying the development of EGPA remains unclear. In EGPA, eosinophils are the most dominant cells in the blood and extravascular tissues and are known to release cytotoxic products such as major basic proteins, eosinophil-derived neurotoxins, and cytokines-including TGF-β [6,7].
We previously reported a murine model of pulmonary allergic vasculitis, in which the pathology was induced by repeated inhalation of OVA [8]. We demonstrated that the small pulmonary arteries of the OVA-exposed mice were occluded due to accumulation of myofibroblasts and collagen deposition. As such, the lung pathology in this murine model is similar to that observed in EGPA patients.
Via activation downstream of the signal transducer and activator of transcription (STAT), phosphatidylinositol 3-kinase (PI3K), and mitogen-activated protein kinase pathways, the Janus kinase (JAK) pathway plays an important role in cytokine receptor-mediated signal transduction. Tofacitinib selectively inhibits JAK1 and JAK3 and suppresses the production of both Th2 and inflammatory cytokines [9]. It has proved highly effective on rheumatoid arthritis, psoriatic arthritis and ulcerative colitis [9]. The efficacy of tofacitinib in the treatment of hypereosinophilic syndrome (HES) and asthma has also been demonstrated [9-11]. In this regard, tofacitinib holds promise as a therapeutic drug for other allergic diseases, including vasculitis.
However, few studies have reported the effects of tofacitinib in the treatment of EGPA. We therefore examined the effects of the JAK1 and JAK3 inhibitor, tofacitinib, on the histological changes in our model of allergic pulmonary vasculitis. Our current results provide a framework for future studies aimed at establishing a therapy for allergic vasculitis.
Animals: We purchased female C57/BL6 mice (aged 8 weeks) from Japan SLC (Shizuoka, Japan). We housed the mice under specific pathogen-free conditions following a 12-hour light-dark cycle, fed them a standard laboratory diet, and gave them water ad libitum. In our experiments we did not synchronize the estrous cycle of the female mice. We regarded the obtained results as the average from all female mice in various stages of the estrous cycle. We performed all experiments described in this study according to the guidelines for the care and use of experimental animals as determined in 1987 by the Japanese Association for Laboratory Animals Science.
Administration of tofacitinib: Tofacitinib was synthesized by Med Chem Express (USA). We administered 2 mg of tofacitinib (10 mg/mL) suspended in 0.025% Tween 20 methylcellullose (Wako, Japan) as a solid dispersion through a flexible feeding tube (for mice, size: φ 0.9×70 mm, CL-4563) (CLEA Japan, Inc). We therefore reliably delivered tofacitinib (100 mg/kg) to the stomach of each mouse. We administered tofacitinib once daily during 1 week of OVA inhalation. Appropriate doses of tofacitinib were determined by performing a dose-response evaluation with the drug using TGF-β as the readout (Supplementary Figure 1).
Immunization and aerosolization protocol: We sensitized the mice according to the methods described in a previous paper [12]. We placed each mouse of the positive control group in a plastic chamber (10 cm×15 cm×25 cm) and exposed them to aerosolized OVA (5 mg/mL in 0.9% saline) for 1 h daily until day 7, as shown in figure 1. We administered the aerosolized OVA using a Pulmo-Aide Compressor/Nebulizer (Devilbiss) (Sunrise Medical HHG, Inc. Somerset, PA, USA) at a flow rate of 5–7 L/min. We exposed the tofacitinib-treated group of mice to aerosolized OVA for 1 h daily until day 7 and in parallel, administered tofacitinib as described above (Figure 1).
