Hearing loss is the most common form of sensory impairment in humans, affecting 5.3% worldwide population. Although approximately 1 in 500 children are born with impaired hearing, sudden or progressive forms of hearing loss can manifest at any age. Hearing impairment following cochlear damage due to noise trauma, ototoxicity or age‐related cochlear degeneration was linked to a common pathogenesis involving the formation of reactive oxygen species (ROS). This review summarizes the current data suggesting a role of mitochondrial ROS overproduction in hearing loss and the molecular mechanism involved in hair cell apoptosis responsible of this disorder. Because increasing number of studies demonstrated that antioxidants and free radical scavengers may serve as effective compounds to block the activation of cochlear hair cell death, targeting members of antioxidant pathways and in the breakdown of superoxide anions and hydrogen peroxidase, could be feasible options for the treatment of several types of hearing loss.
Highlight
- ROS overproduction plays a key role in hearing loss
- ROS induce mitochondrial damage and cochlear hair cell apoptosis
- Antioxidants as effective therapeutic agents for hearing disorders
The mammalian cochlea is the sensory organ capable of perceiving sound over a range of pressure, and discriminating both infrasonic and ultrasonic frequencies in different species. The organ of Corti is located in the cochlea of the inner ear and is responsible for the detection of sound. This organ harbours the auditory sensory epithelium, which, in humans, contains approximately 16,000 hair cells that are patterned into three rows of outer hair cells (OHCs) and one row of inner hair cells (IHCs) [1,2]. Each hair cell contains, at its apical surface, a mechanically sensitive organelle that consists of rows of actin-filled stereocilia that increasein height. An extracellular matrix, the tectorial membrane, covers the apical surface of the organ of Corti and is attached to the hair bundles of OHCs. The cell bodies of hair cells form specialized adhesive contacts with supporting cells that adhere at their basolateral surfaces to the basilar membrane, an extracellular matrix assembly with a different molecular composition from the tectorial membrane [3,4].
Hearing is initiated when sound waves that reach the outer ear travel through the ear canal to the tympanic membrane. Then, the sound energy is transferred, via the bony ossicles of the middle ear, to the oval window at the base of the fluid-filled cochlea. The motions of the oval window are converted into fluid pressure waves that induce vibrations in the basilar membrane. Then, the vibrations are transferred onto the hair cells, leading to the deflection of the hair cell stereocilia [5]. This deflection causes stretch-sensitive ion channels to open. These are non-selectively permeable to cations and are located at the base of the tip links, with 1 or 2 channels per tip link. Stereocilia bathe in endolymph, which is rich in potassium and characterized by an endocochlear potential of +80 mV, but the hair cell body has a potential between -70 mV and -55 mV. As a result, the electric potential between the endolymph and the hair cell body (between 135 and 150 mV) causes a massive influx of potassium ions from the endolymph to the hair cell when the mechanically-sensitive ion channels open [6]. The depolarization of the IHCs causes L-type voltage-sensitive calcium channels located near the regions of afferent synapses to open. Each active region in characterized by the presence of an electron-dense structure called a synaptic ribbon. This synaptic ribbon is anchored to the plasma membrane and surrounded by synaptic vesicles which contain glutamate.
In the same way as for the IHCs, acoustic vibrations cause motion of the stereocilia of the OHCs, which in turn causes a modulation in cellular potential. However, in contrast to what happens in the IHCs, variations in the OHC's potential causes them to change shape. When they depolarize they become shorter, and become longer at repolarization. These changes in the length of the OHCs are caused by voltage-sensitive changes in the shape of prestin that is abundant in the cellular membrane [7]. OHCs therefore have a role of oscillators that cause localized amplification of the basilar membrane. The opening of these transduction channels leads to hair cell depolarization and to the release of neurotransmitters onto afferent neurons, which form synapses with hair cells. The electrical signals are propagated through the nervous system and processed in the brainstem and auditory cortex [7].
Mitochondria are the main ATP power house in eukaryotic cells. This is not the only cellular way to obtain ATP as the glycolytic process in the cytosol also produces it, albeit with less efficiency [8]. Beside this ATP production, mitochondria are used for other cellular functions: they participate in programmed cell death [9], the homeostatic control of calcium and iron concentration [10], and the control of autophagy [11].
