Abstract
Diabetic Ketoacidosis (DKA) is an endocrine disorder characterized by pronounced hyperglycemia, occurring in individuals with pre-existing diabetes mellitus. Emerging evidence has delineated a complex spectrum of cardiac manifestations of DKA leading to challenges in management. The term “pseudo-infarction” has often been used as an umbrella term to clarify these complex cardiac manifestations. This case-series highlights three separate cases of DKA with concomitant myocardial injury and the difficulty and complexity it poses to clinicians when deciding on management and treatment.
Keywords
DKA, myocardial injury
Introduction
Diabetic Ketoacidosis (DKA) is an endocrine pathology commonly associated with profound hyperglycemia in the setting of underlying uncontrolled diabetes mellitus. This metabolic derangement can manifest in both Type 1 Diabetes Mellitus (T1DM) and Type 2 Diabetes Mellitus (T2DM), constituting a hyperglycemic emergency characterized by a triad of hyperglycemia, ketosis, and anion gap metabolic acidosis [1]. Inciting factors often encompass medication noncompliance or the presence of secondary triggers such as underlying infection [1]. Standard diagnostic protocols typically entail close monitoring of blood glucose levels, assessment of serum ketones, determination of anion gap, and comprehensive blood gas analysis. Management strategies conventionally entail stringent glycemic control, aggressive fluid resuscitation, and vigilant monitoring of ketosis resolution, anion gap normalization, and correction of electrolyte imbalances, as well as addressing any possible underlying infectious etiology [1,2].
There is pre-existing literature highlighting a relationship between DKA and its effect on the cardiovascular system including several acute EKG changes [3]. Among these are acute pseudo-myocardial infarction patterns which can result from multiple alternative cardiac pathologies such as myocarditis, myocardial necrosis, and electrolyte disturbances [4-7]. A paper from Nunes, et al. [8] attempted to redefine the clinical meaning of pseudo-myocardial infarction to better align with modern standards of myocardial infarction. The use of cardiac troponin and high sensitivity troponin when diagnosing acute myocardial infarctions (AMI) has prompted the need for a change in definition [8,9]. Whereas the older definition entails utilizing chest pain in conjunction with EKG changes, Nunes postulates that the definition should be broadened to include troponin elevation in the absence of definitive CAD or ischemic infarction [10]. For this case report, the latter definition will be used.
Carrizales-Sepúlveda, et al. [11] conducted a literature review examining the cardiovascular implications of DKA, examining 20 cases of pseudo-myocardial infarction in patients with DKA. In 18 of these cases with concomitant hyperkalemia, 85% showed EKG changes showing ST elevations in the anteroseptal leads. Eight of these cases underwent cardiac catheterization which showed no evidence of obstructive coronary disease, and these EKG changes corrected in all these patients after DKA treatment and resolution [12]. Interestingly, this does not seem to be exclusive to patients with hyperkalemia as there has also been a reported case of pseudo-infarction in a patient that was normokalemic [4,6]. This case series attempts to discuss the complex relationship between DKA and myocardial injury and the challenges it poses to a clinician.
Case presentation
Case 1:
The patient is a 19-year-old non-obese black male with a past medical history of T1DM on an insulin at home who was admitted to the medical intensive care unit (MICU) for DKA. The patient presented with a two-day history of generalized weakness. The patient had not taken the insulin at home for a week due to not receiving medications from the pharmacy. The patient had never been in DKA prior to this admission. Family history was significant for an MI in the patient’s father at an early age (30s), otherwise no history of diabetes or any other family members having DKA. Social history was positive for daily vape and marijuana use. The patient also stated that he recently had a sore throat, however, did not see a provider for this. Review of systems was negative aside from increased urinary frequency. Physical exam was within normal limits.
On initial vitals the patient met SIRS criteria without evidence of infection (Table 1). In the ED the patient received a dose of piperacillin-tazobactam. Blood cultures and urine cultures obtained were negative. A diagnosis of DKA was established following initial laboratory results (Table 2). Pertinent labs for diagnosis included high anion gap metabolic acidosis, positive urine and serum ketones, severe hyperglycemia, and multiple electrolyte disturbances. Initial EKG showed ST repolarization patterns and peaked T waves suggestive of hyperkalemia but initial Troponin I was negative (Table 1 and Figure 1). Calcium gluconate and sodium bicarbonate were administered in the ED. The patient was started on DKA protocol with serial monitoring of labs and venous blood gas to evaluate acid-base status as well as close monitoring of anion gap until closure. The treatment regimen consisted of aggressive fluid resuscitation, insulin drip, electrolyte replenishment and telemetry monitoring. The patient continued to improve both clinically and chemically. Eventually the patient had a negative anion gap x 1, serum blood glucose <200, and negative ketones about 24 hours after admission.
