Herpes simplex virus (HSV) infects epithelial cells and establishes lifelong latency in neuronal nuclei of the regional ganglion. Although active replication of HSV can be treated effectively with anti-HSV drugs, control of the reactivated HSV in the regional ganglion is a difficult task: Reactivated HSV in the trigeminal ganglion (TG) comes down via axon and infect the cornea Some patients eventually develop vision threatening herpetic stromal keratitis (HSK) that is the second leading cause of corneal blindness after trauma. Corneal herpes continues to cause blindness in developed countries. In this review, we describe initial attachment of HSV, entry and infection at peripheral epithelial cells, establishment of latency in neuron, reactivation and egress, corneal infection and immunopathology of HSK. We also discuss prevention and treatment of the disease.
Worldwide, more than 1 billion individuals carry a latent HSV-1 in sensory neurons, predominantly in the trigeminal ganglion (TG) [1,2]. Primary infection of HSV-1 occurs in the orolabial mucosa without any symptom [1-3]. When overt disease is elicited, infected epithelial cells are eliminated rapidly by the host immunity (4). When the infected area is innervated with the trigeminal nerve, HSV becomes latent in the TG [5,6]. Axons consist of microtubules oriented toward the plus-end terminus. Entering viruses are transported retrograde via microtubules by dynein while kinesin conveys cargos anterogradely [7-9]. This initial movement develops lifelong HSV-1 latency in nerve growth factor (NGF) reactive A5+ neurons while HSV-2 in contrast is latent in KH10+ neurons [10,11]. The majority of infected neurons survive without apoptosis or necrosis so that sensitivity of the corneal surface remains unchanged during HSV latency in TG [12]. Reactivated HSVs are frequently detectable in tears, saliva or in secretions of the genital tract. These are major routes of virus spread and the source of primary infection of HSV [13]. A small population of people have developed recurrent diseases such as ulcers in the oral mucosa or lips, dendritic keratitis and genital herpes even if they were immunologically educated [14]. After repeated episodes, corneal herpes eventually develops to an immune mediated herpetic stromal keratitis (HSK). HSK is still an important cause of blindness in developed countries [15].
The eye is an immunologically privileged site [16]. It strives to maintain transparency of the cornea to preserve the pathway of light to the retina. Microbial invasion on the corneal surface is prevented by washing out infectious agents with tears containing inhibitors such as lactoferrin, lysozyme, secretory IgA plus complement, type 1 interferon and suppressing factors on the growth of new blood vessels. These sustain inflammatory cell infiltrations [17-20].
However, primary ocular HSV-1 infection does occur. It causes acute kerato-conjunctivitis with hyperemia, edema and swollen lymphoid follicles in the conjunctiva which is often indistinguishable from acute adenovirus infection [21]. Corneal ulcers extend to dendritic figures. It may spread to cause a geographic ulceration [21, 22] (Figures 1 and 2).
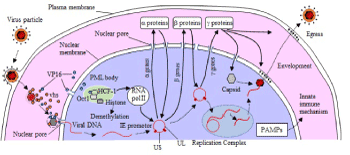
Figure 1. Entrance and proliferation of HSV
HSVs enter epithelial cells following the regulated processes: attachment, binding fusion, and penetration into cytoplasm. Entered capsid and tegument proteins like VP16 trafficked toward the nucleus. Through the nuclear pore, viral genomes are injected into the host cell nucleus. Immediately, viral DNAs associate with host heterochromatin in the host cell nuclei. After de-repressing process, HCF-1, Oct-1 and VP-16 start replication processes of HSV, namely immediate early α, early β and late γ genes transcriptions. Newly synthesized genome forms a rolling circle and cleaved into equimolar unit. It is packaged into a newly made capsid. Mature enveloped viral particles are bud from the cellular membrane.
VP-16: virus protein 16, HCF-1: host cell factor 1, vhs protein: virion host shut off protein, PML body: promyelocytic leukemia body UL and US: unique long and short region, PAMPS: pathogen associated molecular patterns
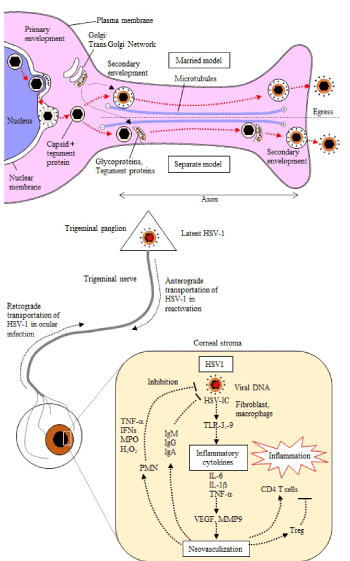
Figures 2 and 3. Egress of HSVs via the anterograde transport on axonal flow
Two egress processes have been observed. HSV capsids enveloped at inner nuclear membrane where HSV capsids obtain tegument proteins VP 16, UL11 and glycoproteins and released into the cytoplasm. They de-envelope at the outer nuclear membrane and the naked capsids released in the cytoplasm. They entered cytoplasmic membrane and mature with a secondary envelope. This mode of maturation and egress is called a married model. Another mod of egress is a separate model. Naked capsids with tegument protein are transported through the axon. Envelope with viral glycoproteins is transported separately and these components are assembled at the sites of egress where epithelial or fibroblastic cells are adjacent of nerve endings. Both models are observed, and the married model may be the dominant one. Long distance transport is depending on the kinesin motors. Microtubules composed of head-to-tail association of α and β tubulin transport virus to the cellular periphery. Reactivated virus transported to the peripheral tissues. In the cornea HSV DNA or viral proteins are detected but they are reactivated virus in trigeminal ganglion transported via axonal Fflow.
Reactivated virus causes unilateral corneal herpes, although rarely it can occur bilaterally [22-25]. The cornea tries to maintain status quo, thus a few patients develop stromal haze namely herpetic stromal keratitis (HSK) after repeated episodes of recurrences [26]. HSK has been studied using the patient’s excised corneas obtained at corneal transplantations [26] and/or in experimental animal models [28-34].
To infect the mouse or the rabbit cornea, a virulent RE or Mckrae strain of HSV [34,35] is instilled onto the scratched cornea. In mice, HSV replicate for 4-6 days in the cornea, then the virus disappears. Following a short quiet period [47], inflammatory cells infiltrate the corneal stroma at 8 to 10 days post infection (PI) with rapid neovascularization of the entire cornea proceeding to the complete loss of clarity of the visual axis. This immune-mediated pathogenesis has been extensively studied as a model of HSK [47, 48]. However, no experimental animal model of HSK has been developed as an outcome of repeated recurrences of reactivated HSV [32-34]. Two to three days PI of HSV on the mouse cornea, progeny viruses are transported to the TG via retrograde axonal flow [5,7,8]. After a short period of active growth in the TG, HSV establishes lifelong latency in the neurons with occasional episodes of reactivation [36]. In mice, spontaneous reactivation is rare [37-39]. Stimuli such as UV irradiation [40], heat [41], tattoo [42], chemotherapy and steroid [43], epinephrine iontophoresis [44], T cell and γIFN removal [45], psychological stress [46] etc. have been used to reactivate HSV, however, results are inconsistent in mice.
The rabbit establishes HSV latency in the TG after acute corneal infection. Reactivation is easier than the mouse; however, immunological analyses of the results in the rabbit are difficult [48]. Guinea pig is a model of genital HSV-2 infection with spontaneous reactivations [49]. When attenuated HSV-2 is inoculated intra-nasally, educated CD4+T cells migrate to the vaginal mucosa and secretory anti-herpes IgA is produced locally by activated B cells [49,50]. Preventive effects are being investigated in several laboratories [51]. The effects of this immunization for corneal herpes has yet to be studied.
Viral envelope glycoproteins gB, gC, gD, gH and gI attach onto the plasma membrane [52-54]. These glycoproteins fuse at the lipid rafts and dynamin on host cell membrane [56]. HSV-1 gB and gC (HSV-2 ligands are gB and gG) adhere to the cell surface glycosoaminoglycans [52-55]. Following entry steps, three gD receptors for HSV-1 [56]: herpes virus entry mediator (HVEM) [58,59];nectin-1 and nectin-2 [60,61];and 3-0-sulfated heparan sulfate [62];are required [56,57]. Nectin-1 is expressed on the corneal epithelium, conjunctiva, lens epithelium, ciliary body, iris, choroid and retina. PTLR α is an inhibitory receptor paired with an activating PTLR β receptor. The cellular ligand for both PTLRs is D99 expressed on activated T cells [63-65]. It deactivates NK cells and prevents HSV infected cell lysis. Glycoproteins gD and gB bind to cell surface receptors: non-muscle myosin IIA (NM-IIA) [63]; paired immunoglobulin-like type 2 receptor α (PILR α) [64, 65]; myelin associated glycoprotein (MAG) [66]; with membrane fusion of gH [57]. When NMHC-IIA, a subunit of NM-IIA [67], is expressed on the cell surface, gB is attached and fused with the cell membrane. NMHC-IIA expression on the corneal and conjunctival cell and NMHC-IIB on neuronal cell are scarce [67,68], however upon HSV entry, they are protruded into microvilli, filopodia [69] etc. by muscle myosin movement [68]. The corneal surface is washed with microbial inhibitors in tears [17-20] by constant blinking. In this environment, NM-IIA offers a way for the HSV infection to the cornea [67]. HSV also enters cells by endocytosis [70]. HSV-1 gH/gL binds to αvβ3-integrin [56,71] and enters HeLa cells and CHO cells by endocytosis on lipid rafts [56]. HSVs are degraded by lysosomal enzymes released in endosome. Macrophages phagocytize HSV when pseudopods are extended by activated TLR2. HSV DNA in the phagosome are degraded by lysosomal enzymes and recognized by TLR 3 and 9 [28,72-75]. Signals of activated TLR3 and 9 are transduced to the NF-κB [76]. When HSV passes through the cytoplasmic membrane, activated Ca++ ion [77], necessary for the transport of viral capsids to the nuclear pore, flows into the cytoplasm from the endoplasm [78]. Nearly 20 tegument proteins are dispersed into the cytoplasm. Inner tegument proteins complexed with capsid are transported to the nuclear pore by the motor dynein in microtubule [78]. Polyamines in the virion neutralize the negative charges of the viral DNA [79]. It is packed tightly in the capsid where the inner pressure reaches about 20 atmospheres [80]. Host nuclear factor importin β, nuclear pore complex proteins and UL36 serve to dock HSV capsids to the nuclear pore [81]. Viral genome is ejected into the nucleoplasm with the high inner pressure [80].