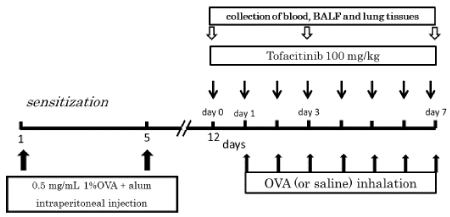
Figure 1. Experimental protocol. We sensitized mice on days 0 and 5 of the protocol by an intraperitoneal injection of 0.5 mL aluminum hydroxide-precipitated antigen containing 8 μg OVA. We exposed the mice daily to aerosolized OVA produced by a Pulmo-Aide Compressor/Nebulizer at a flow rate of 5–7 L/min in a plastic chamber. We administered tofacitinib (100 mg/kg) orally once daily over the course of one week
Collection and measurement of specimens: After we had exposed the mice to aerosolized OVA every day over a period of 1 week in the presence or absence of tofacitinib, we sacrificed them 24 h after the final inhalation (either day 3 or day 7) by cutting the femoral artery and collected the BALF and lung tissue, as shown in figure 1. As a control, we collected samples after exposing mice to aerosolized saline instead of OVA every day over 1 week. We collected and analyzed the BALF according to the methods described in a previous paper [8]. Numbers of cells recovered per mouse were expressed as the mean and standard deviation (SD) for each treatment group. After harvesting the BALF, we fixed the lungs with 10% neutral-buffered formalin and embedded them in paraffin. We then stained 3 μm-thick sections with hematoxylin and eosin (HE) and Elastica-Masson (EM).
Semi-quantitative analysis of pathological changes in the pulmonary arteries: We assessed the extent of the histological changes in the pulmonary arteries as described previously [13]. In brief, 3 μm-thick sections were stained with HE and EM, and evaluated by light microscopy. We determined histological scores according to the criteria outlined previously [13]. We scored five vessels of the lung tissue sections for each mouse, with diameters ranging from 20–50 μm, and reported the average value for each mouse as the histological index.
Immunohistochemical staining for smooth muscle actin and Ki-67: For the immunohistochemical staining, we adopted the biotin-streptavidin system using a Histofine Kit (Nichirei, Tokyo, Japan). We used anti-α smooth muscle actin (α-SMA) (Spring Bioscience, Fremont, CA, USA) and anti-mouse Ki-67 (Biolegend, San Diego, CA, USA), as previously reported [8,13].
Cytokine measurement: Using a commercially available enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA), we assessed BALF for the measurement of IL-4, IL-5, IL-6, IL-13, TNF-α and TGF-β concentrations.
Counting Ki-67 positive cells: We counted cells with brown-colored nuclei as Ki-67 positive. We also counted cells that had accumulated in the intraluminal space in the pulmonary arteries. We calculated the ratio of Ki-67-positive cells as the number of brown nuclei/number of whole cells stained with hematoxylin (blue nuclei) in five arteries of each mouse.
Statistical analysis: We used the Mann–Whitney U Test in the analysis of results. All values are expressed as means±SD. We considered values of P<0.05 to be statistically significant.
Effects of tofacitinib on total cell number and eosinophil numbers in BALF: In the tofacitinib-treated group with OVA inhalation, the total cell number and the number of eosinophils in BALF on day 7 increased dramatically compared with mice exposed only tofacitinib (Figure 2). On day 7, the total cell number and the number of eosinophils in the BALF of the tofacitinib-treated mice decreased significantly compared with those of the control mice (Figure 2).
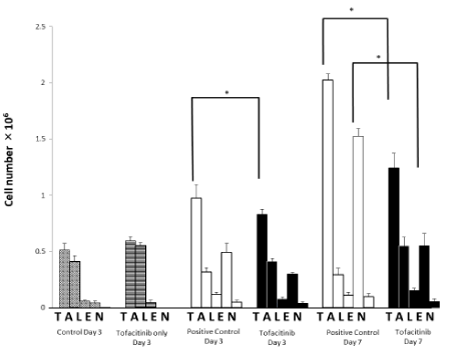
Figure 2. The effect of tofacitinib on differential count of leukocytes in the BALF. Hatched columns: OVA-sensitized mice (n=9) exposed to saline and treated with tofacitinib (control). Open columns: OVA-sensitized mice (n=9) exposed to OVA (positive control). Closed columns: OVA-sensitized mice (n=9) exposed to OVA and treated with tofacitinib. T: total cells; A: alveolar macrophages; L: lymphocytes; E: eosinophils; N: neutrophils. Data are means±SD, and ∗p<0.05
Cytokine concentration in BALF: On days 3 and 7, we measured cytokine concentrations in the BALF collected from the lungs of mice treated with OVA inhalation and tofacitinib. As shown in Figures 3A, 3D, 4A and 4B, the concentrations of IL-5, IL-6, IL-13, TGF-β in BALF from OVA-sensitized mice that were not treated with tofacitinib (i.e., the positive control group) peaked on day 3 and decreased on day 7. The concentrations of IL-4 and TGF-β in the BALF of the positive control group were highest on day 7 after OVA inhalation. On days 3 and 7, the concentrations of IL-4 and IL-6 in the BALF of the tofacitinib-treated group were significantly lower than those in the BALF of the positive control group. Tofacitinib treatment significantly suppressed the peak concentration of IL-5 in the BALF on day 3. The concentration of IL-13 in BALF did not significantly differ between the two groups. However, IL-13 concentration in the tofacitinib-treated group tended to be low when compared with that of the positive control on day 3.