However, while monitoring the patient the telemetry strip on the monitor showed ST elevations. An EKG was performed which showed ST elevations in multiple leads (Figure 2). A troponin level was immediately ordered, and high sensitivity Troponins were ~4000. Despite this, the patient remained asymptomatic, and the physical exam did not show any evidence of pleural rub or abnormal heart sounds. Cardiology was consulted and further labs were obtained including erythrocyte sedimentation rate. The patient was started on treatment for acute coronary syndrome (ACS) including aspirin, clopidogrel, and heparin drip. Transthoracic echocardiography (TTE) showed an ejection fraction of 60%-65% without wall motion abnormalities or effusion. After careful consideration of the full clinical picture, coronary angiography was deferred. It was suspected that the cardiac changes were due to demand ischemia from overwhelming metabolic stress. There was also suspicion of myopericarditis due to the patient testing positive for Coxsackie virus later in the course. The patient was ultimately discharged with routine follow up.
Labs/Vitals |
Values |
Temp (36-38°C) |
35.2°C (36-38°C) |
HR |
107 bpm |
RR |
23 breaths/min |
BP |
167/106 mmHg |
SpO2 |
99%-100% on room air |
WBC (4-10 × 109/L) |
43.86 × 109/L |
H&H (Hgb, Hct) (14-17 g/dL and 41%-51% respectively) |
16.6 g/dL & 51.8% |
Platelets (150-350 × 109/L) |
342 × 109/L |
Table 1. Initial vitals and CBC. Per SIRS criteria, the patient was positive for 3 out of 4 criteria (Temp, WBC, HR). Values in parentheses are normal values as per guidelines from American College of Physicians
|
Initial |
24 h after admission |
Na (136-145 mEq/L) |
121 mEq/L |
137 mEq/L |
K (3.5-5 mEq/L) |
6.5 mEq/L |
4.2 mEq/L |
Cl (98-106 mEq/L) |
89 mEq/L |
108 mEq/L |
HCO3 (23-28 mEq/L) |
<12 mEq/L |
13 mEq/L |
Mg (1.5-2.4 mg/dL) |
2.6 mg/dL |
1.9 mg/dL |
Phosphorus (3-4.5 mg/dL) |
7.8 mg/dL |
1.7 mg/dL |
Anion Gap |
22 (<12) |
16 |
Serum Glucose (70-100 mg/dL when fasting) |
600 mg/dL |
218 mg/dL |
ALT/AST (0-35 units/L) |
43/40 units/L |
31/24 units/L |
Alk Phos (36-92 unit/L) |
254 units/L |
128 units/L |
BUN/Cr |
29/1.8 mg/dL |
11/1 mg/dL |
Lactate (0.67-1.8 mmol/L) |