In the nucleus, HSV DNA is linearized and circularized [82] before the viral protein production (Figure 1) [83,84]. When replicating, it forms a rolling circle and extended genomes are packaged into a newly made capsid after cleavage into equimolar monomeric units [84]. In the epithelial cell, 103-104 virus particles are produced in 8-10 hours [85]. Immediately after the entrance into the nucleus, promoter regions of HSV genome; α (immediate early), β (delayed early) and γ (late) gene associate epigenetically on the methylated histone H3-Lysine 9 and 27 host heterochromatin [86,87] .
Sixty eight percent of the HSV-1 genome consists of (G+C) [88] Speckled protein of 100KDa (Sp100) binds hypo-unmethylated CpG islands [88-94]. In addition, the HSV genome triggers innate anti-DNA responses with interferon α or γ by promyelocytic leukemia (PML) protein bodies in nuclear lamina or nuclear domain (ND10) [95,96] locating in the inter-chromosomal spaces or near the nuclear membrane. Upon HSV infection, ND 10 constituents are degraded, dispersed [97-101] and provided places for the latency or replication of HSV genome [102,103,104]. RING finger domain of ICP 0 [99] confers E3 ubiquitin ligase activity in its N-terminal region [100] and triggers the proteasome dependent degradation of PML and Sp100 [101]. PML proteins contain repressive cofactors Sp100, Daxx and ATRX [99]. Upon sensing foreign DNA, nuclear interferon inducible protein 16 (IFI 16) binds to the transcriptional activators of viral genome and links to the heterochromatic suppression of HSV genome [104-106]. Depletion of IFI16 in epithelial cells or fibroblasts results in histone modifications of decreased H3K9me3, increased H3K4me3 of ICP4 promotor and increased expression of IE genes [105,107]. They also increased RNA polymerase II loading on IE genes in HSV infected cells [108]. Corepressor element-1 silencing transcription factor (CoRest)/REI-silencing transcription factor (REST) repressor complex consists of histone deacetylase (HDAC) and CoRest binds HDACs and Rest [109]. This complex functions to repress neuronal genes in non-neuronal cells [110]. CoRest interacts ICP0 which colonizes with ND10 in the nucleus and blocks the repressive action of HCLR [109,111]. In epithelial cells, ND10 disassembles in a few hours and dispersed in the nuclear microenvironment [112]. These heterochromatin repressions are counteracted by viral (VP16 and ICP0) and host cell factor 1 (HCF1), lysine specific demethylase1 (LSD1), octamer protein-1(Oct-1), specificity protein 1 (Sp1) and GA binding protein (GABP) transcription factors. LSD1 plays a key role in the de-repression of α genes [113]. Alpha HSV-1 gene promotors contain binding sites for Oct-1, VP16 and a cellular protein designated HCF1 [114-117]. Oct-1 and VP-16 bind to the promotor domains. VP16 assembles HCF1, Oct-1 and lysin specific demethylase 1 (LSD1) changes heterochromatin to euchromatin. LSD1 is recruited by VP16 from cytoplasm to nucleus. ICP0 is dispersed in the cytoplasm. On productive infections of HSV-1, after entering viral DNA into nucleus, HCF1, Oct-1 and other transcription factors reduce heterochromatin in an hour and activate transcription of α-genes [118]. With these de-repressing processes, viral immediate early genes (α genes: ICP-0, -4, -22, -27, -47, Us1.5) start to be transcribed. After α gene expression, β1→ β2 genes and γ1→γ2 genes are expressed in cascade fashion [1,119]. β genes produce enzymes for the DNA replication and γ genes make structural proteins and assembly of infectious viruses [1]. Immediate early protein ICP-0 and ICP-4 activate β and γ gene expression [1]. ICP-0 is related to the transcription, interferon response, cell cycle and degradation of ND10 [120]. It also plays a key role of reactivation from latency [120]. ICP-27 inhibits mRNA translation by disrupting a splicing of RNA and helps transport viral mRNAs to the nucleus [121]. ICP-47 interrupts TAP conjugation of viral antigens with MHC at the endoplasmic reticulum and hinders transportation to the cell surface. Hence CD8+T cells can not recognize viral antigen and do not activate [122].
When HSV enters the ganglionic neurons, viral lytic genes are repressed associating with the host cell heterochromatin and latency associated transcripts (LATs) promotor and enhancer bind to the euchromatin [92,123]. To be functional in neurons, HSV DNA requires several days more than in epithelial cells for several reasons. First, the pool of histones is much less in the nucleus of sensory ganglionic neurons. Second, most of the HCF-1 is retained in the cytoplasm and is scarce in neuronal nuclei. Third, after the virus passes through the cytoplasmic membrane, envelope protein VP16 remains in the cytoplasm [114,124-126]. LATs expression in neurons reduce lytic gene expression during the acute phase of infection [127,128]. These characteristics of neurons contribute to HSV latency in neuronal cells. Some infected TG neurons produce progeny HSVs but do not cause apoptosis or necrosis [129,130]. The majority of infected ganglionic neurons survive harboring the circular HSV genome as episomes [69,131]. All these outcomes are supported by epigenetic regulations and innate or acquired immune responses [132-135]. Functioning neurons in TG are detectable by sensing stimuli on the corneal surface [12]. In mice, HSV DNAs in the TG are detected by in situ hybridization. They are distributed to neighboring cells via dendrites in all three branches of TG several days after corneal infection [136]. Any ganglionic neurons may harbor up to a thousand copies of HSV DNA (137). These results indicate that infiltrating CD8 T cells don’t induce apoptosis in TG [133-135, 137,138]. Release of exosomes from neurons containing mRNA, microRNA (miRNA), viral proteins, HSV DNAs and/or infectious viruses might contribute wide spread of HSV [139-140]. Neutrophils break up these multiple apoptotic bodies containing live viruses. They are phagocytized by macrophages and spread virus to neighboring ganglionic neuronal cells [141,142].
During latency, HSVs transcribe stable two major antisense RNAs, latency associated transcript (LATs), from the unique region of the viral genome. HSVs also transcribe minor LAT only detectable with sensitive methods [138-139]. LAT is an 8.3kb primary transcript, which is spliced into stable 1.5 and 2.0 kb major LAT introns, as well as a 6.3 kb minor LAT exon [143,144]. LATs stabilize latent HSVs [145,146] and downregulate genes required for a lytic infection. Discovery of the gene regulation by small RNAs (sRNAs) was swiftly extended to the study of herpesviruses. HSVs encode many miRNAs within or adjacent to the LAT locus. Two sRNA1 and 2 are encoded in the first 1.5kb LAT. They inhibit productive infection in tissue culture [147,148]. sRNA2 suppresses ICP4 and increase HVEM expression on neurons [149] without affecting cognate mRNA levels [148]. In addition, LATs transcribed twenty-seven miRNAs [1,149-152]. Eleven miRNAs are expressed in the related region of LATs and their functions have been studied in neurons and epithelial cells [149,150]. Primary LAT gene encodes seven miRNAs, while the eighth is derived from a transcript upstream of the LAT promotor [153]. Less abundant potential miRNAs originating from LAT region are identified by deep sequencing. Most of these miRNAs suppress immediate early gene expression by their antisense sequences [152]. Six miRNAs: miR-H2-3P, H3-3P, H4-3P, H5-3P, H6-3P and H7-5P accumulate in latently infected neurons [149,150]. miR-H2 is complementary to ICP0 mRNA and regulates ICP0 expression [149,153]. It promotes latency by globally repressing lytic gene expression [154]. However recent results suggest that H2 is less effective in inhibiting expression of ICP0 than neuronal host miR-138 which inhibits lytic gene expression in ganglia by targeting ICP0 mRNA [155,156]. Mutated miR-138 target sites increased α-gene expression in the infected eye and TG [155, 156]. H-4 downregulates neurovirulent ICP34.5 expression [157]. Except H4-3P, all miRNAs are expressed in infected cells in which progeny viruses are produced. miR-H8-5P, H15, H17, H18, H26 and H27 accumulate during reactivation (158). miR-H3-3P, H13 and H27 enhance growth of HSV [158]. H5-3P and H27 are produced without viral protein synthesis similar to the viral α-gene products [158]. The synthesis of H1-5P, H3-5P, H6-3P, H7-5P, H16 and H26 require prior protein synthesis like those of viral β and γ-mRNAs [158]. In mouse studies, miR-155, a host’s miRNA, controls HSV latency and prevents spread of HSV from TG to the brain. It also suppresses zosteriform spread of skin lesion by intradermal HSV infection [159] LATs have been strongly implicated in the global control of HSV gene expression during latency through the post-translational modification (PTM) by methylation on histone H3 associated with viral promoters [160,161]. As described in the previous section, IFI16 is an innate sensor of the foreign DNA in the nucleus. In the cytoplasm, cGAS enzyme binds STING (Stimulator of Interferon Genes) and activates the IRF-3 signaling pathway [162-164]. STING and cGAS sense HSV DNA as foreign and synthesizes cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). Together with IRF3 and IFI16, cGAMP induces IFN-β in HSV infected cells [162-164].
