A variety of factors, whether extracellular (mutagens/carcinogens and viruses in the environment, chronic inflammation and radiation associated with the environment and/or electronic devices/machines) and/or intracellular (oxidative metabolites of food, oxidative stress due to inflammation, acid production, replication stress, DNA replication/repair errors, and certain hormones, cytokines, growth factors), pose a constant threat to the genomic integrity of a living cell. However, in the normal cellular environment multiple biological pathways including DNA repair, cell cycle, apoptosis and the immune system work in a precise, regulated (tightly controlled), timely and concerted manner to ensure genomic integrity, stability and proper functioning of a cell. If damage to DNA takes place, it is efficiently and accurately repaired by the DNA repair systems. Homologous recombination (HR) which utilizes either a homologous chromosome (in G1 phase) or a sister chromatid (in G2) as a template to repair the damage, is known to be the most precise repair system. HR in G2 which utilizes a sister chromatid as a template is also called an error free repair system. If DNA damage in a cell is so extensive that it overwhelms the repair system/s, the cell is eliminated by apoptosis. Thus, multiple pathways ensure that genome of a cell is intact and stable. However, constant exposure to DNA damage and/or dysregulation of DNA repair mechanism/s poses a risk of mutation and cancer. Oncogenesis, which seems to be a multistep process, is associated with acquisition of a number of genomic changes that enable a normal cell to progress from benign to malignant transformation. Transformed/cancer cells are recognized and killed by the immune system. However, the ongoing acquisition of new genomic changes enables cancer cells to survive/escape immune attack, evolve into a more aggressive phenotype, and eventually develop resistance to therapy. Although DNA repair (especially the HR) and the immune system play unique roles in preserving genomic integrity of a cell, they can also contribute to DNA damage, genomic instability and oncogenesis. The purpose of this article is to highlight the roles of DNA repair (especially HR) and the immune system in genomic evolution, with special focus on gastrointestinal cancer.
DNA repair, homologous recombination, genomic evolution, immune system, gastrointestinal cancer, esophageal adenocarcinoma
Homologous recombination plays a key role in the maintenance of genomic integrity of a cell: Double stranded breaks (DSBs), which can be generated by a number of intrinsic and extrinsic factors, are deleterious for a cell and must be accurately repaired. DSBs are mostly repaired by either homologous recombination (HR) or non-homologous end joining. Repair through HR depends on strict sequence homology and is known to be the most precise repair mechanism, whereas non-homologous end joining (NHEJ) directly ligates broken DNA ends and is known to be a more error prone mechanism [1,2]. HR plays important roles during both mitosis and meiosis. Mitotic HR occurs in both the germline and somatic cells and plays a key role in the maintenance of genomic integrity by ensuring precise and timely repair of DNA damage. HR is closely linked to cell cycle. Cell cycle checkpoints ensure that damage to DNA is repaired prior to entry of cell into mitosis (G2/M checkpoint) or S phase (G1 checkpoint). Proteins such as p53 regulate both the cell cycle checkpoints and HR. The G2 checkpoint plays a critical role in the maintenance of genomic integrity because if DSBs are not repaired before entry of the cell into mitosis, they can lead to rearrangements or loss of chromosomal fragments in the subsequent G1 phase of the cell cycle. In late S and G2 phases, HR can use the sister chromatid as a template to accomplish error free repair of DNA damage. Homologous recombination in G1, which utilizes homologous chromosome as a template for repair synthesis, is suppressed at multiple levels. This ensures that the mitotic homologous recombination machinery uses only the sister chromatid as a template so that repair synthesis is error free [3-5]
Meiotic HR ensures genetic diversity and proper chromosomal segregation: Meiotic homologous recombination is not only a repair mechanism but also serves as a tool to generate diversity through exchange of genetic information between homologous chromosomes and also contributes to evolution of genes and proper chromosomal segregation [1,4,6]. Meiotic recombination in the first nuclear division is initiated by the introduction of double-strand breaks (DSBs) by Spo11, a topoisomerase-like enzyme, at specific sites known as hotspots, which range in number from 10,000-40,000 in mammalian cells [7]. Induction of DNA breaks is followed by repair synthesis using a homologous chromosome or a sister chromatid as a template. It seems multiple mechanisms ensure usage of a homologous chromosome as the more frequent/preferred choice over sister chromatid during meiotic HR in yeast [8]. This is important to ensure exchange of genetic material between homologous chromosomes and accurate chromosomal segregation.
Repetitive sequences are protected from HR to avoid genomic instability. Repetitive sequences, comprising nearly 50% of human genome, pose a threat to genome stability. This is because these sequences can potentially promote unnecessary/unscheduled homologous recombination and may also induce non-allelic homologous recombination leading to deleterious genomic changes (deletions, insertions, inversions, translocations) and instability. Repetitive sequences involving transposan elements (TEs) can undergo transposition activity leading to genomic rearrangements. Moreover, repetitive sequences can also interfere in proper chromosomal segregation. Therefore, as a safety mechanism, regions of genome containing repetitive sequences are made inaccessible through the formation of heterochromatin. Histone hypoacetylation, DNA methylation and trimethylation of lysine 9 of histone H3 (H3K9me3) are prominent epigenetic marks of heterochromatin. A non-histone protein heterochromatin protein 1 (HP1) recognizes and interacts with these marks leading to further compaction of heterochromatin [9]. Heterochromatin formed at pericentromeric regions is associated with H4K20me3 [10]. H3K9me3 and DNA methylation are prominent marks at subtelomeric and telomeric regions. Other important marks of heterochromatin include H3K9me3 in endogenous retroviral sequences (classes I- II) in embryonic stem cells [11]and DNA methylation and SUV39H in LINE elements (non-LTR retrotransposons) in embryonic stem cells of mouse [12].