2.6 mmol/L |
1.1 mmol/L |
Plasma Ketones |
Small |
Small |
Serum Osm |
323 mOsm/kg/H2O |
|
Urine Osm |
492 mOsm/Kg/H20 |
|
High sensitivity troponin (<14 ng/mL) |
7.58 ng/L |
|
CKMB (Index) |
1.35 |
|
Creatine Kinase |
163 units/L |
|
HbA1c |
>14% (<5.7%) |
|
Arterial Blood Gas: |
pH |
<6.818 (7.35-7.45) |
7.348 (7.35-7.45) |
pCO2 |
32.5 |
40.9 |
pO2 |
<30.7 |
<30.7 |
HCO3- |
Unable to result |
22.5 |
Table 2. Lab values at initial visit, 24 hours after admission. Numbers in parentheses denote normal values as per American College of Physicians.
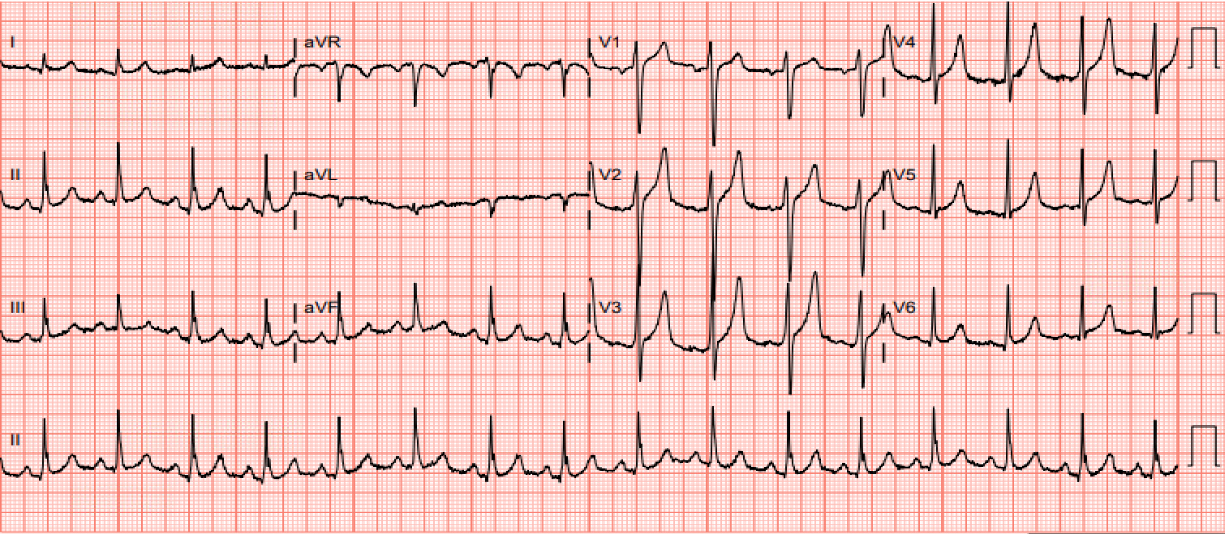
Figure 1. EKG on admission. Spiked T-waves in multiple leads indicative of hyperkalemia
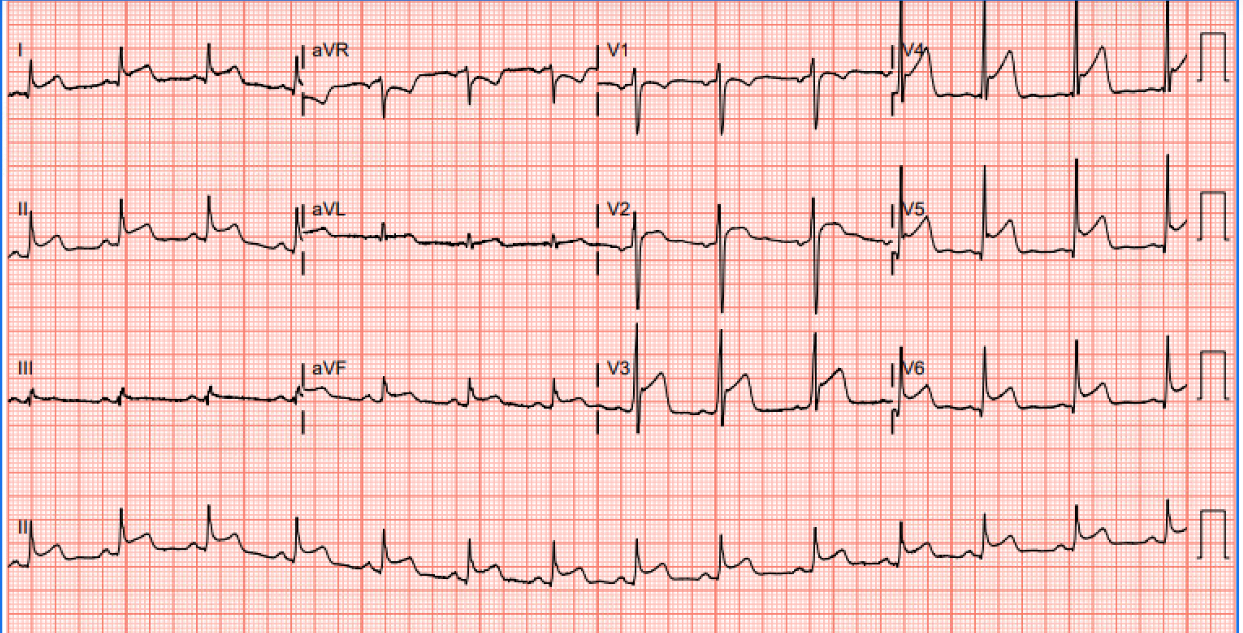
Figure 2. EKG prior to transfer: ST elevations in multiple leads, notably in leads I, II, V2-V6, and T-wave inversions in lead V1
Case 2:
Patient is a 34-year-old non-obese male with past medical history of T1DM who was initially brought in via EMS for two days of nausea and vomiting and altered mental status in the setting of elevated finger sticks. On initial evaluation in the emergency room, the patient was found to be hypotensive and had lab work suggesting diabetic ketoacidosis including elevated anion gap, acidosis, and the presence of serum ketones (Table 3 and Table 4). The patient also met SIRS criteria of unknown source and as such, the patient was admitted to the MICU for further management of DKA and suspected sepsis. The patient was subsequently started on an insulin drip for management of DKA with serial arterial blood gases (ABG) every 4 hours. Anion gap and serum ketones were also trended at a similar frequency. Given the patient was in DKA and placed on an insulin drip, hourly fingerstick blood glucose monitoring was performed. The patient continued to receive insulin drip, was started on standing nebulizers, and given calcium gluconate. The patient continued to receive aggressive IV fluid resuscitation. Nephrology was consulted for recommendations regarding initiation of hemodialysis given the hyperkalemia. Hemodialysis was ultimately deferred as the patient’s potassium showed improvement on the insulin drip and nebulizers.