When HSV infected, elicited stress leaks mitochondrial DNA (mtDNA) into the cytosol [165]. cGAS and STING sense the situation and elevate a type 1 interferon response [164]. Foreign DNA and viral proteins induce expression of IFNs, inflammatory cytokines and chemokines that recruit inflammatory immune cells to TG [132, 166]. Neutrophils are scarce in TG but monocyte, mast cells, macrophages, γδT cells are infiltrated and surround infected neurons [167,168]. γδT cells induce IFN-γ and activated macrophages secret NO and TNF-α [169, 170]. These inhibit viral growth. Eventually, circulating CD8+T cells and CD4+T cells, but not tissue resident T cells, accumulate in inflammatory sites in TG [133]. Neurons usually do not express MHC-I; however, in TG, neurons contain latent HSV [171]. These neurons express small amount of viral proteins such as gC, gB498-505 with MHC-I [133,135]. CD8+T cells recognize them with direct contact. In TG, LAT positive neurons are surrounded by primed CD8+, T cells at the periphery [170-172]. LAT positive neurons are protected from apoptosis by granzyme B released from CD8+T cells [135,173]. However, recent evidence suggests that CD11c+, CD8α+ monocytes play the major role to establish HSV latency and reactivation in neurons [174]. CD8+ T cells are eventually exhausted with programmed death-1 (PD-1) expression with increased latency [175]. In neurons, autophagy is another important defense mechanism [176].
Various stimuli in TG shift a micro-environmental balance to a favorable one for viral α gene expression rather than for cellular repression [160,177]. An in vitro study of HSV reactivation indicates that the initial step is induced following a removal of nerve growth factor (NGF) in culture medium [178-180]. In vivo, tyrosine kinase A (TrkA) receptor activates PI3-K (phosphatidylinoisitol 3-kinase) p110 alpha catalytic subunit with NGF [181-183]. When NGF is withdrawn in culture medium, 3-phosphoinositide-dependent protein kinase-1 (PDK1) is recruited to the plasma membrane and phosphorylates serine/threonine kinase Akt [179,180,183]. The NGF removal inactivates mTORC-1, blocks 4E-BP1 phosphorylation, suppresses cap-dependent mRNA translation and induces HSV-1 reactivation [181]. External stimulations sensed by trigeminal nerve endings distributed in orolabial epithelia or ocular surface are immediately delivered to the TG neurons. When latent HSV DNAs in neurons sensed the reduced level of NGF, initial steps taken by silenced HSV DNA are different from a well-ordered cascade fashion replication [184,185]. They are controlled by infiltrated CD8+ T cells, NK cells and γδT cells [135,186,187]. IFN-γ is produced by them, then HSV replication is halted and there is a return to the latency (187). For the initial 24 hours, VP16/UL48 translocates from the cytoplasm to the nucleus [124,188] and guides Oct-1 and HCF-1 [182,189]. Only in a small number of neurons, ICP0 and ICP4 are expressed and degrade ND10 [190]. Progeny virions are made in cascade fashion and they are transported to the axonal termini [182]. Virus gene transcription does not follow with any specific kinetic schedules seen in the epithelial cells, but reactivated virus released to the peripheral tissues at 14 to 48 hours after stimulation signals received [182]. Most of the released viruses don’t produce overt disease. They are shed silently in the tears, nasal or genital secretions and/or saliva, but they may be a source of virus spread [191].
Following reactivation, newly synthesized concatemeric virus genome is processed as a rolling circle mode described in the previous section [1,192]. Reactivated virus spreads from TG to peripheral tissues via axons. As ganglionic neurons are bipolar cells, HSV can spread in both directions [136]. It can cause encephalitis in rare instances [8]. Initial innate immunity and subsequent specific immune responses prevents viral invasion in the brain but latent HSV can be detected in the neurons in the brain [193].
Packaged viral nucleocapsids in the nucleus bud through inner nuclear membrane where they obtain a primary envelop (Figures 2 and 3) [194]. During this primary envelopment, they have obtained tegument protein VP16, UL11 and membrane glycoproteins [195,196]. Egress of viral capsid through the nuclear pore by fusion of gB with outer nuclear membrane is dependent upon the viral proteins UL31, UL34 and Us3 kinase [197]. At the outer nuclear membrane, capsids de-envelope and naked capsids are released into the cytoplasm [197]. The virions are processed for assembly and maturation in the cytoplasmic transport vesicles where capsids coated with tegument, then obtain a secondary envelope containing markers of trans-Golgi network (TGN) and viral glycoproteins [198]. Capsids move toward transport vesicles. UL36, UL37 and Us11 in the tegument interact with kinesin motors [199,200]. Hollow 25nm diameter microtubules composed of 13 protofilaments are tied in a head-to-tail association of α-tubulin and β-tubulin in the axon and used for a long distance transport of virus to the periphery [201]. They have a ‘plus’ end oriented towards the cellular periphery [202,203]. The cargo of virus particles egresses from axons with three viral envelope proteins gI, gE and Us9 [204]. Currently there are two models of transportation (Figure 2) [205]: “The married model” tells us that virus particles are assembled and enveloped in the neuronal cell bodies before entering axons and transported to egress at the peripheral distal ending [5]. “The separate model” indicates that naked capsids with tegument proteins but without envelop are transported through the axon. Envelope with viral glycoproteins are transported separately and these components are assembled at near the sites of egress [206]. Both models are observed, and the married model may be the predominant one. Virions are released into an extra-neuronal environment by fusion of transport vesicles with an epithelial cell or fibroblast plasma membrane. As described in the first section of this review, most of them cause asymptomatic shedding.
In HSK, infiltrated cells release angiogenic factors, EGF via IL-6, Robo 4, MMP-2 and MMP-9. Leaky new vessels grow from the limbus to the corneal center. Inflammatory cells infiltrate via the new vessels. Visual acuity is severely hampered by the cloudiness of the corneal stroma. This is called HSK [24-28] (Figure 3).
The immunopathological nature of HSK is recognized by a ring-shaped haze where IgM, IgG and IgA are deposited together with virus particles [207]. Viral antigens deposited in keratocytes induced anti-HSV antibodies plus complement deposition or antibody-dependent cell mediated cytotoxicity (ADCC) with inflammatory cellular damage [17,207-209]. The antivirals combined with steroid keeps the affected cornea clear. This supports the immunological nature of the disease [210]. Rabbits showed immune complexes play a role in the pathogenesis of HSK [207]. In HSK, aberrant Th1 type responses mediate chronic immuno-inflammation [25,31,211]. When the mouse corneal epithelium is inoculated with HSV-1, it occasionally invades the upper corneal stroma until 4–5 days post infection (PI) [35]. During this initial phase of infection, cytokines (IL-1α, IL-1β, TNF-α, IL-6, IL-12, IFN-γ ), chemokines (IL-8, MIP-1α, MIP-1β, MCP-1, MCP-2, RANTES.), and other mediators such as nitric oxide, COX 2, prostaglandin E2 are produced either by resident corneal cells or infiltrating cells [31,47,76,212-215]. Neutrophils and macrophages are detected in the active lesion. After a 3-4 days interval, robust immuno-inflammatory cells infiltrate in the stroma [31,35]. This infiltration consists of mainly CD4 T cells [47,216], which recognize HSV tegument proteins UL21 and UL49 [217,218]. CD4 T cells, NK cells and Langerhans cells infiltrate via new vessels [31,219-222]. Nude or SCID mice [223,224] do not develop typical HSK. Regulatory T cells (Treg) were found to improve clinical signs in murine chronic HSK [225]. When regulatory CD4 T Foxp3+ cells are dominant over the CD4 Th1 cells, HSK lesion remains mild [226,227]. In current models, herpetic keratitis has been induced in the naïve mouse by the inoculation of a large amount of virus in the scarified cornea. However, the frequent virus reactivation does not occur in animal model of HSK [32,34,35,228]. Instead, in humans, repeated or continual recurrent episodes of virus reactivation from the ganglion are observed in a person who has sufficient innate and adaptive immunity. Further investigation requires to answer what viral or immune molecules drive stromal keratitis in the chronic phase without active viral replications in the current mouse model.
