Abstract
Thévenard's disease is a rare familial ulcero-mutilating acropathology (UMA), autosomal dominant involving the peripheral nervous system. It typically located especially on the feet and characterized by cutaneous painless ulcerations and bone mutilations leading to secondary osseous deformations and osteoarticular destruction (mimicking Charcot’s food) associated with distal peripheral sensory neurological impairment, initially affecting the lower limbs with sock-like paresthesia and then extending to the upper limbs. The worsening of trophic disorders is progressive. The diagnosis of this condition can be challenging due to the rarity of the disease often unrecognized.
Keywords
thevenard's disease, familial ulcero-mutilating acropathy, osteo-articular destruction,; sensory neuropathy
Introduction
Thevenard's disease (TD) is a rare familial ulcerative-mutilating acropathy (UMA) associated with distal peripheral sensory neurological impairment, initially affecting the lower limbs with sock-like paresthesia and then extending to the upper limbs. One of the first clinical descriptions comes from Thévenard [1]. Its adverse course most often results in amputation, causing severe disability.
TD is classified among the hereditary sensory and autonomic neuropathy type I corresponding a motor and sensory nerve disorder characterized by the early loss of deep sensation and the onset of paresthesia, primarily in the form of sudden attacks of pain [2-4]. It’s mode of transmission is autosomal dominant, but recessive forms have been recently described [2-4]. Genetic confirmation involves identifying mutations in the SPTLC1 gene. The loss of deep sensation quickly leads to painless lesions of the limbs which, if not recognized early, may require amputation, the ultimate treatment for osteitis. TD is characterized by axonal degeneration of unmyelinated fibers and small-caliber myelinated fibers in the cords, roots and posterior ganglia of the spinal cord (Van Bogaert and Denny-Brown) [4].
Case presentation
This is the case of a 47-year-old female patient with no particular medical or surgical history—aside from trophic disorders of the ear at the age of 27—who was investigated for a perforating plantar ulcer in the heel with an adverse course leading to phlegmon in the metatarsals despite local care and appropriate broad-spectrum antibiotic therapy.
The patient’s parents we aren’t a consanguineous couple with negative family history (particularly any similar of trophic acropathy cases in family was reported). Physical examination confirmed a two deep and large ulcer with local signs of infection and widespread necrosis were observed in the soles of right foot and a cubic deformity of the left foot (Figure 1, 2 and 3).
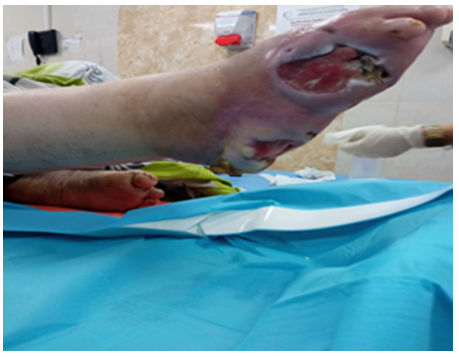
Figure 1. Ulceration of the Right foot
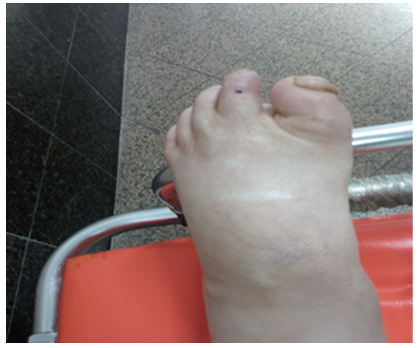
Figure 2. Cubic deformity of the left foot
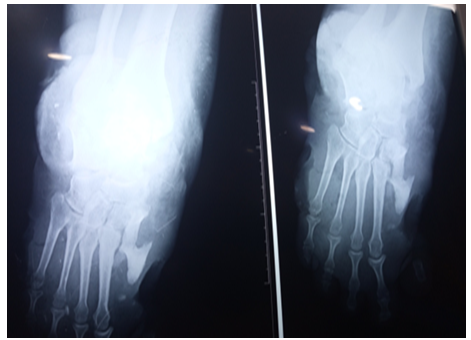
Figure 3. X-ray showing osteitis
Neurologically examination showed bilateral and symmetrical hypoesthesia involving the extremities and decreased sensation to pain and temperature in her lower limb and diminishes of the Achilles reflexes were. No cataract deafness or signs of cranial nerve damage or signs of myelitis were found. The laboratory assessment was normal, particularly with regard to infection and metabolism.
Fasting blood glucose and HbA1c levels, kidney function, lipid profile, and liver function tests were normal. There was no inflammatory syndrome (normal ESR, CRP, and serum protein electrophoresis profile…) and the complete blood count, TSH us, and electrolytes were unremarkable. There was no indication of a systemic cause markers of autoimmunity (antinuclear antibodies, antineutrophil cytoplasmic antibodies antiphospholipid antibodies and rheumatoid factor were negative). Cardiovascular exploration was unremarkable (vascular Doppler echo of the lower limbs, supra-aortic trunks, echo Doppler cardiography).