The patient underwent full sepsis workup including imaging to locate sources of infection, urinalysis, and blood cultures. The patient was hemodynamically unstable despite fluid resuscitation; therefore, a right femoral central venous catheter and arterial line were placed for vasopressor support and accurate blood pressure monitoring. For vasopressors the patient was initially started on norepinephrine and vasopressin was added to further help increase the patient’s MAP >65. The patient began showing increasing respiratory difficulty and was tachypneic with a respiratory rate in the 40s. Due to concern of impending respiratory failure, the patient was intubated for airway protection and started on sedation with midazolam drip and fentanyl drip. The patient was started on IV pantoprazole every 12 hours after that the patient was intubated and started on hydrocortisone 100 mg every 8 hours to further help achieve a MAP >65. Patient was started on broad spectrum antibiotics with renally dosed cefepime and vancomycin by level given the acute renal failure likely in the setting of shock (Table 4). Urinalysis was positive for a urinary tract infection (UTI) and blood culture grew E. Coli.
Labs/Vitals |
Values |
Temp (36-38°C) |
33.3°C (36-38°C) |
HR |
82 bpm |
RR |
18 breaths/min |
BP |
86/37 mmHg |
spO2 |
100% on room air |
WBC (4-10 × 109/L) |
20.65 × 109/L |
H&H (Hgb, Hct) (14-17 g/dL and 41%-51% respectively) |
12.1 g/dL & 43.7% |
Platelets (150-350 × 109/L) |
303 × 109/L |
Table 3. Initial vitals and CBC. Values in parentheses are normal values as per guidelines from American College of Physicians
|
Initial |
24 h after admission |
Na (136-145 mEq/L) |
116 mEq/L |
133 mEq/L |
K (3.5-5 mEq/L) |
7.8 mEq/L |
5.1 mEq/L |
Cl (98-106 mEq/L) |
75 mEq/L |
94 mEq/L |
HCO3 (23-28 mEq/L) |
<12 mEq/L |
21 mEq/L |
Mg (1.5-2.4 mg/dL) |
2.9 mg/dL |
1.9 mg/dL |
Phosphorus (3-4.5 mg/dL) |
7.8 mg/dL |
1.7 mg/dL |
Anion Gap |
Unable to calculate (<12) |
18 |
Serum Glucose (70-100 mg/dL when fasting) |
>600 mg/dL |
>600 mg/dL |
ALT/AST (0-35 units/L) |
19/21 units/L |
18/23 units/L |
Alk Phos (36-92 unit/L) |
123 units/L |
122 units/L |
BUN/Cr |
100/5.9 mg/dL |
99/5.8 mg/dL |
GFR |
12 (>90) |
|
Lactate (0.67-1.8 mmol/L) |
4.9 mmol/L |
2.7 mmol/L |
Procalcitonin |
9.89 ng/mL (<0.01 ngmL) |
|
Plasma Ketones |
12.89 mmol/L (<0.6 mmol/L) |
4.25 mmoL/L |
High sensitivity troponin (<14 ng/mL) |
87 ng/L |
7008 ng/L |
CKMB (Index) |
1.35 |
4.60 |
Creatine Kinase |
245 units/L |
645 units/L |
HbA1c |
>10.3% (<5.7%) |
|
Arterial Blood Gas: |
pH |
7.045 (7.35-7.45) |
7.341 (7.35-7.45) |
pCO2 |
25.4 |
38.8 |
pO2 |
110 |
115 |
HCO3- |
6.9 |
21 |
Table 4. Lab values at initial visit, 24 hours after admission. Numbers in parentheses denote normal values as per American College of Physicians
During hospitalization, the patient had an initially elevated high sensitivity troponin of 87 ng/dL that continued to uptrend to ~7000 ng/dL. The patient also began to develop EKG changes including ST elevations in the anterior leads (Figure 3). The patient was started on heparin drip and initiated on aspirin and clopidogrel after receiving loading doses. Cardiology was subsequently consulted. A transthoracic echo was performed showing an ejection fraction of 45%-50%, hypokinesis of the basal mid inferoseptal walls. There was also akinesis of the right ventricular wall with McConnell’s sign and RV strain. The patient was started on dobutamine drip due to concern of cardiogenic shock. There was no previous TTE for comparison, however given the continued rising troponin and concerning wall motion abnormalities, the decision was made to have the patient undergo coronary angiography to evaluate for obstructive lesions and the need for PCI versus CABG. Cardiac catheterization showed reduced cardiac output and systemic vascular resistance suggestive of vasodilatory shock and normal, unobstructed coronary arteries. Further investigation revealed gram negative septicemia and pulmonary embolism. Once the infection was treated and the patient continued to improve, the patient was ultimately able to be discharged with routine follow up.