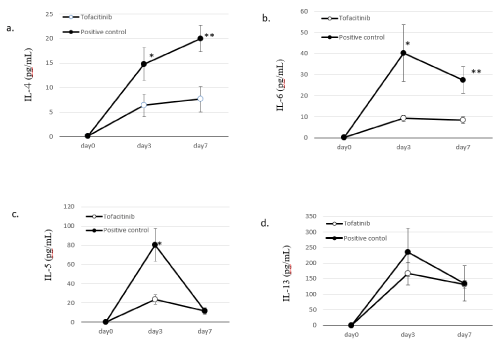
Figure 3. Cytokine concentration in the BALF. Figure A, IL-4; Figure B, IL-6; Figure C, IL-5; Figure D, IL-13. Closed circles: OVA-sensitized mice exposed to OVA (positive control). Open circles: OVA-sensitized mice exposed to OVA and treated with tofacitinib. Data are means±SD, and ∗p<0.05, ∗∗p<0.001
TGF-β concentration in the BALF of the positive control group increased until day 7 after OVA inhalation, whereas that of the tofacitinib-treated group decreased significantly on day 7 (Figure 4A). In contrast, the concentration of TNF-α in the BALF on day 7 was significantly higher in the tofacitinib-treated group than in the positive control group (Figure 4B).
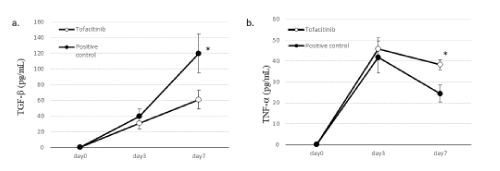
Figure 4. Cytokine concentration in the BALF. Figure A, TGF-β; Figure B, TNF-α, Closed circles: OVA-sensitized mice exposed to OVA (positive control). Open circles: OVA-sensitized mice with exposure to OVA and treated with tofacitinib. Data are means±SD, and ∗p<0.05
Effects of tofacitinib on the histological changes in pulmonary arteries: Almost all small pulmonary arteries in mice from the positive control group were highly obstructed due to the accumulation of cellular infiltrates, which included proliferating myofibroblasts (Figure 5A, 5C). Additionally, we observed increased collagenous materials (Figure 5A, 5C). In contrast, there was a marked reduction in the histological changes of the small pulmonary arteries in the tofacitinib-treated mice when compared with those of the positive controls (Figure 5B, 5D).
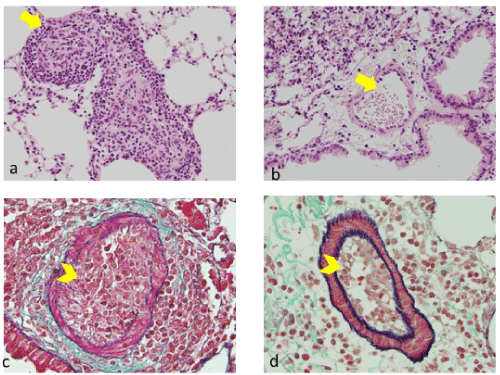
Figure 5. Effects of tofacitinib on allergic pulmonary vascular pathological changes evaluated by hematoxylin and eosin (HE) staining and Elastica-Masson (EM) staining. A: A pulmonary artery totally occluded by intraluminal myofibroblasts (yellow arrow) in the OVA-sensitized mice with exposure to OVA, on day 7 (HE Staining). B. Absence of intraluminal myofibroblast accumulation (yellow arrow) in OVA-sensitized mice exposed to OVA and treated with tofacitinib (Day 7, HE Staining). On day 7 in the positive control, the intraluminal space of the pulmonary artery was occupied (yellow arrowhead) and greenish collagenous material was present. The vascular wall was partially disrupted (EM staining). D: We did not observe intravascular myofibroblasts in the pulmonary artery (yellow arrowhead) and observed only blood cells in the lumen on day 7 in the tofacitinib-treated mice (EM staining)
On day 7, we performed semi-quantitative analysis of the histological vascular changes in the lungs of positive controls and tofacitinib-treated mice. The scoring in this analysis was according to the severity index described in the Methods section.