Repetitive sequences are protected from meiotic HR to avoid missegregation. To safeguard the integrity of germline DNA, multiple mechanisms ensure that meiotic recombination involving homologous chromosome and cross over process is suppressed/prevented at regions of repetitive DNA sequences [13]. These include: 1) epigenetic changes that convert areas containing repetitive sequences into closed (heterochromatin) regions making them inaccessible to recombination machinery; 2), suppression of DSBs at these sites; and 3) ensuring that repair process either uses sister chromatid instead of homologous chromosome or choses mechanisms not involving cross over [13]. Epigenetic changes which contribute to the formation of closed (heterochromatin) areas can include histone modifications such as H4K20me3, H3K9me3 and hypoacetylation of H3 and H4 at telomeric regions [14-16], histone modifications like H3K9me [17,18], H3K9me2 and DNA methylation in pericentric areas [19], usage of alternate histones such as replacement of conventional H3 by CENH3 (CENP-A) in the nucleosomes of centromeres. In expressed areas of the genome, the prominent epigenetic marks include H3K4me3, which is associated with formation of DSBs [20] and meiotic recombination in mouse [21] and yeast [22]; H2A.Z, a variant of histone H2A, and H3K4me3 localized at the sites of homologous recombination cross over in Arabidopsis; and H3K9ac associated with meiotic recombination in yeast and mice [23,24]. DNA methylation, which seems to be guided by histone marks [25], is an important mechanism to suppress gene expression as well as a universal mark associated with heterochromatin and the suppression of meiotic recombination [26].
HR can become deleterious if dysregulated or spontaneously elevated: The ability of a cell to properly perform its functions requires a concerted and coordinated action of multiple biological pathways [27-30]. To ensure the proper functioning, all these pathways are tightly regulated at multiple levels including gene expression, chromatin modification and chromosome maintenance. The regulatory mechanisms ensure that a required task is accomplished at exactly the right pace, place and time [1,9,31]. Failure or any defect/s in the regulatory mechanisms can result in loss of specificity and precision, and unscheduled or unnecessary activity with potentially deleterious consequences [1,27-30]. In the normal cellular environment, HR is tightly regulated and precise. If damage to DNA takes place, it is detected by specific protein/s and depending on type of damage, a specific set of repair genes are either expressed and/or activated through modifications. Right after repair, the repair machinery has to inactivated through degradation and/or other modifications. Dysregulation or increase in HR activity can potentially result in non-allelic homologous recombination (NAHR), homeologous recombination (with reduced dependence on strict sequence homology/identity) and/or unnecessary and untimely recombination events. These aberrant forms of HR can potentially cause all sorts of genomic rearrangements including deletions, amplifications, translocations, and aberrations associated with segregation defects [2,32,33]. It is well documented that NAHR and other error prone forms of HR including BIR and Single Strand Annealing (SSA) play a significant role in generating genomic instability [1, 2,34-36]. Therefore, high (dysregulated or spontaneously elevated) HR does not necessarily mean more repair. In an environment of dysregulated HR, although possibility of accurate repair of DSBs exists, inaccurate repair also takes place causing genomic rearrangements and other aberrations. Using multiple myeloma and/or esophageal adenocarcinoma as model systems, we have demonstrated that spontaneously elevated HR contributes to genomic instability [37,38], development of drug resistance [37], telomere maintenance and growth of cancer cells as tumor in SCID mice [39]. Unpublished data from our laboratory suggest that spontaneously elevated HR may also lead to increase in spontaneous DNA breaks. This is probably because of HR-associated nuclease activity, production of DNA fragments in consequence to HR-mediated deletion events, and/or encapsulation of deleted products within lipid membrane, leading to formation of micronuclei. Cell cycle checkpoint defects are also associated with chromosomal segregation errors and generation of micronuclei. Micronuclei are not only a marker but also a source of genomic instability. Aberrant DNA repair in micronuclei has been associated with chromothripsis, which is characterized by massive genomic rearrangements in a localized area of genome [40-42]. It has been proposed that an alternate (error prone) HR called microhomology-mediated break induced replication (MMBIR) is a key mechanism underlying chromothripsis [43,44]. Dysregulation of cell cycle check points or defects in HR/cell cycle regulatory proteins can also cause unscheduled recombination events leading to deleterious genomic rearrangements [3-5,45,46]. There are evidences that suggest that repetitive sequence like Tandem repeats, Alu sequences and LTRs are hot spot of genomic rearrangements and copy number variations which is mainly triggered by NAHR [33,47]. Moreover, at regions containing repetitive sequences, the involvement of homologous chromosome and the cross over process poses a risk of recombination to become non-allelic. The cross over homologous recombination too close to centromere in meiosis can also adversely affect kinetochore assembly and chromosomal segregation [48]. In fact, several defects in chromosomal segregation have been attributed to cross over recombination at telomeres in human [49], near centromeres in human [50,51] and near centromeres in yeast [52]. Thus, dysregulated HR can mediate genomic instability and evolution contributing to development and/or progression of cancer.