In addition, mitochondrial respiration produces reactive oxygen species (ROS). These free radicals include the superoxide anion (O2·−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH) and have essential role in cell signaling [12]. Under normal conditions, ROS produced by the mitochondria are easily metabolized or scavenged by endogenous antioxidant mechanisms as catalase, superoxide dismutase and glutathione, and balance cell homeostasis. However, aging process, pharmacological treatment or external factors can alter this balance and this imbalance is called oxidative stress [13].
ROS are very unstable species, reacting with other molecules and thereby damaging proteins, lipids and DNA [14]. In this way, ROS generated by mitochondria are hypothesized to damage key mitochondrial components, such as mitochondrial DNA (mtDNA), mitochondrial membranes, respiratory chain proteins, and nuclear DNA that affect mitochondrial function [15]. ROS-induced damage has the potential to alter electron transport chain function and decrease the efficiency of ATP production. Moreover, several studies proposed the free radical theory of ageing, which suggested that free radicals can affect and are possibly the cause of the ageing process of animals [16]. This creates a “vicious cycle” in which ROS production from the mitochondrial electron transport chain is able to cause damage to the mtDNA found in close proximity. Because mtDNA encodes the majority of mitochondrial proteins, errors in gene expression of mtDNA may result in dysfunctional mitochondrial subunits. Dysfunctional mitochondria are then thought to contribute to further ROS leakage due to their inefficiency, which could then exacerbate mtDNA damage in a continuing vicious cycle [17,18].
Hearing loss is the most common form of sensory impairment in humans, affecting 360 million persons worldwide, with a prevalence of 183 million adult males and 145 million adult females. In nonsyndromic deafness, only hearing function is noticeably altered, whereas syndromic deafness is accompanied by other physiological defects. Although approximately 1in 500 children are born with impaired hearing, sudden or progressive forms of hearing loss can manifest at any age [19].
Hearing loss can be caused by environmental factors, such as exposure to noise or ototoxic chemicals, or by aged related senescence. Traumatic injury, such as injury caused by exposure to an explosion or to the firing of a gun, can lead to sudden hearing loss. Sometimes this hearing loss is accompanied by the perception of a constant ringing noise called tinnitus [20]. Moreover, genetic factors as mutations in MT-TS1, MYO7A or ACTG1 genes [21-23], between many others, have already been linked to nonsyndromic hearing loss. Identification of these genetic defects that cause hearing impairment has been instrumental in the discovery of molecular pathways that are involved in the regulation of auditory perception. Knowledge of these pathways provides a starting point for the development of therapeutic options for individuals that are affected by hearing impairment. Exciting advances in regenerative medicine and gene therapy offer potential alternative routes for restoring lost auditory function or for slowing down its progression.
Noise exposure is responsible for approximately 10% of hearing loss in adults, in particular military veterans [24]. Short impulses of high intensity noise such as a gunshot or explosion can trigger sudden hearing loss, which is generally irreversible and associated with structural damage to the auditory system. Susceptibility to damaging effects of noise differs remarkably among individuals, which indicates that genetic factors might be important in disease development. Gene association studies using candidate-gene approaches have focused mostly on genes that are linked to oxidative stress, K+ recycling and the heat shock response. Interestingly, the gene encoding catalase, an enzyme involved in oxidative stress reduction, appears to be directly linked to acquired and inherited hearing loss [25].
On the other hand, various chemical agents as aminoglycoside antibiotics, platinum- containing chemotherapy agents and nonsteroidal anti-inflammatory drugs as aspirin are ototoxic [26,27]. For example, aminoglycosides, antibiotic with broad spectrum activity, cause significant hearing loss, with estimates of a 20–50% chance of incidence when treating acute infections [28,29]. Hair cells are readily damaged by this compound probably due the similarity of hair cell mitochondrial ribosomes to their bacterial counterparts [30]. Also, compounds as cisplatin and carboplatin are extremely ototoxic, particularly in children. The role of platinum drugs is the induction of cancer cell apoptosis by DNA genomic cross-linking. However, hair cells seem to be highly sensitive to this apoptosis, probably due to their selective drug 5 transport [31].