In the tofacitinib-treated group, the severity index was significantly reduced when compared to that in the positive control group (Figure 6).
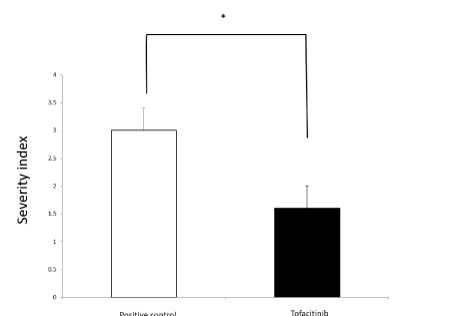
Figure 6. Effects of tofacitinib on the severity of vascular changes. Open circles: histological scores of the positive control on day 7 (n=9). Closed circles: the tofacitinib-treated mice on day 7 (n=9). We present data as mean±SD, and ∗p<0.05
Though α-SMA staining analysis showed myofibroblasts occupied the intravascular space of the pulmonary arteries in the positive control group (Figure 7A-7C), this cell population was absent in the tofacitinib-treated group (Figure 7D).
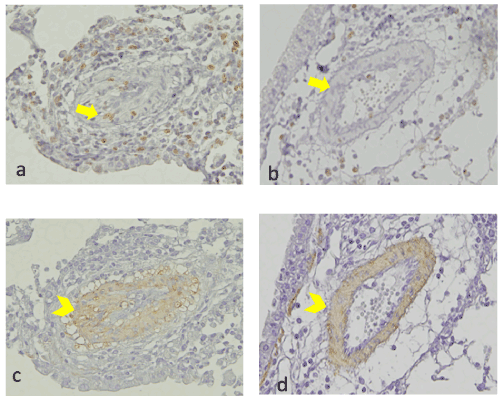
Figure 7. Immunostaining for Ki-67 and α-SMA. a: We observed Ki-67-positive cells in myofibroblasts that had accumulated in the intraluminal space of the pulmonary artery in the OVA-sensitized mice with exposure to OVA, on day 7 (yellow arrow). b: On day 7, Ki-67-positive cells were very sparse in the intraluminal space of OVA-sensitized mice exposed to OVA and treated with tofacitinib (yellow arrow) c: On day 7, the intraluminal myofibroblasts in the OVA-sensitized mice exposed to OVA were positively stained with anti-α-SMA (yellow arrow head). d: Smooth muscle cells in the vascular wall were also positive (yellow arrowhead). We did not observe intraluminal myofibroblast accumulation in the OVA-sensitized mice exposed to OVA and treated with tofacitinib. Only the smooth muscle cells in the vascular wall were positive (yellow arrowhead)
We performed immunohistochemistry for Ki-67 to detect proliferating cells in pulmonary vasculitis [14]. In the positive control group, we detected the expression of Ki-67 in the intraluminal myofibroblasts (yellow arrow) and cells in the vascular wall (Figure 7A). In contrast, Ki-67-expressing cells were sparse in the pulmonary vascular tissue of the tofacitinib-treated group (Figure 7B). Semi-quantitative analysis revealed that there were significantly less intraluminal Ki-67-positive cells in the tofacitinib-treated mice when compared with positive control mice (Figure 8).