Case 3:
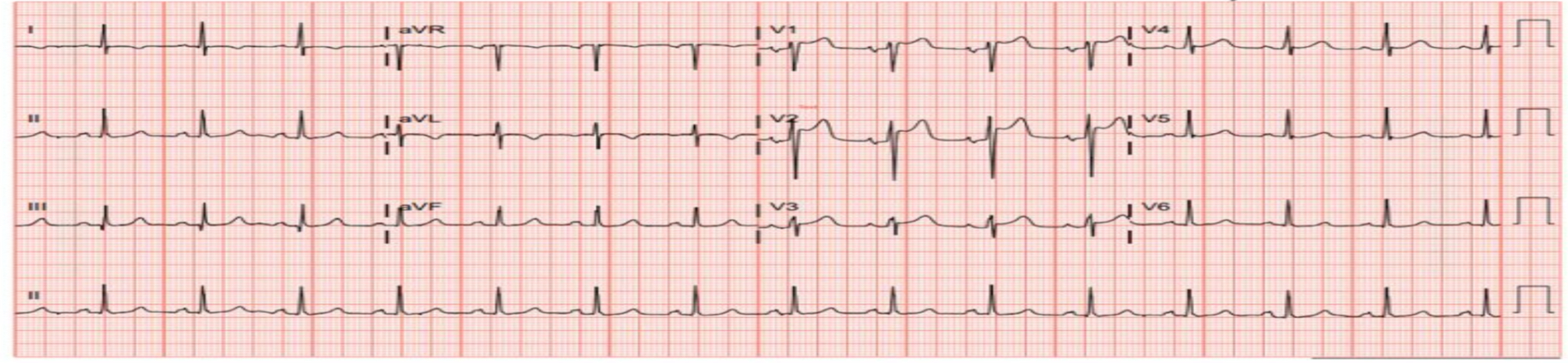
Figure 3. EKG showing ST elevations in the anterior leads, most notably in leads V1-V2
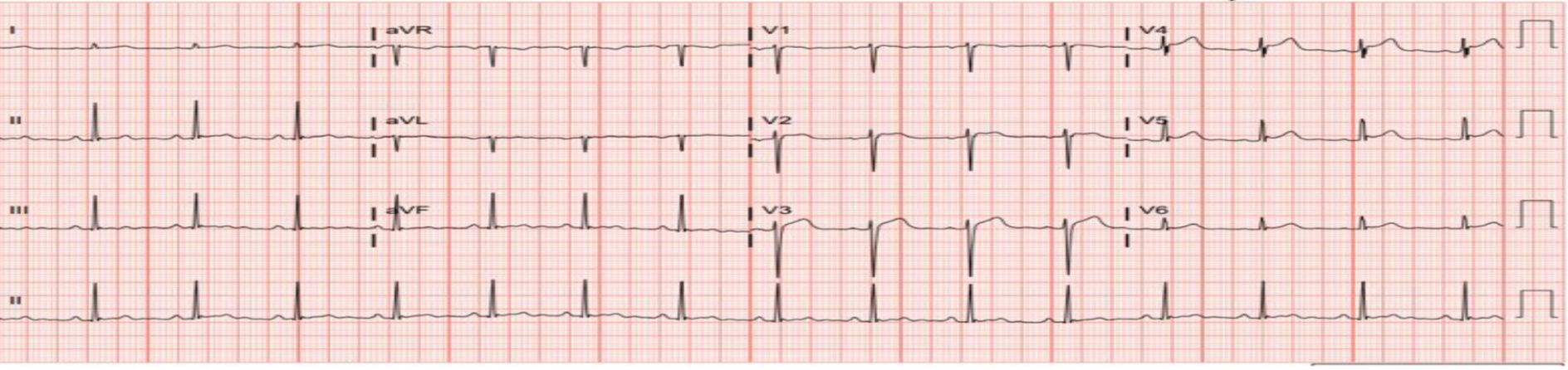
Figure 4. EKG showing ST elevation in anterior leads, primarily in leads V2-V3, however the degree of elevation does not meet STEMI criteria
Patient is a 42-year-old non-obese male with a past medical history of T1DM on insulin, hypertension, and hyperlipidemia who presented to the emergency room with a chief complaint of repeated vomiting and elevated home finger sticks at home (>400 ng/dL). The patient on initial evaluation endorsed inconsistent use of home insulin. The patient’s initial labs showed evidence of diabetic ketoacidosis and as such, the patient was started on an insulin drip (Table 6). The patient also was given aggressive IV fluid administration due to osmotic diuresis in the setting of DKA. The patient was admitted to the MICU for further evaluation and management of DKA.