Role of the immune system in cancer prevention: The host immune system consists of the innate and adaptive arms of immunity (Figure 1). The innate immune system is comprised of cells of myeloid origin (i.e. cells of monocytic lineage including monocytes, dendritic cells and macrophages; granulocytes including neutrophils, eosinophils, basophils, and mast cell; and innate lymphoid cells including natural killer (NK) cells). Adaptive immune cells include cells of lymphoid origin including T (helper CD4+ T and killer CD8+ T) and B cells. Other lymphoid cells such as natural killer T (NKT) and gamma-delta T (γδT) cells are semi-adaptive in nature [53].
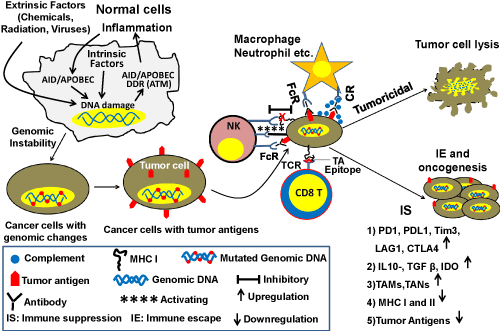
Figure 1. Schematic diagram showing the roles of immune system and DNA repair/HR in genomic instability and oncogenesis. Cancer cells express tumor antigens that are recognized by the immune system. Innate immune cells (macrophages, neutrophils, NK cells) engulf and kill tumor cells by ADCC/CDC pathways by binding to the Fc part of antibodies through Fc receptors (FcR) or binding to complement bound to the tumor antigen-antibody complex by complement receptor (CR). They can kill the tumor cells by secreting toxic elements without direct contact with tumor cells. NK cells have another unique way of killing tumor cells through cytotoxic activity. With progression of cancer, cells downregulate the expression of MHC class I which impairs the inhibitory signal for recognizing tumor cells as self and generate foreignness. This allows activating signal to work between NK cells and tumor cells leading to lysis of the cancer cells. Intracellular tumor antigens are processed and presented by MHC class I of tumor cells to killer CD8 T+ cells in the adaptive arm of the immune system that can specifically kill the tumor cells by their cytotoxic activity. With progression of cancer, the TME becomes more immunosuppressive. DNA damage and genomic instability can cause inflammation and inflammation further increases DNA damage and genomic instability. Symbols used in the figure are explained within the rectangle.
One of the important functions of the immune system is to recognize and eliminate cancer cells. Immune cells recognize cancer cells through specific tumor antigens (TAs) expressed on their surface. These antigens can be classified into several subtypes as follows: 1) tumor associated antigens (TAA); 2) tumor specific antigens (TSA); 3) Oncofetal antigens (OFA); 4) oncoviral antigens and; 5) lineage or differentiation specific antigens 6) cancer testis antigens which are usually expressed in germ cells in adults but a group of these antigens [MAGE(Melanoma-associated antigen 1), NY-ESO (New York- Esophageal cancer 1)] are highly expressed in many cancer cell types (melanoma, liver, lung, bladder). These antigens are harnessed as targets for cancer vaccination. TAAs appear due to overexpression of certain genes in tumor cells and may also be expressed at lower level in normal cells. TSAs arise from mutated proteins, proteins with aberrant glycosylation or unusual lipids on the cell surface and, therefore, are foreign to the host immune system. Oncofetal proteins, alpha-fetoprotein (AFP) and carcinoembryonic antigen (CEA) are usually expressed in embryo before the host immune system is developed and can be re-expressed by some cancer cells, hence they are seen as foreign to the immune system. Oncoviral proteins are antigens expressed in cancers caused by viral infection [54,55].
Dendritic cells (of the innate immune system) play a critical role in priming T cells against TAs through antigen presentation. During the initial stage of tumor development, macrophages and neutrophils kill cancer cells through their cytotoxic activities including antibody-dependent cell-mediated cytotoxicity (ADCC), complement dependent cytotoxicity (CDC) and other pathways. However, with progression of cancer these innate immune cells can transform into tumor-promoting cells such as tumor-associated macrophages (TAMs) or tumor-associated neutrophils (TANs) [53]. The complement system, which increases the ability of antibodies and phagocytic cells to eliminate cancer/damaged cells and microbes through either phagocytosis, induction of inflammation, or direct cellular rupture, has also been shown to play a crucial role in killing tumor cells. In fact, many CD8+ T and NK cells play a crucial role in killing cancer cells in the early stage by recognizing them as foreign. CD8+ T cells recognize epitopes of TAs that are processed in the cancer cell and presented by MHC class I on the cancer cell surface [55]. During progression, cancer cells can downregulate MHC class I expression, although lack of MHC class I expression can trigger their destruction by NK cells [53]. Plasma B cells play an important role in cancer prevention by secreting antibodies to TAs that kill cancer cells either through complement-mediated lysis or by ADCC [53]. Apart from direct engagement of killer cells or antibodies, soluble immunokines like IFNγ, IFNα, IL-2, secreted in the tumor microenvironment (TME) can contribute to the prevention of cancer growth/progression [56].