Large amounts of bioactive CpG motifs in HSV DNA can induce NO and Th1 type cytokines like IL-6, TNF, and IFN- in vivo and in vitro [81,205,224]. Macrophages and dendritic cells recognize them by TLR-9 expressed on the endosome [213,229]. In the human corneal button or experimental mouse cornea, HSVDNA has been detected by in situ hybridization or conventional PCR [26,224]. Real time PCR demonstrated HSVDNA persistence in the corneal buttons obtained from HSK patients [26]. Viral antigen and antibody complexes are deposited in human HSK [207]. Toll-like receptors (TLRs) recognize pathogen associated molecular pattern (PAMPs) of HSV and trigger initial host responses [224,230-232]. TLR-3 is expressed in the endosome and the surface of corneal epithelial cells and fibroblasts and recognizes double stranded RNA produced during the process of HSV replication [233,234]. TLR-9 expressed in the endosome senses non-methylated HSV CpG [213]. HSV DNAs and/or HSV-IC have been detected in the mouse cornea long after the infectious virus has been cleared [215,235,236]. When human corneal epithelial cells (HCE) and human corneal fibroblasts (HCRF) were infected with UV-inactivated HSV, transfected with HSV DNA or treated with HSV-IC, increased expression of TLR-3, -9 gene and IL-6 were observed in HCRFs [213]. Shortly after HSV-1 enters cells, virion shut off protein (vhs, UL41 gene) inhibits cellular RNA function [237,238]. However, IL-1 and IL-6 are still upregulated in the infected cells [239-241]. IL-6 induces expression of vascular endothelial growth factor (VEGF) and potentiates neovascularization [221,242]. Transfection of HCRF and a human macrophage cell line THP-1 [213] with HSV DNA or treatment with HSV-IC produces VEGF. Production of MMP-9 was elevated in THP-1 cells but not in corneal cells [233]. THP-1 cells may produce MMP-9 via TLRs because THP-1 cells inhibited MMP-9 production with anti-TLR-2, -3 and -4 antibody treatment [213,243]. IL-1β, IL-6 and TNF-α also induce VEGF and MMP-9 expression in corneal cells and macrophages. Continued presence of HSV DNAs and HSV-ICs in the cornea may trigger inflammation and angiogenesis in HSK. VEGF and MMP-9 induction by IL-6 may be a therapeutic target via TLRs mediated cytokine pathways [222,244].
In mouse model, new vessels sprout on the cornea after HSV-1 infection [245]. PMNs pass through these leaky vessels to the afflicted cornea [234]. PMNs are the prominent infiltrating cells in acute herpetic keratitis in patients and experimental animals [31,246]. Chemotactic factors, IL-8, Gro-α and granulocyte/macrophage colony-stimulating factor (GM-CSF) were released from corneal cells and macrophages infected with HSV-1 or treated with virus components. They attract and/or hold PMNs at the afflicted site, activates and prolongs survival of them [228,247]. PMNs cocultured with HCE, THP-1 cells or virus components produced highly reactive oxygen species (hROS) [228,247]. Such activated PMNs suppress HSV growth [228]. When they were overlaid onto the HSV-1-infected Vero cells, virus growth was inhibited [229]. Supernatants of the PMNs obtained after cocultured with HSV-infected or virus components treated HCE or THP-1 cells, HSV growth was halted [229]. TNF-α released from the PMNs was not enough for virus inhibition. IFN-α, -β and -γ were not released from PMNs mixed with treated HCE and/or THP-1 cells supernatants. H2O2 was released from PMNs and THP-1 cells infected with Mckrae, transfected with HSVDNA or treated with HSV-IC. H2O2 inhibited HSV-1 growth directly [229]. Myeloperoxidase (MPO) was also released by PMNs, contributing virus inhibition [229]. PMNs released NO in the supernatants when they were mixed with supernatants of HCE or THP-1 cells treated with HSVDNA and HSV-IC. Released NO was significantly higher concentration than those obtained from untreated HCE supernatant, however, it does not directly inhibit viral growth at the range of the concentrations obtained from treated HCE [229]. Low concentration of NO released from PMNs may convert naïve CD4 T cell to Th1 cells [248]. Neutrophils produce other factors like monokine induced by interferon γ (MIG). MIG induces and accumulates CD4+ Th1 cells [249]. When PMNs and CD4+ T cells were mixed and incubated with HCE supernatants treated with HSV components, MIG was released [229]. These local environments may contribute to the peripheral naïve CD4 +T cell differentiation to Th1 but further studies are required for this problem.
Effective HSV vaccine has been tried including a nonvirulent virus, viral proteins, viral DNAs etc. [250-253]. However, none of them were effective in clinical trials. To obtain a better vaccine, delivery of effectors to the target is critical.
We are deeply indebted to Laura C. Hooper for her discussion, advise and technical help. We thank Harutaka Katano for his help to improve figures We also thank Michael Ross and Thomas Tarpley for reviewing the manuscript . We appreciate Kubo Konomi for secretarial help.
- Roizman B, Knipe DM, Whitley RJ (2013) Herpes Simplex Virus. in: D.M. Knipe, and P.M. Howley, (Eds.), Field’s Virology, Walters Kluwer/Lippencott Williams &Wilkins., pp. 1823-1897.
- Dasgupta G, Chentoufi AA, Kalantari M, Falatoonzadeh P, Chun S, et al. (2012) Immunodominant "asymptomatic" herpes simplex virus 1 and 2 protein antigens identified by probing whole-ORFome microarrays with serum antibodies from seropositive asymptomatic versus symptomatic individuals. J Virol 86: 4358-4369. [Crossref]
- Kalantari-Dehaghi M, Chun S, Chentoufi AA, Pablo J, Liang L, et al. (2012) Discovery of potential diagnostic and vaccine antigens in herpes simplex virus 1 and 2 by proteome-wide antibody profiling. J Virol 86: 4328-4339.
- Barber GN (2001) Host defense, viruses and apoptosis. Cell Death Differ 8: 113-126. [Crossref]
- Antinone SE, Smith GA (2010) Retrograde axon tra(nsport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J Virol 84: 1504-1512. [Crossref]
- Baringer JR, Pisani P (1994) Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol 36: 823-829. [Crossref]
- Smith G (2012) Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66: 153-176. [Crossref]
- Steiner I, Kennedy PG, Pachner AR (2007) The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6: 1015-1028. [Crossref]
- Antinone SE, Zaichick SV, Smith GA (2010) Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J Virol 84: 13019-13030. [Crossref]
- Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, et al. (2011) A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol 85: 6669-6677.
- Bertke AS, Ma A, Margolis MS, Margolis TP (2013) Different mechanisms regulate productive herpes simplex virus 1 (HSV-1) and HSV-2 infections in adult trigeminal neurons. J Virol 87: 6512-6516. [Crossref]
- Himmelein S, Lindemann A, Sinicina I, Strupp M, Brandt T, et al. (2015) Latent herpes simplex virus 1 infection does not induce apoptosis in human trigeminal Ganglia. J Virol 89: 5747-5750. [Crossref]
- Koelle DM, Wald A (2000) Herpes simplex virus: the importance of asymptomatic shedding. J Antimicrob Chemother 45 Suppl T3: 1-8.
- Mark KE, Wald A, Magaret AS, Selke S, Olin L, et al. (2008) Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis 198: 1141-1149. [Crossref]
- Liesegang TJ (2001) Herpes simplex virus epidemiology and ocular importance. Cornea 20: 1-13. [Crossref]
- Streilein JW (2003) Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol 3: 879-889. [Crossref]
- Fujihara T, Hayashi K (1995) Lactoferrin inhibits herpes simplex virus type-1 (HSV-1) infection to mouse cornea. Arch Virol 140: 1469-1472. [Crossref]
- Zavaro A, Samra Z, Baryishak R, Sompolinsky D (1980) Proteins in tears from healthy and diseased eyes. Doc Ophthalmol 50: 185-199. [Crossref]
- Tang Q, Hendricks RL (1996) Interferon gamma regulates platelet endothelial cell adhesion molecule 1 expression and neutrophil infiltration into herpes simplex virus-infected mouse corneas. J Exp Med 184 1435-1447. [Crossref]
- Hendricks RL, Weber PC, Taylor JL, Koumbis A, Tumpey TM, et al. (1991) Endogenously produced interferon alpha protects mice from herpes simplex virus type 1 corneal disease. J Gen Virol 72: 1601-1610. [Crossref]
- Darougar S, Wishart MS, Viswalingam ND (1985) Epidemiological and clinical features of primary herpes simplex virus ocular infection. Br J Ophthalmol 69: 2-6.
- Holland EJ, Schwartz GS (1999) Classification of herpes simplex virus keratitis. Cornea 18: 144-154.
- KRW (1998) Epidemiology of ocular infection Lippincott Williams & Wilkins, Philadelphia.