Finally, the most common form of sensory impairment in older people is the age-related hearing loss [32]. This disorder is characterized by symmetric sensorineural hearing loss that starts at high frequencies with a prevalence of 35% of individuals over 65 years of age [33]. Although hearing loss has been considered to be part of a natural ageing process, not all humans suffer from age-related hearing loss; heritability studies suggest that the source of variability is both genetic and environmental. Interestingly, statistical studies have established associations between age-related hearing loss and genes linked to ROS detoxification, such as arylamine N-acetyltransferase 2 and glutathione S-transferase [34-36]. Other mitochondrial genes as Mitochondrial Uncoupling Protein 2 have also be linked to this disorder [37]; strongly confirming an essential role of mitochondria in hearing loss.
As mentioned before, mitochondria play a key role in ROS generation, and mutations in genes that affect mitochondrial function have been linked to genetic forms of hearing loss. Increasing number of studies demonstrated that hearing impairment following cochlear damage due to noise trauma, drug ototoxicity or age‐related cochlear degeneration is directly linked to a common pathogenesis involving the formation of ROS [38-41]. In this way, some studies demonstrated that age-related loss of cochlear hair cells was accelerated in Sod1 mutant mice [42], whereas guinea pig overexpressing catalase in their cochlea significantly protected hair cells and hearing thresholds after ototoxic treatment [43].
Cochleae are extremely vulnerable to oxidative stress because of the high metabolic demands of their mechanosensory hair cells in response to sound stimulation. Normally, ROS produced by hair cell mitochondria during physiological conditions are scavenged by hair cell endogenous antioxidant mechanisms. However, when excess ROS following noise overstimulation or ototoxic drug insults overwhelm the hair cell antioxidant defenses, elevated ROS concentration leads to genetic and cellular alterations which cause cellular dysfunctions such as lipid peroxidation, polysaccharide depolymerization, nucleic acid disruption, oxidation of sulfhydryl groups, and enzyme inactivation, consequently leading to permanent cochlear degeneration [44,45]. This increase of ROS also induces impaired blood flow to the cochlea, fused hair cell stereocilia and degeneration of supporting structures and nerve fibers; degenerative changes are also observed in the stria vascularis [46].
It is well known that mtDNA is particularly sensitive to oxidative damage because of the lack of protective histones. The mtDNA mutations will accumulate and expand with cell division, thus contributing to age-related diseases [47]. And evidence shows mtDNA mutations and mitochondrial dysfunction are involved in the aging and several diseases among which we find hearing loss. MtDNA mutations comprise point mutations, insertions and deletions. The constant mitochondrial turnover is crucial for maintaining normal function of mitochondria with age, especially for the post-mitotic nature of cells in cochlea. An essential role of mtDNA is the balance regulation of mitochondrial biogenesis, degradation, and fusion/fission ratio [48], among other functions. In this way, the oxidative damage of hair cell mtDNA could be the responsible of mitochondrial dysregulation and then the induction of hair cell apoptosis [49,50] (Figure 1). Corroborating this hypothesis, Menardo and collaborators demonstrated that mitochondrial biogenesis, characterized by ratio of mtDNA/nuclear DNA and activity of mitochondrial citrate synthase, was increased at young age and decreased at old age in senescence-accelerated prone mouse 8 (SAMP8), quite opposite to the senescence- accelerated resistant mice 1 (SAMR1) [40], suggesting that hair cells attempted to maintain normal mitochondrial function by strong early but limited stimulation of mitochondrial biogenesis.