Role of the immune system in etiology of cancer in the context of the GI tract: The gastrointestinal (GI) tract is lined with epithelial cells that come in direct contact with a variety of substances/agents that can potentially disrupt genomic integrity and/or stability. These substances/agents, which may include mutagens and carcinogens present in certain foods, the low pH of certain dietary products (especially sodas), high temperature of some foods (more relevant to the mouth and esophagus), oxidative metabolites of food, microbiome dysbiosis, and likely others, make the epithelial barrier vulnerable. If this barrier is compromised, chronic inflammatory conditions may be initiated. Chronic inflammation in the esophagus, stomach, pancreas, intestine, colon, liver, and bile duct is a potent etiologic agent for corresponding tissue specific precancerous conditions such as inflammatory bowel disease, gastritis, pancreatitis, esophagitis, hepatitis, and cholangitis [57]. Inflammation is known to induce oxidative stress as well as APOBEC deaminase activity, which can potentially disrupt genomic integrity and stability leading to the development of cancer (Figures 1 and 2). Although chronic inflammation (caused by intrinsic or extrinsic factors) can potentially contribute to oncogenesis, inflammation can also be induced by cancer [58] thus giving rise to a vicious cycle. In the cancer environment, inflammation is impacted by the interplay of immunne cells, cancer cells and other stromal cells in the TME. Moreover, with progression to cancer, soluble factors in the TME can create an immune-suppressive and cancer promoting milieu.
Figure 1. Schematic diagram showing the roles of immune system and DNA repair/HR in genomic instability and oncogenesis. Cancer cells express tumor antigens that are recognized by the immune system. Innate immune cells (macrophages, neutrophils, NK cells) engulf and kill tumor cells by ADCC/CDC pathways by binding to the Fc part of antibodies through Fc receptors (FcR) or binding to complement bound to the tumor antigen-antibody complex by complement receptor (CR). They can kill the tumor cells by secreting toxic elements without direct contact with tumor cells. NK cells have another unique way of killing tumor cells through cytotoxic activity. With progression of cancer, cells downregulate the expression of MHC class I which impairs the inhibitory signal for recognizing tumor cells as self and generate foreignness. This allows activating signal to work between NK cells and tumor cells leading to lysis of the cancer cells. Intracellular tumor antigens are processed and presented by MHC class I of tumor cells to killer CD8 T+ cells in the adaptive arm of the immune system that can specifically kill the tumor cells by their cytotoxic activity. With progression of cancer, the TME becomes more immunosuppressive. DNA damage and genomic instability can cause inflammation and inflammation further increases DNA damage and genomic instability. Symbols used in the figure are explained within the rectangle.
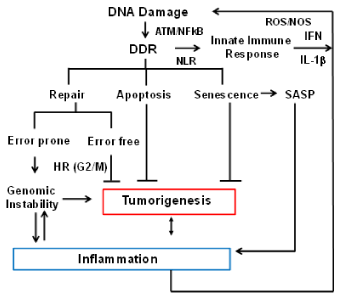
Figure 2. Crosstalk between DNA damage response (DDR) and the immune response. The flow chart indicates how the immune system and DNA repair are interconnected, and how aberrant functions of these pathways impact each other and genome stability. DNA damage caused by various extrinsic and intrinsic factors leads to activation of DNA damage response (DDR). A number of proteins involved in DDR have also been shown to function by sensing cytoplasmic DNA and activating inflammatory cytokines and chemokines resulting in inflammation. Inflammation itself generates oxidative stress leading to DNA damage and further increase in genomic instability.
Role of immune system in cancer growth and progression: Cancer advances when immune surveillance and immunoediting fails (Figure 1). This occurs in three consecutive stages called elimination, equilibrium and escape (3Es) [59].
Figure 1. Schematic diagram showing the roles of immune system and DNA repair/HR in genomic instability and oncogenesis. Cancer cells express tumor antigens that are recognized by the immune system. Innate immune cells (macrophages, neutrophils, NK cells) engulf and kill tumor cells by ADCC/CDC pathways by binding to the Fc part of antibodies through Fc receptors (FcR) or binding to complement bound to the tumor antigen-antibody complex by complement receptor (CR). They can kill the tumor cells by secreting toxic elements without direct contact with tumor cells. NK cells have another unique way of killing tumor cells through cytotoxic activity. With progression of cancer, cells downregulate the expression of MHC class I which impairs the inhibitory signal for recognizing tumor cells as self and generate foreignness. This allows activating signal to work between NK cells and tumor cells leading to lysis of the cancer cells. Intracellular tumor antigens are processed and presented by MHC class I of tumor cells to killer CD8 T+ cells in the adaptive arm of the immune system that can specifically kill the tumor cells by their cytotoxic activity. With progression of cancer, the TME becomes more immunosuppressive. DNA damage and genomic instability can cause inflammation and inflammation further increases DNA damage and genomic instability. Symbols used in the figure are explained within the rectangle.
Elimination: In the elimination stage, the transformed cells are eliminated by the immune system through recognition of TAs as well as other mechanisms as discussed above.