- Sundmacher RA (1980) Clinic-virologic classification of herpetic anterior segment diseases with special reference to intraocular herpes. in p.203-210 Herpetishe Augener Krankungen Freiburg. Munich Bergman, 1981.
- Streilein JW, Dana MR, Ksander BR (1997) Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol Today 18: 443-449. [Crossref]
- Shimomura Y, Deai T, Fukuda M, Higaki S, Hooper LC, et al. (2007) Corneal buttons obtained from patients with HSK harbor high copy numbers of the HSV genome. Cornea 26: 190-193. [Crossref]
- Liesegang TJ (1999) Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18: 127-143. [Crossref]
- Hayashi K, Hooper LC, Hooks JJ (2008) Who (what) pays toll for the development of herpetic stromal keratitis (HSK). Semin Ophthalmol 23: 229-234. [Crossref]
- Banerjee K, Biswas PS, Rouse BT (2005) Elucidating the protective and pathologic T cell species in the virus-induced corneal immunoinflammatory condition herpetic stromal keratitis. J Leukoc Biol 7724-7732.
- Stulting RD, Kindle JC, Nahmias AJ (1985) Patterns of herpes simplex keratitis in inbred mice. Invest Ophthalmol Vis Sci 26: 1360-1367. [Crossref]
- Biswas PS, Rouse BT (2005) Early events in HSV keratitis--setting the stage for a blinding disease. Microbes Infect 7: 799-810. [Crossref]
- Shimeld C, Hill T, Blyth B, Easty D (1989) An improved model of recurrent herpetic eye disease in mice. Curr Eye Res 8: 1193-1205. [Crossref]
- Stuart PM, Keadle TL (2012) Recurrent herpetic stromal keratitis in mice: a model for studying human HSK. Clin Dev Immunol 2012: 728480. [Crossref]
- Tullo AB, Shimeld C, Blyth WA, Hill TJ, Easty DL (1983) Ocular infection with herpes simplex virus in nonimmune and immune mice. Arch Ophthalmol 101: 961-964. [Crossref]
- Babu JS, Thomas J, Kanangat S, Morrison LA, Knipe DM, et al. (1996) Viral replication is required for induction of ocular immunopathology by herpes simplex virus. J Virol 70: 101-107. [Crossref]
- Efstathiou S, Minson AC, Field HJ, Anderson JR, Wildy P (1986) Detection of herpes simplex virus-specific DNA sequences in latently infected mice and in humans. J Virol 57: 446-455. [Crossref]
- Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, et al. (2002) Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. ProcNatl Acad Sci U S A 99: 978-983. [Crossref]
- Gebhardt BM, Halford WP (2005) Evidence that spontaneous reactivation of herpes virus does not occur in mice. Virol J 2: 67.
- Margolis TP, Elfman FL, Leib D, Pakpour N, Apakupakul K, et al. (2007) Spontaneous reactivation of herpes simplex virus type 1 in latently infected murine sensory ganglia. J Virol 81: 11069-11074.
- Laycock KA, Lee SF, Brady RH, Pepose JS (1991) Charactrizatrion of a murine model of recurrent herpes simplex viral keratitis induced by ultraviolet B radiation. Invest Ophthalmol Vis Sci 32: 2741-2746. [Crossref]
- Moriya A, Yoshiki A, Kita M, Fushiki S, Imanishi J (1994) Heat shock-induced reactivation of herpes simplex virus type 1 in latently infected mouse trigeminal ganglion cells in dissociated culture. Arch Virol 135: 419-425. [Crossref]
- Begolli Gerqari A, Ferizi M, Kotori M, Daka A, Hapciu S, et al. (2018) Activation of herpes simplex infection after tattoo. Acta Dermatovenerol Croat 1: 75-76.
- Saito M, Kiyozaki H, Obitsu T, Imoto H, Taniyama Y, et al. (2016) Herpes simplex virus-1 encephalitis induced by chemoradiotherapy and steroids in an esophageal cancer patient: a case report. BMC Cancer 16: 233. [Crossref]
- Shimomura Y, Gangarosa LP Sr, Kataoka M, Hill JM (1983) HSV-1 shedding by lontophoresis of 6-hydroxydopamine followed by topical epinephrine. Invest Ophthalmol Vis Sci 24: 1588-1594. [Crossref]
- Liu T, Khanna KM, Carriere BN, Hendricks R (2001) Gamma interferon can prevent herpes simplex virus type1 reactivation from latency in sensory ganglions. J Virol 75: 11178-11184. [Crossref]
- Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL (2007) Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J Immunol 179: 322-328.
- Araki-Sasaki K, Tanaka T, Ebisuno Y, Kanda H, Umemoto E, et al. (2006) Dynamic expression of chemokines and the infiltration of inflammatory cells in the HSV-infected cornea and its associated tissues. Ocul Immunol Inflamm 14: 257-266. [Crossref]
- Webre JM, Hill JM, Nolan NM, Clement C, McFerrin HE, et al. (2012) Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J Biomed Biotechnol 2012: 612316. [Crossref]
- Stanberry LR, Kern ER, Richards JT, Overall JC, Jr. (1985) Recurrent genital herpes simplex virus infection in guinea pigs. Intervirology 24: 226-231.
- Sato A, Suwanto A, Okabe M, Sato S, Nochi T, et al. (2014) Vaginal memory T cells induced by intranasal vaccination are critical for protective T cell recruitment and prevention of genital HSV-2 disease. J Virol 88: 13699-13708. [Crossref]
- Shin H, Kumamoto Y, Gopinath S, Iwasaki A (2016) CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat Commun 7: 13346. [Crossref]
- Pertel PE, Fridberg A, Parish ML, Spear PG (2001) Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279: 313-324. [Crossref]
- Herold BC, Visalli RJ, Susmarski N, Brandt CR, Spear PG (1994) Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J Gen Virol 75: 1211-1222. [Crossref]
- Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, et al. (1998) Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol 72: 6119-6130. [Crossref]
- Spear PG, Eisenberg RJ, Cohen GH (2000) Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275: 1-8. [Crossref]
- Gianni T, Gatta V, Campadelli-Fiume G (2010) {alpha}V{beta}3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc Natl Acad Sci U S A 107: 22260-22265. [Crossref]
- Montgomery RI, Warner MS, Lum BJ, Spear PG (1996) Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87: 427-436. [Crossref]
- Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG (1998) Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280: 1618-1620. [Crossref]
- Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, et al. (1998) A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246: 179-189. [Crossref]
- Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grünwald K, et al. (2009) Infection of neurons and encephalitis after intracranial inoculation of herp4es simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci USA 106: 17916-17920. [Crossref]
- Valyi-Nagy T, Sheth V, Clement C, Tiwari V, Scanlan P, Kavouras JH, et al. (2004) Herpes simplex virus entry receptor nectin-1 is widely expressed in the murine eye. Curr Eye Res 29: 303-309.
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, et al. (1999) A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99: 13-22. [Crossref]
- Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, et al. (2010) Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467: 859-862. [Crossref]
- Satoh T, Arii J, Suenaga T, Wang J, Kogure A, et al. (2008) PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132: 935-944. [Crossref]
- Shiratori I, Ogasawara K, Saito T, Lanier LL, Arase H (2004) Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J Exp Med 199: 525-533. [Crossref]
- Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, et al. (2010) Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107: 866-871.
- Arii J, Hirohata Y, Kato A, Kawaguchi Y (2015) Nonmuscle myosin heavy chain IIb mediates herpes simplex virus 1 entry. J Virol 89: 1879-1888.
- Li D, Miller M, Chantler PD (1994) Association of a cellular myosin II with anionicphospholipids and the neuronal plasma membrane. Proc Natl Acad Sci U S A 91: 853-857. [Crossref]
- Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W (2005) Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170: 317-325. [Crossref]
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, et al. (2001) Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell 8: 169-179. [Crossref]
- Parry C, Bell S, Minson T, Browne H (2005) Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J Gen Virol 86: 7-10. [Crossref]
- Nicola AV, Straus SE (2004) Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78: 7508-7517. [Crossref]
- Nicola AV, Hou J, Major EO, Straus SE (2005) Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol 79: 7609-7616. [Crossref]
- Hayashi K, Hooper LC, Chin MS, Nagineni CN, Detrick B, et al. (2006) Herpes simplex virus 1 (HSV-1) DNA and immune complex (HSV-1-human IgG) elicit vigorous interleukin 6 release from infected corneal cells via Toll-like receptors. J Gen Virol 87: 2161-2169. [Crossref]
- Blander JM, Medzhitov R (2004) Regulation of phagosome maturation by signals from toll-like receptors. Science 304: 1014-1018. [Crossref]
- Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2: 675-680. [Crossref]
- Cheshenko N, Liu W, Satlin LM, Herold BC (2007) Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol Biol Cell 18: 3119-3130. [Crossref]
- Dohner K, Radtke K, Schmidt S, Sodeik B (2006) Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J Virol 80: 8211-8224. [Crossref]
- Gibson W, Roizman B (1971) Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc Natl Acad Sci U S A 68: 2818-2821. [Crossref]
- Bauer DW, Huffman JB, Homa FL, Evilevitch A (2013) Herpes virus genome, the pressure is on. J Am Chem Soc 135: 11216-11221. [Crossref]
- Copeland AM, Newcomb WW, Brown JC (2009) Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J Virol 83: 1660-1668. [Crossref]
- Strang BL, Stow ND (2005) Circularization of the herpes simplex virus type 1 genome upon lytic infection. J Virol 79: 12487-12494. [Crossref]
- Lehman IR, Boehmer PE (1999) Replication of herpes simplex virus DNA. J Biol Chem 274: 28059-28062.