The electromyogramme showed a sensory peripheral neuropathy. Cubic deformity of the left foot and osteitis were observed in X-ray of the foot [5]. At the end of his explorations no specific pathology was detected to explain these torpid lesions. The diagnosis of acrodystrophic neuropathy linked to TD was established based on the distal peripheral sensory neurogenic syndrome and the result of the electromyogram and after exclusion in the others etiologies (diabetes, Charcot-Marie-Tooth disease, Bureau and Barriere’s Disease linked to alcoolic intoxication, amyloidosis...) [6,7]. The treatment was limited to local care, bandages, immobilization and antibiotic therapy. Despite the local care and the appropriate broad-spectrum antibiotic therapy, and dump shoes we deplored a torpid progression of the lesions resulted in osteitis and warranted elective amputation of right mid-leg (Figure 1, 2 and 3) [7].
Results and discussion
TD is a purely neurological disorder causing torpid ulcerations and is classified among hereditary sensory and dysautosomal neuropathies of type 1. TD constitutes a complex and rare pathology characterized by the progressive degeneration of sensory neurons causing torpid ulcerations. The mode of transmission of TD is autosomal dominant, but recessive forms have been described. In our case, we observed thermoalgesic sensitivity disorders with painless ulcers located on the soles of the feet, progressing to perforating plantar ulcers, later complicating with osteitis and then amputation down to the mid-leg due to resistance to treatment this despite multidisciplinary support many specialties (orthopedist, dermatologist, internist….).
Our case remained sporadic. This rare condition is often misunderstood and a source of diagnostic error. A few Maghreb and African publications are reported in the literature [8,9]. Algerian authors reported a family case (8) and some authors signaling hand involvement in TD [10]. The literature reported also an exceptional dramatic case of TD of a Syrian 6 years’ old female child, with no family history, characterized by the extension to the upper limbs complicated by lytic lesion in the radial bone and loss of both legs due to gangrenous ulcers and self-amputations. This case report was remarkable by the association with congenital hypodontia. [11].
Conclusion
TD is rare and disabling and invalidating disease with high morbidity [12]. Treatment is purely preventive in the absence of curative treatment based on lifestyle changes, foot off-loading, and psychological support. The outcome of TD was often chronic and have poor prognosis. The involvement of the upper limb reported by some authors is exceptional and occurs later after the involvement of the lower limbs [10-12].
Conflicts of interest
The author declares no conflict of interest.
References
- Thevenard AL (1953) Acropathie ulcero-mutilantefamiliale. Acta Neurol Belg 52: 1-24.
- Alberca R, Albert P, Arjona V, Miranda-Nieves G (1973) Sporadic acrodystrophic neuropathy. J Neurol Sci 20: 85-95. [Crossref]
- Hicks EP (1922) Hereditary perforating ulcer of the foot The Lancet, London.
- Denny-Brown D (1951) Hereditary sensory radicular neuropathy. J Neurol Neurosurg Psychiatry14: 237-252. [Crossref]
- Allmann KH, Leu H, Burg G, Hodler J (1996) Hereditary sensory and autonomic neuropathy type I (Thévenard’s disease). Skeletal Radiol 25: 501-504. [Crossref]
- Borut Nikolić, Milan Mišović, Miodrag Zorić, Dušica Matović, Aleksandra Aleksić (2010) Thevenard’s disease - a hereditary sensory and autonomic neuropathy type I. Serb J Dermatol Venereol 2: 59-64
- Chabli H, Akhdari N, Hocar O, Amal S (2015) Acrodystrophic neuropathy: Four case reports and review of the litterature. Médecine et chirurgie du pied 31: 59-63.
- N Amiri, M Necib, H Filleli, M Louanchi, N Toubal () L’acropathie ulcéro-mutilante familiale : Une affection rare et souvent méconnue ! service de Neurologie, CHU Ibn Sina, Annaba Algérie.
- Allognon C, Akagha C, OU BB, LF SL, Boguikouma JB (2022) Takayasu's disease: an underdiagnosed entity in Sub-Saharan Africa. Report of five Gabonese cases. Med Trop Sante Int 2: mtsi-v2i3. [Crossref]
- Facca S, Choughri H, Liverneaux P (2006) Hand involvement in Thevenard's disease: A new" phlegmonous" form. An exceptional case report. Chir Main 25: 175-178. [Crossref]
- Turk T, Haitham Al Husseini MH, Al Assil H (2020) A recessive case of Thévenard’s disease, aka. ulcero-mutilating acropathology syndrome with hypodontia. Oxf Med Case Reports 7: omaa055. [Crossref]
- Sana S, Mourad F, Nadia L, Soumaya B, Houda M, et al. (2014) L’amputation Une issue fatale pour Une Maladie rare. L’acropathie Ulceromutilante De Thevenard. Tunis Med.