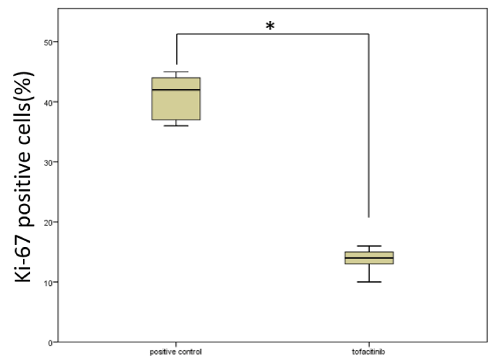
Figure 8. Effects of tofacitinib on the number of Ki-67-positive cells. Intraluminal Ki-67-positive cells of the OVA-sensitized mice exposed to OVA (positive control) (n=5) or treated with tofacitinib (n=5) on day 7. We present data as mean±SD and *p<0.01
Our study demonstrated that tofacitinib ameliorated vasculitis in the pulmonary arteries of OVA-exposed mice. Tofacitinib also reduced the number of eosinophils and the concentration of IL-4, IL-5, IL-6, and TGF-β1 in the BALF of OVA-sensitized mice.
We observed a marked reduction in the eosinophil number in the BALF of the tofacitinib-treated group when compared with that of the positive control mice. We assumed that the reduction in eosinophil counts in the BALF was caused by tofacitinib via at least two mechanisms. First, the drug reduces the levels of Th2 cytokines such as IL-4, IL-5, and IL-13. These cytokines are positive regulators of eosinophilopoiesis in bone marrow, the recruitment of eosinophils from the bone marrow to the inflammatory site, and the survival and activation of eosinophils. Second, tofacitinib inhibits JAK-STAT intracellular signal transduction through the activity of eosinophilic-promoting cytokine.
Tofactinib reduces the level of cytokines by inhibiting Th2 cell differentiation. King et al. reported that tofacitinib and another JAK inhibitor, ruxolitinib, decreased blood eosinophil counts in patients with HES [10]. Pathological amounts of eosinophil-promoting Th2 cytokines-such as IL-4, IL-5, and IL-13-are involved in the pathogenesis of HES, and are thought to play critical roles in eosinophilopoiesis, chemotaxis, survival, and activation of eosinophils. Kudlacz et al. reported that IL-4 knockout mice showed a marked reduction in pulmonary eosinophilia, and that tofacitinib reduced the number of BAL eosinophils in OVA-sensitized mice [15]. IL-4 plays an essential role in the differentiation of Th2 lymphocytes, and JAK1/JAK3 signaling is critical for its production. Impaired Th2 lymphocyte differentiation elicited by tofacitinib might cause a decrease in a wide range of Th2 cytokines, including IL-4, IL-5, and IL-13.
Tofacitinib can also directly inhibit cytokine-dependent JAK signaling triggered by IL-4, IL-5 and IL-13. IL-4 plays a critical role in eosinophilopoiesis in the bone marrow, IgE production, and eosinophil accumulation in the pulmonary tissue [16]. IL-5 promotes eosinophil survival, differentiation, and migration [17], while IL-13 induces recruitment of eosinophils to the airways [18]. Although it is a non-selective JAK inhibitor, tofacitinib preferentially inhibits JAK1 and JAK3, rather than JAK2. In this regard, activation of the IL-4 receptor is potently inhibited by tofacitinib because JAK1 and JAK3 combination are involved in the phosphorylation of the intracellular portion of the IL-4 receptor [19]. On the other hand, the IL-5 receptor utilizes JAK2/JAK2 combination for its phosphorylation. Tofacitinib may therefore be a less potent inhibitor of IL-5 receptor signaling when compared to its effects on IL-4-driven events [20].
IL-6 plays important roles in inflammation, hematopoiesis, and the immune response [21], and is present in high concentrations in the airways of asthmatics and in mice used for modeling asthmatic disease [22,23]. IL-6 reportedly promotes the differentiation of Th2 cells by enhancing endogenous IL-4 production by CD4 T cells [24]. Our study demonstrated a decrease in IL-6 concentrations in the BALF of OVA-sensitized mice treated with tofacitinib after inhalation of OVA. IL-6 is produced not only by immune cells (for instance, macrophages, dendritic cells, mast cells, and B cells) but also by non-immune cells such as epithelial cells and fibroblasts [25]. Because we used OVA inhalation as the trigger of allergic reactions in the lung, we assumed that this stimulated airway epithelial cells to produce significant levels of IL-6, although airway dendritic cells and macrophages are other possible sources of this cytokine. Although we speculate that IL-6 production from these cells might be inhibited by tofacitinib, further studies will be required in order to determine whether tofacitinib targets other cell types.