- Conway JF, Homa FL (2011) Nucleocapsid structure, assembly and DNA of herpes simplex virus in: S.K. Weller, (Ed.), Alphaherpesviruses: MolecularVirology Caster Academic Press, Norfolk UK pp. 75-193.
- Suspe`ne R, Aynaud MM, Koch S, Pasdeloup D, Labetoulle M, et al. (2011) Genetic editing of herpes simplex virus 1 and Epstein–Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J Virol 85: 7594-7602. [Crossref]
- Herrera FJ, Tiezenberg SJ (2004) VP16-dependent association of chromatoin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters. J Virol 78: 9689-9696. [Crossref]
- Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A (2010) The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 24: 1253-1265. [Crossref]
- Isaac A, Wilcox KW, Taylor JL (2006) SP100B, a repressor of gene expression preferentially binds to DNA with unmethylated CpGs. J Cell Biochem 98: 1106-1122. [Crossref]
- Sahin U, Lallemand-Breitenbach V, de The H (2014) PML nuclear bodies: regulation, function and therapeutic perspectives. J Pathol 234: 289-291.
- Gu H, Zheng Y (2016) Role of ND10 nuclear bodies in the chromatin repression of HSV-1. Virol J 13: 62.
- Chee AV, Lopez P, Pandolfi PP, Roizman B (2003) Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol 77: 7101-7105.
- Chelbi-Alix MK, de The H (1999) Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18: 935-941. [Crossref]
- Saffert RT, Kalejta RF (2006) Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80: 3863-3871. [Crossref]
- Barlow PN, Luisi B, Milner A, Elliott M, Everett R (1994) Structure of the C3HC4 domain by 1H-nuclear magnetic resonance spectroscopy. A new structural class of zinc-finger. J Mol Biol 237: 201-211.
- Gu H (2016) Infected cell protein 0 functional domains and their coordination in herpes simplex virus replication. World J Virol 5: 1-13.
- Everett RD, Parada C, Gripon P, Sirma H, Orr A (2008) Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82: 2661-2672. [Crossref]
- Gu H, Zheng Y, Roizman B (2013) Interaction of herpes simplex virus ICP0 with ND10 bodies: a sequential process of adhesion, fusion, and retention. J Virol 87: 10244-10254. [Crossref]
- Lukashchuk V, Everett RD (2010) Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J Virol 84: 4026-4040. [Crossref]
- Maul GG, Guldner HH, Spivack JG (1993) Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J Gen Virol 74: 2679-2690. [Crossref]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, et al. (2014) IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog 10: e1004503. [Crossref]
- Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM (2013) Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110: E4492-E4501. [Crossref]
- Li T, Diner BA, Chen J, Cristea IM (2012) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109: 10558-10563. [Crossref]
- Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, et al. (2014) Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289: 23568-23581. [Crossref]
- Gu H, Liang Y, Mandel G, Roizman B (2005) Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A 102: 7571-7576. [Crossref]
- Song X, Zhao Z, Barber B, Gregory C, Wang PF, et al. (2011) Treatment patterns and metastasectomy among mCRC patients receiving chemotherapy and biologics. Curr Med Res Opin 27: 123-130. [Crossref]
- Gu H, Roizman B (2007) Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104: 17134-17139. [Crossref]
- Maul GG (1998) Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20: 660-667. [Crossref]
- Kalamvoki M, Roizman B (2008) Nuclear retention of ICP0 in cells exposed to HDAC inhibitor or transfected with DNA before infection with herpes simplex virus 1. Proc Natl Acad Sci U S A 105: 20488-20493.
- Kristie TM, Roizman B (1987) Host cell proteins bind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1 alpha genes. Proc Natl Acad Sci U S A 84: 71-75. [Crossref]
- McKnight JL, Kristie TM, Roizman B (1987) Binding of the virion protein mediating alpha gene induction in herpes simplex virus 1-infected cells to its cis site requires cellular proteins. Proc Natl Acad Sci U S A 84: 7061-7065. [Crossref]
- Pellett PE, McKnight JL, Jenkins FJ, Roizman B (1985) Nucleotide sequence and predicted amino acid sequence of a protein encoded in a small herpes simplex virus DNA fragment capable of trans-inducing alpha genes. Proc Natl Acad Sci U S A 82: 5870-5874. [Crossref]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM (2009) Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15: 1312-1317. [Crossref]
- Efstathiou S, Preston CM (2005) Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res 111: 108-119. [Crossref]
- Honess RW, Roizman B (1974) Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 14: 8-19. [Crossref]
- Boutell C, Everett RD (2013) Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94: 465-481.
- Smith RW, Malik P, Clements JB (2005) The herpes simplex virus ICP27 protein: a multifunctional post-transcriptional regulator of gene expression. Biochem Soc Trans 33: 499-501. [Crossref]
- Hill A, Jugovic P, York I, Russ G, Bennink J, et al. (1995) Herpes simplex virus turns off the TAP to evade host immunity. Nature 375: 411-415. [Crossref]
- Conn KL, Hendzel MJ, Schang LM (2011) Core histones H2B and H4 are mobilized during infection with herpes simplex virus 1. J Virol 85: 13234-13252.
- Kristie TM, Vogel JL, Sears AE (1999) Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci U S A 96: 1229-1233. [Crossref]
- Kolb G, Kristie TM (2008) Association of the cellular coactivator HCF-1 with the Golgi apparatus in sensory neurons. J Virol 82: 9555-9563.
- Lakin ND, Palmer R, Lillycrop KA, Howard MK, Burke LC, et al. (1995) Down regulation of the octamer binding protein Oct-1 during growth arrest and differentiation of a neuronal cell line. Brain Res Mol Brain Res 28: 47-54. [Crossref]
- Henderson G, Jaber T, Carpenter D, Wechsler SL, Jones C (2009) Identification of herpes simplex virus type 1 proteins encoded within the first 1.5 kb of the latency-associated transcript. J Neurovirol 15: 439-448. [Crossref]
- Thomas SK, Lilley CE, Latchman DS, Coffin RS (2002) A protein encoded by the herpes simplex virus (HSV) type 1 2-kilobase latency-associated transcript is phosphorylated, localized to the nucleus, and overcomes the repression of expression from exogenous promoters when inserted into the quiescent HSV genome. J Virol 76: 4056-4067. [Crossref]
- Knotts FB, Cook ML, Stevens JG (1974) Pathogenesis of herpetic encephalitis in mice after ophthalmic inoculation. J Infect Dis 130: 16-27. [Crossref]
- Hamza MA, Higgins DM, Feldman LT, Ruyechan WT (2007) The latency-associated transcript of herpes simplex virus type 1 promotes survival and stimulates axonal regeneration in sympathetic and trigeminal neurons. J Neurovirol 13: 56-66.
- Deshmane SL, Fraser NW (1989) During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 63: 943-947.
- Halford WP, Gebhardt BM, Carr DJ (1996) Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol 157: 3542-3549.
- Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL (2003) Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18: 593-603. [Crossref]
- Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, et al. (2003) Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol 163: 2179-2184. [Crossref]
- Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, et al. (2008) Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322: 268-271. [Crossref]
- Ekstrand MI, Enquist LW, Pomeranz LE (2008) The alpha-herpesviruses: molecular pathfinders in nervous system circuits. Trends Mol Med 14: 134-140. [Crossref]
- Sawtell NM (1997) Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol 71: 5423-5431. [Crossref]
- Held K, Junker A, Dornmair K, Meinl E, Sinicina I, Brandt T, et al. (2011) Expression of herpes simplex virus 1-encoded microRNAs in human trigeminal ganglia and their relation to local T-cell infiltrates. J Virol 85: 9680-9685.
- Kalamvoki M, Du T, Roizman B (2014) Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc Natl Acad Sci U S A 111: E4991-E4996. [Crossref]
- Kalamvoki M, Deschamps T (2016) Extracellular vesicles during Herpes Simplex Virus type 1 infection: an inquire. Virol J 13: 63. [Crossref]
- Johnson KE, Chikoti L, Chandran B (2013) Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87: 5005-5018.
- Ennaciri J, Menezes J, Proulx F, Toledano BJ (2006) Induction of apoptosis by herpes simplex virus-1 in neonatal, but not adult, neutrophils. Pediatr Res 59: 7-12. [Crossref]
- Heilingloh CS, Kummer M, Muhl-Zurbes P, Drassner C, Daniel C, et al. (2015) L particles transmit viral proteins from herpes simplex virus 1-infected mature dendritic cells to uninfected bystander cells, inducing CD83 downmodulation. J Virol 89: 11046-11055.
- Javier RT, Stevens JG, Dissette VB, Wagner EK (1988) A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology 166: 254-257.