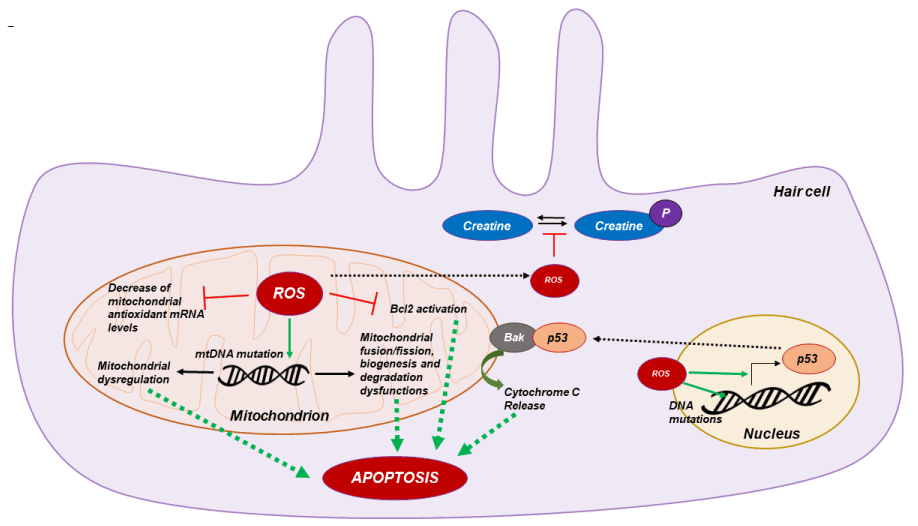
Figure 1. Molecular mechanism of mitochondrial ROS apoptosis. ROS overproduction in hear cell induce mtDNA deletion leading to mitochondrial dysfunctions. Also increase of ROS concentration impair hair cell metabolism by phospho-creatine imbalance and increase p53 transcription factor expression leading to mitochondrial cytochrome C release and hair cell apoptosis
The most important proteins mediating cell survival decisions at the level of the mitochondria are the members of the Bcl-2 family. Bcl-2 family members act upstream of caspase activation and serve as checkpoints in the regulation of apoptosis. Bcl-2 is localized primarily to the outer mitochondrial membrane, where it functions to inhibit cytochrome c release and preventing optotic pathway activation [51]. Cunningham and collaborators demonstrated that bcl-2 protein is present in the hair cells of the inner air and that overexpression of bcl-2 in transgenic mice increased hair cell survival after neomycin treatment. Furthermore, Bcl-2 overexpression reduced active caspase-9 positive hair cells and apoptotic pathway activation compared to wild type [52]. ROS overproduction leads to a down-regulation of bcl-2 [53], for this reason several groups proposed that the oxidative stress could also induce apoptotic pathway activation in cochlear hair cell by bcl-2 inhibition [54] (Figure 1).
As mentioned before the hearing system involves high-energy-demanding metabolic processes and creatine plays an important role in maintaining this energy supply in the cochlea as demonstrated using a creatine kinase knockout mouse model that exhibited preferential high-tone hearing loss [55]. Besides its role in energy replenishment, creatine also exerts a strong antioxidant effect by reducing the intra-mitochondrial production of ROS, as well as elevating and preserving the mitochondrial membrane potential [56]. Moreover, increase of ROS production in hair cells leads to the inhibition of enzymatic creatine kinase activity leading to an imbalance of creatine phosphorylation (Figure 1) and deficiencies in energy supply in hair cell stereocilia. Since oxidative damage caused by ROS has become a common athological cause involved in several types of hearing loss, creatine supplements are believed to improve the mitochondrial antioxidant defense system and maintain optimal energy homeostasis [57].
Finally, some studies proposed that a nuclear pro-apoptotic signaling pathway could also play a key role in hair cell apoptosis. It is well known that the nuclear transcription factor p53 is activated by oxidative DNA damage and that activation of p53 can trigger apoptosis in a wide range of cell types [58]. In response to oxidative cell stress, p53 rapidly translocates to mitochondria and directly binds to Bak and induces its oligomerization, leading to cytochrome c release and then apoptotic pathway induction [59]. Dr Salvi group demonstrated that p53 is activated in hair cells following cisplatin exposure, while deletion of p53 protects hair cells from the same drug-induced cell death [60,61]. So, in response to oxidative DNA damage caused by mitochondria-derived ROS in the aged cochlea and other target tissues, p53 may translocate to mitochondria and activate Bak, leading to Bak-mediated hair cell mitochondrial apoptosis (Figure 1).
Accumulating evidence demonstrated that antioxidants and free radical scavengers may serve as effective therapeutic agents to block the activation of death mechanisms induced by Ros overproduction, including the auditory system [62]. Therapy for hearing loss mediated by ROS overproduction is most likely to be successful during the early stages of disease progression, when structural damage and hair cell loss is limited. Some clinical and military trials have been carried out for temporary threshold shift, in which administration of antioxidant nutritional supplements, such as Mg2+ or vitamin B12, before moderate noise exposure showed some beneficial effects [63, 64]. Animal models have been useful for assessing whether counteracting the effects of ROS might be of therapeutic value. Antioxidants, such as glutathione, d-methionine, resveratrol and ascorbic acid, all attenuated noise-induced hearing loss in animal models when given before noise exposure [65].