Labs/Vitals |
Values |
Temp (36-38°C) |
36.3°C (36-38°C) |
HR |
101 bpm |
RR |
26 breaths/min |
BP |
80/27 mmHg |
spO2 |
100% on room air |
WBC (4-10 × 109/L) |
21.68 × 109/L |
H&H (Hgb, Hct) (14-17 g/dL and 41%-51% respectively) |
14.5 g/dL & 46.4% |
Platelets (150-350 × 109/L) |
186 × 109/L |
Table 5. Initial vitals and CBC. Values in parentheses are normal values as per guidelines from American College of Physicians
|
Initial |
24 h after admission |
Na (136-145 mEq/L) |
128 mEq/L |
154 mEq/L |
K (3.5-5 mEq/L) |
7.9 mEq/L |
4 mEq/L |
Cl (98-106 mEq/L) |
86 mEq/L |
122 mEq/L |
HCO3 (23-28 mEq/L) |
<12 mEq/L |
24 mEq/L |
Mg (1.5-2.4 mg/dL) |
3.9 mg/dL |
2.5 mg/dL |
Phosphorus (3-4.5 mg/dL) |
11.4 mg/dL |
1.6 mg/dL |
Anion Gap |
21 |
8 |
Serum Glucose (70-100 mg/dL when fasting) |
>600 mg/dL |
105 mg/dL |
ALT/AST (0-35 units/L) |
44/27 units/L |
44/94 units/L |
Alk Phos (36-92 unit/L) |
122 units/L |
80 units/L |
BUN/Cr |
98/6.9 mg/dL |
41/2.2 mg/dL |
GFR |
12 (>90) |
|
Lactate (0.67-1.8 mmol/L) |
4.2 mmol/L |
1.5 mmol/L |
Procalcitonin |
9.25 ng/mL (<0.01 ngmL) |
|
Plasma Ketones |
>13.50 mmol/L (<0.6 mmol/L) |
0.54 mmoL/L |
High sensitivity troponin (<14 ng/mL) |
194 ng/L |
20456 ng/L |
CKMB (Index) |
2.48 |
5.12 |
Creatine Kinase |
230 units/L |
1173 units/L |
HbA1c |
10 (<5.7%) |
|
Arterial Blood Gas: |
pH |
7.093 (7.35-7.45) |
7.456 (7.35-7.45) |
pCO2 |
20.7 |
34 |
pO2 |
42.6 |
101 |
HCO3- |
6.3 |
23.9 |
Table 6. Lab values at initial visit, 24 hours after admission. Numbers in parentheses denote normal values as per American College of Physicians
The patient underwent serial ABG monitoring and glucose monitoring while on insulin drip. The patient’s blood glucose eventually dropped below 200 ng/dL and insulin was transitioned to subcutaneous insulin sliding scale with long-acting insulin for coverage. Although initially hyperkalemic on the patient’s initial complete metabolic panel, the patient’s potassium remained within normal limits on subsequent lab work.
During the hospital course the patient had an initial high sensitivity troponin of 194 ng/dL, however denied anginal symptoms. Although asymptomatic, the patient had a troponin trend that eventually plateaued at ~20,500. The patient also had new changes on EKG showing ST elevation in the anterior leads (Figure 4). The patient also received an initial TTE which showed preserved ejection fraction and did not show any new wall motion abnormalities. Cardiology was consulted and given the markedly up trending troponins, the patient was initiated on treatment for ACS including dual antiplatelet therapy and heparin drip. The patient underwent a coronary angiogram which revealed non-obstructing coronary artery disease. A stress echocardiogram was later performed which was significant for an EF of 40% w/ diffuse hypokinesis. Given ACS was ruled out, there was suspicion that the patient had perimyocarditis given the new echo findings of reduced LV function. Once DKA resolved, the patient was discharged on goal directed medical therapy for new-onset heart failure with reduced ejection fraction and given outpatient follow up with cardiology.
Discussion
To understand the complex relationship between DKA and myocardial injury, it is important to understand the role of uncontrolled diabetes and its effect on the heart. Although the mechanism continues to remain unclear, literature postulates that DKA can cause acute myocardial injury [1]. One potential cause for EKG findings is related to underlying metabolic derangements in DKA contributing to EKG changes as opposed to coronary obstruction. [13] Prior literature shows that EKG changes seen in these cases tend to be reversible [2]. Tretjak, et al. discuss a case with a 28-year-old female in DKA which had a presentation and findings similar to our patients. Like our patients, there were ST elevations on EKG, however in their patient they also had hypokinetic findings and lowered EF (45%) found on echo similar to case 2 & 3. Repeat echocardiogram findings showed reversibility of the hypokinetic cardiac findings as well as normalization of the EF. Coronary angiography at that time showed patent coronaries with no evidence of obstructive CAD. [2]
Literature discussing myocardial injury in DKA appears scarce, however there are some case reports detailing similar presentations to ours. One case discussed by Shim, et al. [13] discusses a pediatric case of a 12-year-old female that presented with elevated troponin and EKG changes. Cardiac workup also revealed LV dysfunction on echocardiogram, however the patient had patent coronary arteries. EKG changes were consistent with a prolonged QTc. This QTc prolongation in other studies was postulated to be a result of ketotic conditions in the pediatric population [14]. Troponin levels appeared to mirror the efficacy of optimizing blood glucose control. In other words, once the DKA resolved and blood glucose normalized, it appears the troponin began to downtrend toward normal limits and the EKG changes had resolved. There was also improvement of the LV dysfunction. This case is important due to it occurring in a pediatric patient since cardiac decompensation is less pronounced in children with diabetes compared to adults with diabetes. This is because in adults with diabetes, these patients may also have early-onset atherosclerosis which can play a role in cardiac decompensation [13]. This important facet is absent in a majority of pediatric diabetic patients, thus suggesting that severe hyperglycemia secondary to DKA in of itself can cause myocardial injury in the acute setting [12]. Some studies suggest the mechanism of injury is a result of severe acidemia in DKA which can lead to myocardial stunning by disrupting the activation of intracellular calcium and contractile proteins [15,16]. This study appeared to contrast what happened in cases 1 and 3 where the troponin leak and EKG changes occurred as the DKA and electrolyte disturbances were resolving.