Equilibrium: When immune elimination is not complete, yet the tumor does not have the capacity to evade the immune system and progress, a steady state can ensue where a viable nascent tumor exists in an equilibrium phase. Escape: With acquisition of new genomic changes, a subset of cancer cells are able to equip themselves for protection against immune attack by: 1) acquiring surface expression of inhibitory molecules such as programmed cell death protein 1 (PD1), programmed cell death protein-ligand 1 (PDL1), lymphocyte-activation gene 3 (LAG3), T-cell immunoglobulin, mucin-domain containing-3 (Tim3), cytotoxic T-lymphocyte-associated protein 4 (CTLA4) on cells in the TME; 2) inducing secretion of immune-suppressive agents such as TGFβ, IL-10, indoleamine 2,3 dehydrogenase (IDO) and inducing expression of cancer growth promoting immune cytokines such as TGFβ, IL-6, IL-17A [60], IL-22 and IL-23 as well as chemokines such as CCL2, CCL5/RANTES, CCL20, CXCL8/IL-8 and CXCL12 [61]. TGFβ and IL-10 are known to be important factors contributing to the suppressive function of regulatory CD4+ T cells (Tregs) [60]. IL-17A, which is secreted mostly by Th17 cells, is associated with poor survival of gastrointestinal cancers. These cytokines/chemokines are known to promote gastrointestinal cancers by allowing growth, angiogenesis and metastasis. In fact, Th17 and Treg cells are very plastic and switch between the two cell types depending on the relative abundance of TGFβ, IL-10, IL-6 and IL-23. Despite the abundance of literature on the tumor-promoting role of IL-17A, in some cancer models, it has been shown to have a role in the cytotoxic activity of CD8+ T cells in anti-tumor responses. Hence the role of IL-17A in cancer needs to be investigated further [62]. The majority of immune cells and gastrointestinal epithelial cells produce TGFβ, which is a master regulator in the gut immune system. The overexpression of TGFβ may also drive the growth of tumor cells. IL-6 is produced by multiple types of cells in the body as well as tumor cells and is also documented to allow/promote tumor cell growth. In addition to the expression of immune suppressive or inflammatory molecules, cancer cells can down-regulate expression of TAs and/or reduce antigen presentation by lowering the expression of MHC class I on cell surface to evade immune detection [60]. Moreover, gastric and pancreatic cancer cells expressing FasL (which engages with FasR on infiltrating leukocytes), can kill the cytotoxic NK and CD8+ T cells [63] as an immune-escape mechanism. Cancer cells can also activate the complement pathway promoting the infiltration immune-suppressive myeloid cells by complement byproducts (C3a, C5a). This infiltration of immune-suppressive myeloid cells has been shown to impair immune clearance of cancer cells and to promote cancer cell survival and proliferation leading to immune escape. Innate cells like tumor-associated macrophages (TAMs), neutrophils (TANs) and myeloid derived suppressor cells (MDSCs) can all contribute to the creation of an immune-suppressive TME allowing growth and metastasis of tumor cells [64].
Prominent defects of immune system in esophageal, colon and other GI cancers: Since the gastrointestinal (GI) tract is naturally exposed to a variety of potentially harmful intrinsic and extrinsic factors, the immune system in the GI tract is appropriately balanced to avoid unnecessary inflammation. A natural semi immune-suppressive environment prevails to ensure that no adverse autoimmune responses are activated. However, the semi-suppressive environment also poses a risk that mutated/transformed cells may go unchecked. Additionally, epithelial cells in GI tract are of a hyperproliferative nature. Once a tumor develops, it can gradually progress and generate a more immune-suppressive and/or inflammatory environment as mentioned above. Additionally, microbiomes play a crucial role in maintaining immune balance and changes in the microbiota could disturb the balance to promote cancer. For example, Fusobacterium nucleutum has been shown to be associated with both esphageal and colorectal cancer, possibly through inducing the production of CCL20. Inflammaton induced by chronic Helicobactor pylori infection can lead to gastric cancer. The efficacy of checkpoint inhibitors has also been shown to depend on the composition of the microbiota [65].
Crosstalk between DNA damage response (DDR) and Immune response: The DNA damage response (DDR) is a critical signaling pathway triggered by damage caused by intrinsic and extrinsic genotoxic agents and replication stress [66]. The DDR signaling pathway consists of sensors that recognize DNA lesions (RPA, MRN, PARP1, Ku70/80 etc.), transducer kinases (ATM, ATR, DNA-PK etc.) that convey the signal downstream, and effector proteins (Chk1, Chk2, P53, PCNA etc.) that induce cell cycle arrest allowing initiation of DNA repair pathways. Figure 2 provides an overview of crosstalk between immune system and genome stability. The DDR is coupled with the innate immune signaling in cells like DCs and macrophages. One of the key components of this signaling system is pattern recognition receptors (PRRs) that recognize foreign nucleic acids as well as damaged host DNA. Toll-like receptors (TLRs) are among the best-characterized PRRs [67]. For example, TLR9 recognizes unmethylated CpG motifs in endosomes. Activated TLR9 recruits MyD88 leading to the induction of inflammatory genes through the transcription of nuclear factor kappa B (NF-κB) and IFN-regulatory factor 7 (IRF-7).
Figure 2. Crosstalk between DNA damage response (DDR) and the immune response. The flow chart indicates how the immune system and DNA repair are interconnected, and how aberrant functions of these pathways impact each other and genome stability. DNA damage caused by various extrinsic and intrinsic factors leads to activation of DNA damage response (DDR). A number of proteins involved in DDR have also been shown to function by sensing cytoplasmic DNA and activating inflammatory cytokines and chemokines resulting in inflammation. Inflammation itself generates oxidative stress leading to DNA damage and further increase in genomic instability.
In recent years a number of studies have highlighted the interplay between DNA damage response and immune response [68,69]. A number of proteins involved in DDR such as Ku70, DNA-PK, and MRE11 have been shown to function as pattern recognition receptors (PRRs) for sensing cytoplasmic DNA and activating inflammatory cytokines and chemokines [70-74]. ATM kinase, a key player in DDR signaling, has been shown to regulate the expression and activation of key transcription factors in immune signaling, such as IRFs and NF-κB [75]. These transcription factors induce the expression of various immune genes, including inflammatory cytokines and chemokines. Activation of NF-κB in turn, has been shown to enhance the repair of double strand breaks by homologous recombination (HR) [76]. In addition, DDR genes such as Ku-70, Ku-80, ATM, and BRCA2 have also been identified as transcriptional targets of NF-κB in tumor cells [77]. In addition to inducing production of pro-inflammatory signals such as IFNs, replicative stress and DDR can also induce the expression of a number of ligands for activating immune receptors such as natural killer group 2, member D (NKG2D) and DNAX accessory molecule 1 (DNAM-1), which are mainly expressed by cytotoxic immune cells such as T cells and natural killer (NK) cells [78]. This upregulation requires the activation of ATM or ATR protein kinases and Chk1 kinase. These reports demonstrate the ability of DNA damage sensors and DDR to directly activate immune signaling, clearly indicating crosstalk between DDR pathway and the innate immune response.