Interestingly, as mentioned before polymorphisms in the gene encoding catalase have been linked to an increased susceptibility to hearing loss in humans, and mice that are heterozygous for a mutation in Sod1 gene show an increased vulnerability to hearing loss induced by noise exposure [42]. These genetic findings provide additional evidence that antioxidant treatment might be crucial for maintaining and recovery of normal hearing under loud noise conditions. In this way, targeting members of antioxidant pathways, including the enzymes that are involved in glutathione metabolism and in the breakdown of superoxide anions and hydrogen peroxidase, could be feasible options for the treatment of several types of hearing loss.
Increasing number of publications demonstrated that mitochondrial ROS production plays a key role in hearing loss by hair cell apoptotic pathway activation. Major ROS production pathway include oxidative phosphorylation dysfunction, nuclear transcription factors activation, mtDNA damage, decreased anti-ROS enzyme activity, etc. Other pathways, such as endoplasmic reticulum stress and necrotic cell death could also be involved in this disorder, but further studies of each type of hearing loss are required including the investigation of ROS mechanism, apoptotic pathway and other types of cell death.
Moreover, the effect of antioxidant supplementation in the prevention of hearing loss is presently controversial and inconclusive. Development of effective technique for antioxidant therapy in hearing loss requires an understanding of hair cell oxidative stress mechanisms underlying this hearing loss that may overcome this obstacle by opening doors to new treatment options.
The author confirms being the sole contributor of this work and approved it for publication.
Author declares no competing interests.
Thanks to Languedoc Roussillon Incubation and La Région Occitanie for funding this review.
- Schwander M, Kachar B, Müller U (2010) The cell biology of hearing. J Cell Biol 190: 9-20. [Crossref]
- Goutman, JD, Elgoyhen AB, Gómez-Casati, ME (2015) Cochlear hair cells: The sound-sensing machines. FEBS Lett.589: 3354-3361. [Crossref]
- McGrath J, Roy P, Perrin BJ (2016) Stereocilia morphogenesis and maintenance through regulation of actin stability. Semin Cell Dev Biol 30255-30255.
- Mann ZF, Kelley MW (2011) Development of tonotopy in the auditory periphery. Hear Res 276: 2-15. [Crossref]
- Hudspeth AJ (1997) How hearing happens. Neuron 19: 947-950. [Crossref]
- Wangemann P (2006) Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol 576: 11-21. [Crossref]
- Raphael Y, Altschuler RA (2003) Structure and innervation of the cochlea. Brain Res Bull 60: 397-422. [Crossref]
- Jones RG, Thompson CB (2009) Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 23: 537-548. [Crossref]
- Borutaite V (2010) Mitochondria as decision-makers in cell death. Environ Mol Mutagen 51: 406-416. [Crossref]
- Contreras L, Drago I, Zampese E, Pozzan T (2010) Mitochondria: the calcium connection. Biochim Biophys Acta 1797: 607-618. [Crossref]
- Scherz-Shouval R, Elazar Z (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17: 422-427. [Crossref]
- Niethammer P, Grabher C, Look AT, Mitchison TJ (2009) A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459: 996-999. [Crossref]
- Du Z, Yang Q, Liu L (2015) NADPH oxidase 2-dependent oxidative stress, mitochondrial damage and apoptosis in the ventral cochlearnucleus of D-galactose-induced aging rats. Neuroscience 286: 281–292.
- Jones DP (2008) Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 295: 849-868. [Crossref]
- Seidman MD, Ahmad N, Joshi D, Seidman J, Thawani S, et al., (2004) Age-related hearing loss and its association with reactive oxygen species and mitochondrial DNA damage. Acta Otolaryngol Suppl 552:16-24.