- Zabolotny JM, Krummenacher C, Fraser NW (1997) The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J Virol 7: 4199-4208. [Crossref]
- Deatly AM, Spivack JG, Lavi E, Fraser NW (1987) RNA from an immediate early region of the type 1 herpes simplex virus genome is present in the trigeminal ganglia of latently infected mice. Proc Natl Acad Sci U S A 84: 3204-3208. [Crossref]
- Goldenberg D, Mador N, Ball MJ, Panet A, Steiner I (1997) The abundant latency-associated transcripts of herpes simplex virus type 1 are bound to polyribosomes in cultured neuronal cells and during latent infection in mouse trigeminal ganglia. J Virol 71: 2897-2904. [Crossref]
- Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C (2008) Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol 14: 41-52. [Crossref]
- Shen W, Sa e Silva M, Jaber T, Vitvitskaia O, Li S, et al. (2009) Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J Virol 83: 9131-9139. [Crossref]
- Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, et al. (2014) Interactions between herpesvirus entry mediator (TNFRSF14) and latency-associated transcript during herpes simplex virus l latency. J Virol 88: 1961-1971. [Crossref]
- Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, et al. (2010) Numerous conserved and divergent micro RNAs expressed by herpes simplex viruses 1 and 2. J Virol 84: 4659-4672. [Crossref]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, et al. (2008) MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate mRNAs. Nature 454: 780-783. [Crossref]
- Sun L, Li Q (2012) The miRNAs of herpes simplex virus (HSV). Virol Sin 27: 333-338. [Crossref]
- Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, et al. (2006) Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol 80: 5499-5508.
- Tang S, Patel A, Krause PR (2009) Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol 83: 1433-1442. [Crossref]
- Jiang X, Brown D, Osorio N, Hsiang C, Li L, et al. (2015) A herpes simplex virus type 1 mutant disrupted for microRNA H2 with increased neurovirulence and rate of reactivation. J Neurovirol 21: 199-209. [Crossref]
- Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, et al. (2014) A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 15: 446-456.
- Pan D, Pesola JM, Li G, McCarron S, Coen DM (2017) Mutations inactivating herpes simplex virus 1 microRNA miR-H2 do not detectably increase ICP0 gene expression in infected cultured cells or mouse trigeminal ganglia. J Virol 91: e02001-e02016. [Crossref]
- Randall G, Roizman B (1997) Transcription of the derepressed open reading frame P of herpes simplex virus 1 precludes the expression of the antisense gamma (1) 34.5 gene and may account for the attenuation of the mutant virus. J Virol 71: 7750-7757. [Crossref]
- Du T, Han Z, Zhou G, Roizman B (2015) Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc Natl Acad Sci U S A 112: E49-E55. [Crossref]
- Bhela S, Mulik S, Reddy PB, Richardson RL, Gimenez F, et al. (2014) Critical role of microRNA-155 in herpes simplex encephalitis. J Immunol 192: 2734-2743. [Crossref]
- Knipe DM, Cliffe A (2008) Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6: 211-221. [Crossref]
- Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, et al. (2005) Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102: 16055-16059. [Crossref]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786-791. [Crossref]
- Wu J, Sun L, Chen X, Du F, Shi H, et al. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826-830. [Crossref]
- Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, et al. (2015) cAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112: E1773-E1781. [Crossref]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, et al. (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553-557. [Crossref]
- Shimeld C, Whiteland JL, Williams NA, Easty DL, Hill TJ (1997) Cytokine production in the nervous system of mice during acute and latent infection with herpes simplex virus type 1. J Gen Virol 78: 3317-3325. [Crossref]
- Shimeld C, Whiteland JL, Nicholls SM, Grinfeld E, Easty DL, et al. (1995) Immune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J Neuroimmunol 61: 7-16.
- Sciammas R, Kodukula P, Tang Q, Hendricks RL, Bluestone JA (1997) T cell receptor-gamma/delta cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. J Exp Med 185: 1969-1975. [Crossref]
- Heise MT, Virgin HW (1995) The T-cell-independent role of gamma interferon and tumor necrosis factor alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infections. J Virol 69: 904-909. [Crossref]
- MacMicking J, Xie QW, Nathan C (1997) Nitric oxide and macrophage function. Annu Rev Immunol 15: 323-350. [Crossref]
- Goldsmith K, Chen W, Johnson DC, Hendricks RL (1998) Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med 187: 341-348. [Crossref]
- Liu T, Tang Q, Hendricks RL (1996) Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol 70: 264-271.
- Jiang X, Chentoufi AA, Hsiang C, Carpenter D, Osorio N, et al. (2011) The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol 85: 2325-2332. [Crossref]
- Mott KR, Allen SJ, Zandian M, Konda B, Sharifi BG, et al. (2014) CD8alpha dendritic cells drive establishment of HSV-1 latency. PLoS One 9: e93444. [Crossref]
- Mott KR, Allen SJ, Zandian M, Ghiasi H (2014) Coregulatory interactions among CD8alpha dendritic cells, the latency-associated transcript, and programmed death 1 contribute to higher levels of herpes simplex virus 1 latency. J Virol 88: 6599-6610
- Rogov V, Dotsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167-178. [Crossref]
- Du T, Zhou G, Roizman B (2013) Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc Natl Acad Sci U S A 110: E2621-E2628.
- Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM (2007) In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol 81: 13248-13253. [Crossref]
- Wilcox CL, Johnson EM, Jr (1987) Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J Virol 61: 2311-2315. [Crossref]
- Wilcox CL, Smith RL, Freed CR, Johnson EM, Jr (1990) Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J Neurosci 10: 1268-1275. [Crossref]
- Kobayashi M, Kim JY, Camarena V, Roehm PC, Chao MV, et al. (2012) A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J Vis Exp 2: 3823. [Crossref]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC (2012) Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog 8: e1002540. [Crossref]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, et al. (2010) Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8: 320-330. [Crossref]
- Kobayashi M, Wilson AC, Chao MV, Mohr I (2012) Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev 26: 1527-1532. [Crossref]
- Penkert RR, Kalejta RF (2011) Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2: 3. [Crossref]
- St Leger AJ, Hendricks RL (2011) CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J Neurovirol 17: 528-534. [Crossref]
- Bigley NJ (2014) Complexity of interferon-gamma interactions with HSV-1. Front Immunol 5: 15. [Crossref]
- Sawtell NM, Triezenberg SJ, Thompson RL (2011) VP16 serine 375 is a critical determinant of herpes simplex virus exit from latency in vivo. J Neurovirol 17: 546-551. [Crossref]
- Liu Y, Gong W, Huang CC, Herr W, Cheng X (1999) Crystal structure of the conserved core of the herpes simplex virus transcriptional regulatory protein VP16. Genes Dev 13: 1692-1703. [Crossref]
- Everett RD (2000) ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioassays 22: 761-770. [Crossref]
- Koelle DM, Corey L (2008) Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med 59: 381-395. [Crossref]
- Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, et al. (2013) The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol 87: 7140-7148. [Crossref]
- Fraser NW, Lawrence WC, Wroblewska Z, Gilden DH, Koprowski H (1981) Herpes simplex type 1 DNA in human brain tissue. Proc Natl Acad Sci U S A 78: 6461-6465. [Crossref]
- Newcomb WW, Fontana J, Winkler DC, Cheng N, Heymann JB, et al. (2017) The primary enveloped virion of herpes simplex virus 1: Its role in nuclear egress. MBio 8: e00825-e00817. [Crossref]
- Baines JD, Jacob RJ, Simmerman L, Roizman B (1995) The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J Virol 69: 825-833. [Crossref]
- Naldinho-Souto R, Browne H, Minson T (2006) Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J Virol 80: 2582-2584. [Crossref]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD (2002) Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76: 8939-8952. [Crossref]
- Owen DJ, Crump CM, Graham SC (2015) Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7: 5084-5114. [Crossref]
- Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79: 9566-9571.
- Loomis JS, Bowzard JB, Courtney RJ, Wills JW (2001) Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J Virol 75: 12209-12219.
- Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13: 83-117. [Crossref]
- Luders J, Stearns T (2007) Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol 8: 161-167. [Crossref]
- Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL (2008) Transport and egress of herpes simplex virus in neurons. Rev Med Virol 1: 35-51. [Crossref]
- DuRaine G, Wisner TW, Howard P, Williams M, Johnson DC. et al. (2017) Herpes simplex virus gE/gI and US9 promote both envelopment and sorting of virus particles in the cytoplasm of neurons, two processes that precede anterograde transport in axons. J Virol 91: e00050-17. [Crossref]
- Kratchmarov R, Taylor MP, Enquist LW (2012) Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev Med Virol 22: 378-391.
- Snyder A, Wisner TW, Johnson DC (2006) Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J Virol 80: 11165-11177.
- Meyers-Elliott RH, Pettit TH, Maxwell WA (1980) Viral antigens in the immune ring of herpes simplex stromal keratitis. Arch Ophthalmol 98: 897-904. [Crossref]
- Pepose JS, Leib DA, Stuart M, Easty DL (1996) Herpes simplex virus disease: anterior segment of the eye Mosby, St. Lois, Mo.
- McBride BW, Ward KA (1987) Herpes simplex-specific IgG subclass response in herpetic keratitis. J Med Virol 21: 179-189. [Crossref]
- Wilhelmus KR, Gee L, Hauck WW, Kurinij N, Dawson CR, et al. (1994) Herpetic Eye Disease Study. A controlled trial of topical corticosteroids for herpes simplex stromal keratitis. Ophthalmology 101 1883-1895: discussion 1895-1896. [Crossref]
- Thomas J, Rouse BT (1997) Immunopathogenesis of herpetic ocular disease. Immunol Res 16: 375-386.