Activation of DDR in response to oncogenic stress induces cellular senescence. Cellular senescence is considered to be a tumor-suppressive mechanism that prevents malignant transformation [79,80]. However, senescent cells develop an altered pro-inflammatory secretory phenotype, called senescence-associated secretory phenotype (SASP) [81,82]. p53, one of the main downstream effectors of the DDR pathway, has been shown to drive this inflammatory phenotype contributing to tumor clearance [83]. A causal role for inflammation in tumorigenesis is well established [84]. Thus, inflammation driven by DDR signaling can also promote tumorigenesis [85]. There is growing evidence showing that chronic inflammation induces production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that drive tumorigenesis. For example, chronic inflammation induced by Helicobacter pylori and hepatitis virus infection have been shown to cause gastric cancer [86] and liver cancer respectively. Furthermore, chronic inflammation and resulting ROS induced by Crohn’s disease (CD) can promote colorectal cancer [87].
Figure 2 summarizes how HR and the immune system impact each other and genome stability. Briefly, DDR signaling can activate an innate immune response that helps in clearing of damaged cells that cannot be repaired, thereby preventing malignant transformation. However, chronic inflammatory responses if unchecked can promote cancer through a further increase in DNA damage. Thus, proper coordination between DDR and the immune response is critical for organismal homeostasis and effective tumors surveillance.
Figure 2. Crosstalk between DNA damage response (DDR) and the immune response. The flow chart indicates how the immune system and DNA repair are interconnected, and how aberrant functions of these pathways impact each other and genome stability. DNA damage caused by various extrinsic and intrinsic factors leads to activation of DNA damage response (DDR). A number of proteins involved in DDR have also been shown to function by sensing cytoplasmic DNA and activating inflammatory cytokines and chemokines resulting in inflammation. Inflammation itself generates oxidative stress leading to DNA damage and further increase in genomic instability.
In the process of oncogenesis, a normal cell, through a variety of genomic changes, acquires new characteristics that enable it to divide indefinitely, escape the immune system, progress to an advanced and more aggressive phenotypes and develop resistance to treatment. It seems that genomic instability, the ability of a cell to constantly acquire genomic changes, arises at an early (precancerous) state and underlies clonal evolution associated with progression to advanced stages of disease. We have previously shown that homologous recombination (HR) is spontaneously elevated (dysregulated) in cancer cells (including esophageal adenocarcinoma) [37,38] and contributes to acquisition of ongoing genomic changes [37,38], telomere elongation and tumor growth [39]. Dysregulated HR can potentially cause amplifications, deletions, translocations, inversions, chromothripsis, LOH and segregation defects, leading to genomic instability which is associated with clonal evolution and oncogenesis. Genomic instability can also cause inflammation (Figure 2). Inflammation is associated with the production of certain factors/activities (such as growth factors, cytokines and deaminase) that further dysregulate DNA repair (including HR) and genome stability. Therefore, in cancer environment, aberrant function of HR and the immune system can interact in causing genomic instability and evolution (Figure 2). Components of dysregulated HR and the immune system (as described in Figures 1 and 2) may, therefore, provide useful targets to inhibit/reduce genomic instability and inhibit/delay genomic evolution and progression in cancer.
Figure 1. Schematic diagram showing the roles of immune system and DNA repair/HR in genomic instability and oncogenesis. Cancer cells express tumor antigens that are recognized by the immune system. Innate immune cells (macrophages, neutrophils, NK cells) engulf and kill tumor cells by ADCC/CDC pathways by binding to the Fc part of antibodies through Fc receptors (FcR) or binding to complement bound to the tumor antigen-antibody complex by complement receptor (CR). They can kill the tumor cells by secreting toxic elements without direct contact with tumor cells. NK cells have another unique way of killing tumor cells through cytotoxic activity. With progression of cancer, cells downregulate the expression of MHC class I which impairs the inhibitory signal for recognizing tumor cells as self and generate foreignness. This allows activating signal to work between NK cells and tumor cells leading to lysis of the cancer cells. Intracellular tumor antigens are processed and presented by MHC class I of tumor cells to killer CD8 T+ cells in the adaptive arm of the immune system that can specifically kill the tumor cells by their cytotoxic activity. With progression of cancer, the TME becomes more immunosuppressive. DNA damage and genomic instability can cause inflammation and inflammation further increases DNA damage and genomic instability. Symbols used in the figure are explained within the rectangle.
Figure 2. Crosstalk between DNA damage response (DDR) and the immune response. The flow chart indicates how the immune system and DNA repair are interconnected, and how aberrant functions of these pathways impact each other and genome stability. DNA damage caused by various extrinsic and intrinsic factors leads to activation of DNA damage response (DDR). A number of proteins involved in DDR have also been shown to function by sensing cytoplasmic DNA and activating inflammatory cytokines and chemokines resulting in inflammation. Inflammation itself generates oxidative stress leading to DNA damage and further increase in genomic instability.