- Barja G (2013) Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxid Redox Signal 19: 1420-1445. [Crossref]
- Bandy B, Davison AJ (1990) Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic Biol Med 8: 523–539. [Crossref]
- Kandola K, Bowman A, Birch-Machin MA (2015) Oxidative stress - A key emerging impact factor in health, ageing, lifestyle and aesthetics. Int J Cosmet Sci 37: 1–8. [Crossref]
- Thompson DC, McPhillips H, Davis RL, Lieu TL, Homer CJ, et al. (2001) Universal newborn hearing screening: summary of evidence. JAMA 286: 2000–2010. [Crossref]
- Knipper M, Zimmermann U, Müller M (2010) Molecular aspects of tinnitus. Hear Res 266: 60- 69. [Crossref]
- Tiranti V (1995) Maternally inherited hearing loss, ataxia and myoclonus associated with a novel point mutation in mitochondrial tRNASer(UCN) gene. Hum Mol Genet 4: 1421–1427.
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, et al. (1997) Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet 16: 188-190. [Crossref]
- Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, et al. (2003) Mutations in the? -actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet 73: 1082-1091. [Crossref]
- Gordon JS, Griest SE, Thielman EJ (2016) Audiologic characteristics in a sample of recently-separated military Veterans: The Noise Outcomes in Servicemembers Epidemiology Study (NOISE Study). Hear Res 16: 30290-30298.
- Konings A, Van Laer L, Pawelczyk M, Carlsson PI, Bondeson ML, et al. (2007) Association between variations in CAT and noise-induced hearing loss in two independent noise-exposed populations. Hum Mol Genet 16: 1872-1883. [Crossref]
- Crundwell G, Gomersall P, Baguley DM (2016) Ototoxicity (cochleotoxicity) classifications: A review. Int J Audiol 55: 65-74. [Crossref]
- Campo P, Morata TC, Hong O (2013) Chemical exposure and hearing loss. Dis Mon 59: 119-138. [Crossref]
- Moore RD, Smith CR, Lietman PS (1984) Risk factors for the development of auditory toxicity in patients receiving aminoglycosides. J Infect Dis 149: 23-30. [Crossref]
- Schacht J, Talaska AE, Rybak LP (2012) Cisplatin and aminoglycoside antibiotics: hearing loss and its prevention. Anat Rec (Hoboken) 295: 1837-1850. [Crossref]
- Agrawal RK, Sharma MR () Structural aspects of mitochondrial translational apparatus. Curr Opin Struct Biol 22: 797-803. [Crossref]
- Langer T, am Zehnhoff-Dinnesen A, Radtke S, Meitert J, Zolk O (2013) Understanding platinum-induced ototoxicity. Trends Pharmacol Sci 34: 458-469. [Crossref]
2021 Copyright OAT. All rights reserv
- Someya S, Prolla TA (2010) Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech Ageing Dev 131: 480-486. [Crossref]
- Yamasoba T, Lin FR, Someya S, Kashio A, Sakamoto T, et al. (2013) Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hear Res 303: 30-38. [Crossref]
- Unal M, Tamer L, DoÄŸruer ZN, Yildirim H, VayisoÄŸlu Y, et al. (2005) N-acetyltransferase 2 gene polymorphism and presbycusis. Laryngoscope 115: 2238-2241. [Crossref]
- Van Eyken E, Van Camp G, Fransen E, Topsakal V, Hendrickx JJ, et al. (2007) Contribution of the N-acetyltransferase 2 polymorphism NAT2*6A to age-related hearing impairment. J Med Genet 44: 570-578. [Crossref]
- Bared A, Ouyang X, Angeli S, Du LL, Hoang K, et al. (2010) Antioxidant enzymes, presbycusis, and ethnic variability. Otolaryngol. Head Neck Surg 143: 263-268. [Crossref]
- Sugiura S, Uchida Y, Nakashima T, Ando F, Shimokata H (2010) The association between 364 gene polymorphisms in uncoupling proteins and hearing impairment in Japanese elderly. Acta Otolaryngol 130: 487-492.
- Shi X, Nuttall AL (2003) Upregulated iNOS and oxidative damage to the cochlear stria vascularis due to noise stress. Brain Res 967: 1-10. [Crossref]
- Van Campen LE (2002) Oxidative DNA damage is associated with intense noise exposure 369 in the rat. Hear Res 164: 29-38.