Several inflammatory diseases-including rheumatoid arthritis, inflammatory bowel disease, vasculitis, and several types of cancer-have been associated with IL-6 [26]. Furthermore, a recent study has shown that an increased serum IL-6 level occurred parallel to endothelial dysfunction in EGPA [27], suggesting it may play an important role in this disease. A previous study demonstrated that blocking the IL-6R with an antibody led to reduced Th2 cytokines and eosinophils in the lungs of OVA-sensitized mice [28]. In our current experiment, tofacitinib might not only have decreased the level of IL-6 in the lung, but may also have inhibited the actions of IL-6 by blocking JAK1/JAK2 activation downstream of the intracellular portion of the IL-6 receptor [29,30].
TGF-β is a critical mediator of tissue fibrosis, including pulmonary fibrosis and the subepithelial fibrosis that accompanies asthma [31-33]. TGF-β not only promotes the proliferation and activation of myofibroblasts but also induces the exaggerated deposition of the extracellular matrix-including collagen and fibronectin in fibrotic tissues [32,33]. We previously reported a marked increase in TGF-β concentration in the BALF of OVA-sensitized mice after OVA exposure [34]. The present study demonstrated that tofacitinib decreased TGF-β concentration in the BALF of the OVA-sensitized mice after OVA exposure, and attenuated vascular remodeling of the pulmonary artery and the proliferation of myofibroblasts. Amongst others, TGF-β is produced from epithelial cells, immune cells such as macrophages and lymphocytes, and mesenchymal cells such as fibroblasts. Eosinophils are a major source of TGF-β in the lungs of mice used for modeling inflammatory diseases [13]. It is therefore reasonable to assume that the tofacitinib-dependent decrease in eosinophils is one of the major causes of reduced TGF-β concentrations in the BALF of the mice with eosinophilic vasculitis.
In summary, our data show that tofacitinib relieves the symptoms of allergic pulmonary vasculitis through a series of events. Tofacitinib suppresses the production of IL-6 and Th2 cytokines such as IL-4, IL-5, and IL-13 from CD4+ T lymphocytes and IL-6. The attenuated cytokine production leads to reduced eosinophil accumulation in the lungs, which in turn reduces the local TGF-β concentration. It is likely the lack of TGF-β that ultimately inhibits vascular remodeling, since the cytokine is required for this process.
- Savige J, Gillis D, Benson E, Davies D, Esnault V, et al. (1999) International consensus statement on testing and reporting of antineutrophil cytoplasmic antibodies (ANCA). Am J Clin Pathol 111: 507-513.
- Jennette JC, Falk RJ (1995) Clinical and pathological classification of ANCA-associated vasculitis: what are the controversies? Clin Exp Immunol 101: 18-22.
- Hellmich B, Gross WL (2004) Recent progress in the pharmacotherapy of Churg-Strauss syndrome. Expert Opin Pharmacother 5: 25-35.
- Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, et al. (2016) EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis.Ann Rheum Dis75: 1583-1594. [Crossref]
- Wechsler ME, Akuthota P, Jayne D (2017) Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med 376: 1921-1932.
- Klion AD, Bochner BS, Gleich GJ, Nutman TB, Rothenberg ME (2006) The hypereosinophilic syndromes working group: approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J Allergy Clin Immunol 117: 1292-1302.
- Keogh KA, Specks U (2006) Churg-Strauss syndrome: update on clinical, laboratory and therapeutic aspects. Sarcoidosis Vasc Diffuse Lung Dis 23: 3-12.
- Yamauchi K, Sasaki N, Niisato M, Kamataki A, Shikanai T, et al. (2010) Analysis of pulmonary allergic vasculitis with eosinophil infiltration in asthma model of mice. Exp Lung Res 36: 227-236.
- Schwartz DM, Kanno Y, Villarino A, Daniella M, Gadina M, et al. (2017) JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 16: 843-862.
- King B, Lee AI, Choi J (2017) Treatment of hypereosinophilic syndrome with cutaneous involvement with the JAK inhibitors tofacitinib and ruxolitinib. J Invest Dermatol 137: 951-954.