The work in our laboratory is supported in part by Department of Veterans Affairs Merit Review Award I01BX001584-01 (NCM), NIH grants P01-155258 and 5P50 CA100707 (MAS and NCM) and Leukemia and Lymphoma Society translational research grant (NCM).
- Haber JE (2014) Genome Stability: DNA Repair and Recombination. New York, NY: Garland Science
- Aguilera A, Gomez-González B (2008) Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet 9: 204-217. [Crossref]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, et al. (2015) A mechanism for the suppression of homologous recombination in G1 cells. Nature 528: 422-426.
- Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS (2014) Is homologous recombination really an error-free process? Front Genet 5: 170-175.
- Branzei D, Foiani M (2008) Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9: 297-308. [Crossref]
- Lambert S, Lopez BS (2001) Role of RAD51 in sister-chromatid exchanges in mammalian cells. Oncogene 20: 6627-6631. [Crossref]
- Cooper TJ, Garcia V, Neale MJ (2016) Meiotic DSB patterning: A multifaceted process. Cell Cycle 15: 13-21. [Crossref]
- Liu Y, Gaines WA, Callender T, Busygina V, Oke A, et al. (2014) Down-regulation of Rad51 activity during meiosis in yeast prevents competition with Dmc1 for repair of double-strand breaks. PLoS Genet 10: e1004005.
- Nishibuchi G, Déjardin J (2017) The molecular basis of the organization of repetitive DNA-containing constitutive heterochromatin in mammals. Chromosome Res 25: 77-87. [Crossref]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, et al. (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18: 1251-1262. [Crossref]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, et al. (2010) Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464: 927-931.
- Bulut-Karslioglu A, De La Rosa-Velázquez IA, Ramirez F, Barenboim M, Onishi-Seebacher M, et al. (2014) Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55: 277-290.
- Termolino P, Cremona G, Consiglio MF, Conicella C (2016) Insights into epigenetic landscape of recombination-free regions. Chromosoma 125: 301-308. [Crossref]
- García-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA, et al. (2004) Epigenetic regulation of telomere length in mammalian cells by the Suv39 h1 and Suv39 h2 histone methyltransferases. Nat Genet 36: 94-99.
- Gonzalo S, García-Cao M, Fraga MF, Schotta G, Peters AH, et al. (2005) Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 7: 420-428. [Crossref]
- Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, et al. (2010) SIRT1 contributes to telomere maintenance and augments global homologous recombination. J Cell Biol 191: 1299-1313.
- Lachner M, Jenuwein T (2002) The many faces of histone lysine methylation. Curr Opin Cell Biol 14: 286-298. [Crossref]
- Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S (2015) Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol 16: 499-513. [Crossref]
- Simon L, Voisin M, Tatout C, Probst AV (2015) Structure and function of centromeric and pericentromeric heterochromatin in Arabidopsis thaliana. Front Plant Sci 6: 1049.
- Borde V, de Massy B (2013) Programmed induction of DNA double strand breaks during meiosis: setting up communication between DNA and the chromosome structure. Curr Opin Genet Dev 23: 147-155.
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV (2012) Genetic recombination is directed away from functional genomic elements in mice. Nature 485: 642-645. [Crossref]
- Borde V, Robine N, Lin W, Bonfils S, Géli V, et al. (2009) Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J 28: 99-111.
- Yamada S, Ohta K, Yamada T (2013) Acetylated Histone H3K9 is associated with meiotic recombination hotspots and plays a role in recombination redundantly with other factors including the H3K4 methylase Set1 in fission yeast. Nucleic Acids Res 41: 3504-3517.
- Buard J, Barthès P, Grey C, de Massy B (2009) Distinct histone modifications define initiation and repair of meiotic recombination in the mouse. EMBO J 28: 2616-2624.
- Guo X, Wang L, Li J, Ding Z, Xiao J, et al. (2015) Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517: 640-644. [Crossref]
- Zemach A, McDaniel IE, Silva P, Zilberman D (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916-919. [Crossref]
- Hustedt N, Durocher D (2016) The control of DNA repair by the cell cycle. Nat Cell Biol 19: 1-9. [Crossref]
- Mjelle R, Hegre SA, Aas PA, Slupphaug G, Drabløs F, et al. (2015) Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst) 30: 53-67. [Crossref]
- Voss TC and Hager GL (2014) Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Gen 15: 69-81.
- Balomenos D1, Martínez-A C (2000) Cell-cycle regulation in immunity, tolerance and autoimmunity. Immunol Today 21: 551-555. [Crossref]
- Sonawane AR, Platig J, Fagny M, Chen CY, Paulson JN, et al. (2017) Understanding Tissue-Specific Gene Regulation. Cell Rep 21: 1077-1088. [Crossref]
- Wright WD, Shah SS, Heyer WD (2018) Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 293: 10524-10535. [Crossref]
- Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, et al. (2008) Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci U S A 105: 11845-11850. [Crossref]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, et al. (2011) Break-induced replication is highly inaccurate. PLoS Biol 9: e1000594. [Crossref]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, et al. (2011) 1000 Genomes Project Mapping copy number variation by population-scale genome sequencing. Nature 470: 59-65.
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, et al. (2008) Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56-64.
- Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC, et al. (2009) Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 113: 2290-2297.
- Pal J, Bertheau R, Buon L, Qazi A, Batchu RB, et al. (2011) Genomic evolution in Barrett's adenocarcinoma cells: critical roles of elevated hsRAD51, homologous recombination and Alu sequences in the genome. Oncogene 30: 3585-3598.