Moller, et al. [17] also reported two cases of myocardial injury in patients with DKA. In this report, both patients were 33 and 30 years old respectively. Similar to our patients, these patients had elevated cardiac biomarkers and EKG changes suggestive of AMI such as ST elevations and T-wave inversions. Workup including coronary angiography was unremarkable for obstructive coronary lesions. Moller, et al. [17] would attribute these findings to the acidosis in DKA and inferred that the severe acidemia along with high levels of free fatty acids caused membrane instability and biomarker leakage [16,17].
Poojary, et al. [18] also did a case-series of three patients discussing troponinemia in patients with DKA, but without evidence of ACS. All three cases had very similar presentations and hospital course. The three patients had troponinemia in the setting of DKA and ST elevations on EKG and as such, a coronary angiogram was performed but did not show any evidence of obstructive coronary lesions to explain the troponinemia and EKG changes. The third case was the only one that also had diminished LV systolic function which was later attributed to takotsubo cardiomyopathy [18].
When looking at our three cases it is important to note the similarities and differences. Regarding similarities, all three of our patients were relatively younger males with Type 1 DM that all presented due to medication noncompliance. This is important since literature has already established diabetes mellitus as a risk factor for both CAD and AMI [19,20]. As such, it is important to stress medication compliance to mitigate the risk of developing AMI in the future. Our case series and other literature now appears to suggest that uncontrolled diabetes mellitus in the acute setting can cause acute myocardial injury. Another similarity is that all three of our cases had patent coronaries, thus indicating that the cause of troponin elevation was not due to obstructive coronary lesions. We believe that in cases 1 and 3, the cause of troponinemia could be attributed to the acidosis from DKA and membrane instability. Case 2 is more complicated and the cause of elevated troponinemia is likely not solely a result of the DKA but likely multifactorial as the patient had multiple reasons to incur myocardial damage including septic shock and acute pulmonary embolism. Another similarity between the three cases is that the EKG changes appeared to occur in the anterior leads in our patients which is also something that has been noted in the literature described above.
One major difference between our three cases was that in cases 2 & 3 there were echocardiogram findings that showed new onset hypokinetic findings. This appears to mimic the case by Tretjak et al especially considering when both their patient and ours had an unremarkable coronary angiogram. Another significant difference is that in cases 1 and 3, the EKG changes appeared to develop as the DKA was improving, however in case 2 the changes appeared while in DKA. We attribute this because case 2 likely had an NSTEMI secondary to multiple factors aside from the DKA.
We ascertain that DKA due to acidemia and weakening of membrane stability appears to cause myocardial injury and troponin leakage from the myocardium without obstructive coronary lesions. This continues to pose a challenge to clinicians regarding workup and management of DKA patients exhibiting these changes. The decision whether to pursue diagnostic coronary angiography in a patient remains a contentious issue as the risks and benefits of diagnostic coronary catheterization must be weighed while a patient is actively acidotic due to DKA. The prevalence of pseudoinfarct patterns such as in the case of our patient further complicates this issue since AMI is a diagnosis that cannot be missed and must be managed quickly. The question then arises whether pursuing diagnostic coronary angiography should be a part of workup in young patients with no risk factors for ACS. We ascertain that prior to pursuing invasive strategies such as coronary angiography in younger patients, the full clinical picture must be considered. There have been useful algorithmic scales to help aid clinical decision making in these scenarios. Amongst these is the thrombosis in myocardial infarction (TIMI) score (Table 7). It is reasonable to suggest that in patients with a TIMI score of <2, coronary angiography should be deferred, especially if in a younger patient. It is reasonable that in a TIMI score >5 a coronary angiogram should be pursued and in those with a score of 3-5, a coronary angiogram should be considered if suspicion is high.
TIMI score factors |
Points |
Age >65 years old |
1 |
Presence of at least 3 risk factors for CAD |
1 |
Previous history of coronary stenosis of 50% or more |
1 |
Presence of greater than or equal to 2 episodes of angina 24 hours before presentation |
1 |
Aspirin use in the past 7 days |
1 |
ST segment deviations greater than or equal to 0.05 on initial EKG at admission |
1 |
Elevated serum cardiac markers of necrosis |
1 |
Table 7. Factors and scoring of TIMI score algorithm [12]
Conclusion
In young patients with low cardiovascular risk factors and increased troponin levels likely due to non-cardiac causes, decisions on the clinical dilemma presented must be approached carefully and expeditiously. We believe our patients highlight the importance of myocardial injury that occurs in patients with DKA. A clinician should always be mindful of this under-recognized phenomenon and proceed with management and treatment accordingly. In the cases discussed in this report as well as other literature, it appears to suggest that DKA can cause troponinemia with myocardial injury, however in the absence of obstructive lesions causing ischemia.