- Rimar D, Alpert A, Starosvetsky E, Rosner I, Slobodin G, et al. (2016) Tofacitinib for polyarteritis nodosa: a tailored therapy.Ann Rheum Dis75: 2214-2216. [Crossref]
- Ohkawara Y, Lei XF, Stampfli MR, Marshall JS, Xing Z, et al. (1997) Cytokine and eosinophil responses in the lung, peripheral blood and bone marrow compartments in a murine model of allergen- induced airways inflammation. Am J Respir Cell Mol Biol 16: 510-520.
- Koizumi R, Sasaki N, Nakamura Y, Suzuki N, Sawai T, et al. (2014) Rapamycin attenuates pulmonary allergic vasculitis in murine model by reducing TGF-β production in the lung. Allergol Int 63: 457-466.
- Bullwinkel J, Baron-Luhr B, Lüdemann A, Wohlenberg C, Gerdes J, et al. (2006) Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells.J Cell Physiol206: 624-635. [Crossref]
- Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P (2008) The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol 582: 154-161.
- Rankin JA, Picarella DE, Geba GP, Temann UA, Prasad B, et al. (1996) Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: lymphocytic and eosinophilic inflammation without airway hyperreactivity.Proc Natl Acad Sci U S A93: 7821-7825. [Crossref]
- Wills-Karp M (1999) Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol 17: 255-281.
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, et al. (1998) Interleukin-13: central mediator of allergic asthma.Science282: 2258-2261. [Crossref]
- Elwood F, Witter DJ, Piesvaux J, Kraybill B, Bays N, et al. (2017) Evaluation of JAK3 biology in autoimmune disease using a highly selective, irreversible JAK3 inhibitor. J Pharmacol Exp Ther 361: 229-244.
- Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, et al. (2017) JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 17: 78.
- Tanaka T, Narazaki M, Kishimoto T (2014) IL-6 in inflammation, immunity, and disease.Cold Spring Harb Perspect Biol6: a016295. [Crossref]
- Marini M, Avoni E, Hollemborg J, Mattoli S (1992) Cytokine mRNA profile and cell activation in bronchoalveolar lavage fluid from nonatopic patients with symptomatic asthma. Chest 102: 661-669.
- Paul B, Mishra V, Chaudhury B, Awasthi A (2009) Status of Stat3 in an ovalbumin-induced mouse model of asthma: analysis of the role of Socs3 and IL-6. Int Arch Allergy Immunol 148: 99-108.
- Diehl S, Chow CW, Weiss L, Palmetshofer A, Twardzik T, et al. (2002) Induction of NFATc2 expression by interleukin 6 promotes T helper type 2 differentiation.J Exp Med196: 39-49. [Crossref]
- Hirano T (1998) Interleukin 6 and its receptor: ten years later.Int Rev Immunol16: 249-284. [Crossref]
- Calabrese LH, Rose-John S (2014) IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol 10: 720-727.
- Pacholczak R, Bazan-Socha S, Iwaniec T, Zareba L, Kielczewski S, et al. (2019) Endothelial dysfunction in patients with eosinophilic granulomatosis with polyangiitis. Clin Rheumatol 38: 417-424.
- Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, et al. (2005) The IL-6R alpha chain controls lung CD4+CD25+Treg development and function during allergic airway inflammation in vivo. J Clin Invest 115: 313-325.
- Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P, et al. (1989) Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med 170: 655-663.
- Smolen JS, Landewe R, Bijlsma J, Burmester G, Chatzidionysiou K, et al. (2017) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 76: 960-977.
- Minshall EM, Leung DY, Martin RJ, Song YL, Cameron L, et al. (1997) Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol 17: 326-333.
- Raghow B, Irish P, Kang AH (1989) Coordinate regulation of transforming growth factor beta gene expression and cell proliferation in hamster lungs undergoing bleomycin-induced pulmonary fibrosis. J Clin Invest 84: 1836-1842.
- Broekelmann TJ, Limper AH, Colby TV, McDonald JA (1991) Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci USA 88: 6642-6646.
- Suzuki N, Sasaki N, Utsumi Y, Nagashima H, Nakamura Y, et al. (2013) Effects of imatinib mesylate onpulmonary allergic vasculitis in a murine model. Int J Rheum Dis 16: 455-462.