- Lu R, Pal J, Buon L, Nanjappa P, Shi J, et al. (2014) Targeting homologous recombination and telomerase in Barrett's adenocarcinoma: impact on telomere maintenance, genomic instability and tumor growth. Oncogene 33: 1495-505.
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, et al. (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482: 53-58. [Crossref]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, et al. (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27-40.
- Terradas M, Martín M, Tusell L, Genescà A (2009) DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair (Amst) 8: 1225-1234. [Crossref]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, et al. (2015) Chromothripsis from DNA damage in micronuclei. Nature 522: 179-184. [Crossref]
- Hastings PJ, Ira G, Lupski JR (2009) A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327.
- Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O'Neil NJ, et al. (2008) RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135: 261-271.
- Bertrand P, Saintigny Y, Lopez BS (2004) p53's double life: transactivation-independent repression of homologous recombination. Trends Genet 20: 235-243.
- Purandare SM, Patel PI (1997) Recombination hot spots and human disease. Genome Res 7: 773-786. [Crossref]
- Talbert PB, Henikoff S (2010) Centromeres convert but don't cross. PLoS Biol 8: e1000326. [Crossref]
- Oliver TR, Tinker SW, Allen EG, Hollis N, Locke AE, et al. (2012) Altered patterns of multiple recombinant events are associated with nondisjunction of chromosome 21. Hum Genet 131: 1039-1046. [Crossref]
- Lamb NE, Freeman SB, Savage-Austin A, Pettay D, Taft L, et al. (1996) Susceptible chiasmate configurations of chromosome 21 predispose to non-disjunction in both maternal meiosis I and meiosis II. Nat Genet 14: 400-405. [Crossref]
- Ottolini CS, Newnham LJ, Capalbo A, Natesan SA, Joshi HA, et al. (2015) Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat Genet 47: 727-735. [Crossref]
- Hall IM, Noma K, Grewal SI (2003) RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc Natl Acad Sci USA 100: 193-198. [Crossref]
- Palucka AK, Coussens LM (2016) The basis of oncoimmunology. Cell 164: 1233-1247. [Crossref]
- Vigneron N (2015) Human tumor antigens and cancer immunotherapy. Biomed Res Int 2015: 948501. [Crossref]
- Gubin MM, Artyomov MN, Mardis ER, Schreiber RD (2015) Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 125: 3413-3421. [Crossref]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, et al. (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24: 541-550. [Crossref]
- Rasch S, Algul H (2014) A clinical perspective on the role of chronic inflammation in gastrointestinal cancer. Clin Exp Gastroenterol 7: 261-272. [Crossref]
- Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454: 436-444. [Crossref]
- Mittal D, Gubin MM, Schreiber RD, Smyth MJ (2014) New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr Opin Immunol 27:16-25.
- Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25: 267-296. [Crossref]
- Nagarsheth N, Wicha MS (2017) Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 17: 559-572. [Crossref]
- Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-Shakib A, et al. (2017) The paradox of Th17 cell functions in tumor immunity. Cell Immunol 322: 15-25. [Crossref]
- Bennett MW, O'Connell J, O'Sullivan GC, D Roche, C Brady, et al. (1999) Expression of Fas ligand by human gastric adenocarcinomas: a potential mechanism of immune escape in stomach cancer. Gut 44: 156-162.
- Engblom C, Pfirschke C, Pittet MJ (2016) The role of myeloid cells in cancer therapies. Nat Rev Cancer 16: 447-462. [Crossref]
- Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF (2018) The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 359: 1366-1370.
- Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461: 1071-1078. [Crossref]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805-820. [Crossref]
- Nakad R, Schumacher B (2016) DNA damage response and immune defense: Links and mechanisms. Front Genet 7: 147. [Crossref]
- Pateras IS, Havaki S, Nikitopoulou X, Vougas K, Townsend PA, et al. (2015) The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol Ther 154: 36-56. [Crossref]
- Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179-204. [Crossref]
- Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, et al. (2011) Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol 186: 4541-4545. [Crossref]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, et al. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12: 959-965. [Crossref]
- Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL (2012) DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1: e00047. [Crossref]
- Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, et al. (2013) DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci USA 110: 2969-2974.
- Li N, Banin S, Ouyang H, Li GC, Courtois G, et al. (2001) ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem 276: 8898-8903. [Crossref]
- Volcic M, Karl S, Baumann B, Salles D, Daniel P, et al. (2012) NF-κB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res 40: 181-195. [Crossref]
- Lim JW, Kim H, Kim KH (2002) Expression of Ku70 and Ku80 mediated by NF-kappa B and cyclooxygenase-2 is related to proliferation of human gastric cancer cells. J Biol Chem 277: 46093-46100. [Crossref]
- Gasser S, Orsulic S, Brown EJ, Raulet DH (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436: 1186-1190.
- Narita M, Lowe SW (2005) Senescence comes of age. Nat Med 11: 920-922. [Crossref]
- Collado M, Gil J, Efeyan A, Guerra C, Schumacher AJ, et al. (2005) Tumor biology: senescence in premalignant tumours. Nature 436: 642. [Crossref]
- Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, et al. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019-1031.
- Young AR, Narita M (2009) SASP reflects senescence. EMBO Rep 10: 228-230. [Crossref]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, et al. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445 :656-660.
- Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420: 860-867. [Crossref]
- Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumour suppression. Annu Rev Pathol 5:99-118.
- Correa P (1995) Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 19 Suppl 1: 37-43. [Crossref]
- Bartsch H, Nair J (2006) Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg 391: 499-510.