- Kumaraguru U, Davis I, Rouse BT (1999) Chemokines and ocular pathology caused by corneal infection with herpes simplex virus. J Neurovirol 5: 42-47. [Crossref]
- Hayashi K, Hooper LC, Detrick B, Hooks JJ (2009) HSV immune complex (HSV-IgG: IC) and HSV-DNA elicit the production of angiogenic factor VEGF and MMP-9. Arch Virol 154: 219-226. [Crossref]
- Kumaraguru U, Rouse BT (2002) The IL-12 response to herpes simplex virus is mainly a paracrine response of reactive inflammatory cells. J Leukoc Biol 72: 564-570.
- Hayashi K, Hooper LC, Okuno T, Takada Y, Hooks JJ (2012) Inhibition of HSV-1 by chemoattracted neutrophils: supernatants of corneal epithelial cells (HCE) and macrophages (THP-1) treated with virus components chemoattract neutrophils (PMN), and supernatants of PMN treated with these conditioned media inhibit viral growth. Arch Virol 157: 1377-1381. [Crossref]
- Youinou P, Colin J, Ferec C (1986) Monoclonal antibody analysis of blood and cornea T lymphocyte subpopulations in herpes simplex keratitis. Graefes Arch Clin Exp Ophthalmol 224: 131-133. [Crossref]
- Verjans GM, Remeijer L, van Binnendijk RS, Cornelissen JG, Volker-Dieben HJ, et al. (1998) Identification and characterization of herpes simplex virus-specific CD4+ T cells in corneas of herpetic stromal keratitis patients. J Infect Dis 177: 484-488. [Crossref]
- Koelle DM, Reymond SN, Chen H, Kwok WW, McClurkan C, et al. (2000) Tegument-specific, virus-reactive CD4 T cells localize to the cornea in herpes simplex virus interstitial keratitis in humans. J Virol 74: 10930-10938. [Crossref]
- Youinou P, Colin J, Mottier D (1985) Immunological analysis of the cornea in herpetic stromal keratitis. J Clin Lab Immunol 17: 105-106. [Crossref]
- Miller JK, Laycock KA, Nash MM, Pepose JS (1993) Corneal Langerhans cell dynamics after herpes simplex virus reactivation. Invest Ophthalmol Vis Sci 34: 2282-2290. [Crossref]
- Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT (2001) Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol 75: 9828-9835. [Crossref]
- Gimenez F, Suryawanshi A, Rouse BT (2013) Pathogenesis of herpes stromal keratitis--a focus on corneal neovascularization. Prog Retin Eye Res 33: 1-9. [Crossref]
- Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A (2003) Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198: 513-520. [Crossref]
- Lundberg P, Welander P, Han X, Cantin E (2003) Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J Virol 77: 11158-11169.
- Maggs DJ, Chang E, Nasisse MP, Mitchell WJ (1998) Persistence of herpes simplex virus type 1 DNA in chronic conjunctival and eyelid lesions of mice. J Virol 72: 9166-9172.
- Veiga-Parga T, Sehrawat S, Rouse BT (2013) Role of regulatory T cells during virus infection. Immunol Rev 255: 182-196. [Crossref]
- Gaddipati S, Estrada K, Rao P, Jerome AD, Suvas S (2015) IL-2/anti-IL-2 antibody complex treatment inhibits the development but not the progression of herpetic stromal keratitis. J Immunol 194: 273-282. [Crossref]
- Meyers-Elliott RH, Chitjian PA, Dethlefs BA (1983) Experimental herpesvirus keratitis in the rabbit: topical versus intrastromal infection routes. Ophthalmic Res 15: 240-256. [Crossref]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, et al. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408: 740-745. [Crossref]
- Akira S, Hemmi H (2003) Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85: 85-95. [Crossref]
- Iwasaki A, Medzhitov R (2004) Toll-like receptor control of the adaptive immune responses. Nat Immunol 5: 987-995. [Crossref]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M (2004) Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103: 1433-1437
- Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T (2002) Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun 293: 1364-1369.
- Ueta M, Hamuro J, Kiyono H, Kinoshita S (2005) Triggering of TLR3 by polyI:C in human corneal epithelial cells to induce inflammatory cytokines. Biochem Biophys Res Commun 331: 285-294. [Crossref]
- Mitchell WJ, Gressens P, Martin JR, DeSanto R (1994) Herpes simplex virus type 1 DNA persistence, progressive disease and transgenic immediate early gene promoter activity in chronic corneal infections in mice. J Gen Virol 75: 1201-1210. [Crossref]
- Takeda S, Miyazaki D, Sasaki S, Yamamoto Y, Terasaka Y, et al. (2011) Roles played by toll-like receptor-9 in corneal endothelial cells after herpes simplex virus type 1 infection. Invest Ophthalmol Vis Sci 52: 6729-6736. [Crossref]
- Kwong AD, Kruper JA, Frenkel N (1988) Herpes simplex virus virion host shutoff function. J Virol 62: 912-921. [Crossref]
- Taddeo B, Roizman B (2006) The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80: 9341-9345. [Crossref]
- Tran MT, Dean DA, Lausch RN, Oakes JE (1998) Membranes of herpes simplex virus type-1-infected human corneal epithelial cells are not permeabilized to macromolecules and therefore do not release IL-1alpha. Virology 244: 74-78. [Crossref]
- Duan R, Remeijer L, van Dun JM, Osterhaus AD, Verjans GM (2007) Granulocyte macrophage colony-stimulating factor expression in human herpetic stromal keratitis: implications for the role of neutrophils in HSK. Invest Ophthalmol Vis Sci 48: 277-284. [Crossref]
- Kanangat S, Babu JS, Knipe DM, Rouse BT (1996) HSV-1-mediated modulation of cytokine gene expression in a permissive cell line: selective upregulation of IL-6 gene expression. Virology 219: 295-300.
- Biswas PS, Banerjee K, Kinchington PR, Rouse BT (2006) Involvement of IL-6 in the paracrine production of VEGF in ocular HSV-1 infection. Exp Eye Res 82: 46-54.
- Lee S, Zheng M, Kim B, Rouse BT (2002) Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J Clin Invest 110: 1105-1111. [Crossref]
- Kim B, Suvas S, Sarangi PP, Lee S, Reisfeld RA, et al. (2006) Vascular endothelial growth factor receptor 2-based DNA immunization delays development of herpetic stromal keratitis by antiangiogenic effects. J Immunol 177: 4122-4131. [Crossref]
- Zheng M, Schwarz MA, Lee S, Kumaraguru U, Rouse BT (2001) Control of stromal keratitis by inhibition of neovascularization. Am J Pathol 159: 1021-1029. [Crossref]
- Thomas J, Gangappa S, Kanangat S, Rouse BT (1997) On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol 158: 1383-1391. [Crossref]
- Fleetwood AJ, Cook AD, Hamilton JA (2005) Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol 25: 405-428. [Crossref]
- Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T (2003) Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem 278: 3170-3175. [Crossref]
- Minagawa H, Hashimoto K, Yanagi Y (2004) Absence of tumour necrosis factor facilitates primary and recurrent herpes simplex virus-1 infections. J Gen Virol 85: 343-347. [Crossref]
- Lundberg P, Welander PV, Edwards CK, 3rd, van Rooijen N, Cantin E (2007) Tumor necrosis factor (TNF) protects resistant C57BL/6 mice against herpes simplex virus-induced encephalitis independently of signaling via TNF receptor 1 or 2. J Virol 81: 1451-1460. [Crossref]
- Niedbala W, Wei XQ, Campbell C, Thomson D, Komai-Koma M, et al. (2002) Nitric oxide preferentially induces type 1 T cell differentiation by selectively up-regulating IL-12 receptor beta 2 expression via cGMP. Proc Natl Acad Sci U S A 99: 16186-16191. [Crossref]
- Inoue T, Inoue Y, Hayashi K, Yoshida A, Nishida K, et al. (2002) Topical administration of HSV gD-IL-2 DNA is highly protective against murine herpetic stromal keratitis. Cornea 21: 106-110. [Crossref]
- Kuo T, Wang C, Badakhshan T, Chilukuri S, BenMohamed L (2014) The challenges and opportunities for the development of a T-cell epitope-based herpes simplex vaccine. Vaccine 32: 6733-6745. [Crossref]
- Gierynska M, Kumaraguru U, Eo SK, Lee S, Krieg A, et al. (2002) Induction of CD8 T-cell-specific systemic and mucosal immunity against herpes simplex virus with CpG-peptide complexes. J Virol 76: 6568-6576.
- Knipe DM, Corey L, Cohen JI, Deal CD (2014) Summary and recommendations from a National Institute of Allergy and Infectious Diseases (NIAID) workshop on "Next Generation Herpes Simplex Virus Vaccines". Vaccine 32: 1561-1562. [Crossref]
- van Diemen FR, Kruse EM, Hooykaas MJ, Bruggeling CE, Schurch AC, et al. (2016) CRISPR/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog 12: e1005701. [Crossref]