Conflicts of interest
There are no conflicts of interest to disclose.
Funding
There is no funding to disclose.
References
- Sweterlitsch EM, Murphy GW (1996) Acute electrocardiographic pseudoinfarction pattern in the setting of diabetic ketoacidosis and severe hyperkalemia. Am Heart J 132: 1086-1089. [Crossref]
- Ahmad R, Narwaria M, Singh A, Kumar S, Haque M (2023) Detecting diabetic ketoacidosis with infection: Combating a life-threatening emergency with practical diagnostic tools. Diagnostics 13: 2441. [Crossref]
- Moulik PK, Nethaji C, Khaleeli AA (2002) Misleading electrocardiographic results in patient with hyperkalaemia and diabetic ketoacidosis. BMJ 325: 1346-1347. [Crossref]
- Master V, Salah M, Salama A, Feitell S (2023) Abstract# 1411932: Pseudo-myocardial infarction in diabetic ketoacidosis with normokalemia. Endocrine Practice 29: S25.
- Khachatryan A, Chow RD, Harutyunyan H, Tamazyan V, Chow RT (2023) Early repolarization augmentation mimicking pseudo-infarction in a patient with diabetic ketoacidosis and normokalemia. Cureus 15: e41546. [Crossref]
- Ruiz-Morales J, Canha C, Al-Saffar F, Ibrahim S (2018) Anterior myocardial pseudoinfarction in a patient with diabetic ketoacidosis. J Geriatr Cardiol 15: 238. [Crossref]
- Odubanjo AA, Kalisetti R, Adrah R, Ajenifuja A, Joseph B, et al. (2018) Severe myopericarditis in diabetic ketoacidosis-all troponin are not myocardial infarction. Clin Med Insights Case Rep 11: 1179547618763356. [Crossref]
- Thygesen K, Alpert JS, Harvey WD (2009) "Pseudo myocardial infarction – A condition in need to be redefined?" Medical Hypotheses 73: 684-688.
- Carrizales-Sepulveda EF, Vera-Pineda R, Jimenez-Castillo RA, Violante-Cumpa JR, Flores-Ramirez R, et al. (2021) The heart in diabetic ketoacidosis: A narrative review focusing on the acute cardiac effects and electrocardiographic abnormalities. Am J Med Sci 361: 690-701. [Crossref]
- Nunes JP (2010) Pseudo myocardial infarction–A condition in need to be redefined?. Med Hypotheses 74: 219-221. [Crossref]
- Carrizales-Sepúlveda EF, Ordaz-Farías A, Vera-Pineda R, Rodríguez-Gutierrez R, Flores-Ramírez R, et al. (2024) Comprehensive echocardiographic and biomarker assessment of patients with diabetic ketoacidosis. Cardiovasc Diabetol 23: 385. [Crossref]
- Rao SS, Agasthi P (2023) “Thrombolysis in myocardial infarction risk score.” StatPearls.
- Shim HJ, Yoo BM, Jin SM, Kang MJ (2021) Myocardial injury in a pediatric patient with diabetic ketoacidosis: A case report. Medicine 100: e25702. [Crossref]
- Kuppermann N, Park J, Glatter K, Marcin JP, Glaser NS (2008) Prolonged QT interval corrected for heart rate during diabetic ketoacidosis in children. Arch Pediatr Adolesc Med 162: 544-549. [Crossref]
- Orchard CH, Kentish JC (1990) Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol 258: C967-C981. [Crossref]
- Eubanks A, Raza F, Alkhouli M, Glenn AN, Homko C, et al. (2012) Clinical significance of troponin elevations in acute decompensated diabetes without clinical acute coronary syndrome. Cardiovasc Diabetol 11: 154. [Crossref]
- Moller N, Foss AC, Gravholt CH, Mortensen UM, Poulsen SH, et al. (2005) Myocardial injury with biomarker elevation in diabetic ketoacidosis. J Diabetes Complications 19: 361-363. [Crossref]
- Poojary I, Khalid U, Patra T, Giri J, Al Heyasat A, et al. (2024) Troponinemia in patients with diabetic ketoacidosis without acute coronary syndrome. Cureus 16: e61064. [Crossref]
- Hajar R (2017) Risk factors for coronary artery disease: Historical perspectives. Heart Views 18: 109-114. [Crossref]
- Pencina MJ, Navar AM, Wojdyla D, Sanchez RJ, Khan I, et al. (2019) Quantifying importance of major risk factors for coronary heart disease. Circulation 139: 1603-1611. [Crossref]