- Menardo J, Tang Y, Ladrech S, Lenoir M, Casas F, et al., (2012) Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse Cochlea. Antioxid Redox Signal. 16: 263- 74. [Crossref]
- Kamogashira T, Fujimoto C, Yamasoba T (2015) Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed Res Int 2015: 617207. [Crossref]
- Johnson KR, Yu H, Ding D, Jiang H, Gagnon LH, et al., (2010) Separate and combined effects of Sod1 and Cdh23 mutations on age-related hearing loss and cochlear pathology in C57BL/6J mice. Hear Res 268: 85-92. [Crossref]
- Kawamoto K, Sha SH, Minoda R, Izumikawa M, Kuriyama H, et al., (2004) Antioxidant gene therapy can protect hearing and hair cells from ototoxicity. Mol Ther 9: 173-181. [Crossref]
- Fujimoto C, Yamasoba T (2014) Oxidative Stresses and mitochondrial dysfunction in age- related hearing loss. Oxid Med Cell Longev 582-849.
- Chen B, Zhong Y (2011) Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex od D-galactose-induced aging rats. Mol Biol Rep 38: 3635-3642.
- Shi X, Nuttall AL (2003) Upregulated iNOS and oxidative damage to the cochlear stria vascularis due to noise stress. Brain Res 967: 1-10. [Crossref]
- Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6: 389-402. [Crossref]
- Lee H, Yoon Y (2016) Mitochondrial fission and fusion. Biochem Soc Trans 44: 1725-1735.
- Baker K, Staecker H. Low dose oxidative stress induces mitochondrial damage in hair cells. Anat Rec (Hoboken). 295: 1868-1876. [Crossref]
- Du Z, Yang Q, Zhou T, et al. D-galactose-induced mitochondrial DNA oxidative damage in the auditory cortex of rats. Mol Med Rep 10: 2861-2867. [Crossref]
- Gross A (2016) BCL-2 family proteins as regulators of mitochondria metabolism. Biochim Biophys Acta 1857: 1243-1246. [Crossref]
- Cunningham LL, Matsui JI. et al. Overexpression of Bcl-2 prevents neomycin-induced hair cell death and caspase-9 activation in the adult mouse utricle in vitro. J Neurobiol 60: 89-100. [Crossref]
- Pugazhenthi S, Nesterova A, Jambal P, Audesirk G, Kern M, et al., (2003) Oxidative stress-mediated down-regulation of bcl-2 promoter in hippocampal neurons. J Neurochem 84: 982-996. [Crossref]
- Han C, Someya S (2013) Mouse models of age-related mitochondrial neurosensory hearing loss. Mol Cell Neurosci 55: 95-100. [Crossref]
- Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, et al., (2007) Hair bundles are specialized for ATP delivery via creatine kinase. Neuron 53: 371-386. [Crossref]
- Meyer LE, Machado LB, Santiago AP, da-Silva WS, De Felice FG, et al. (2006) Mitochondrial creatine kinase activity prevents reactive oxygen species generation: antioxidant role of mitochondrial kinase-dependent ADP recycling activity. J Biol Chem 281: 37361-37371. [Crossref]
- Minami SB, Yamashita D. et al., (2007) Creatine and tempol attenuate noise-induced hearing loss. Brain Res 1148: 83-89. [Crossref]
- Culmsee C, Mattson MP (2005) p53 in neuronal apoptosis. Biochem Biophys Res Commun 331: 761-777. [Crossref]
- Lindenboim L, Borner C, Stein R (2011) Nuclear proteins acting on mitochondria. Biochim Biophys Acta 1813: 584-96. [Crossref]
- Zhang M, Liu W, Ding D, Salvi R (2003) Pifithrin-alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience 120: 191-120. [Crossref]
- Cheng AG, Cunningham LL, Rubel EW (2005) Mechanisms of hair cell death and protection. Curr. Opin. Otolaryngol. Head Neck Surg. 13: 343-348. [Crossref]
- Takumida M, Anniko M (2005) Radical scavengers: a remedy for presbyacusis. A pilot study. Acta Otolaryngol 125: 1290-1295. [Crossref]
- Attias J, Bresloff I (2003) Preventing noise induced otoacoustic emission loss by increasing magnesium (Mg2+) intake in guinea-pigs. J Basic Clin Physiol Pharmacol. 14: 119-1136.
- Quaranta A, Scaringi A. Bartoli R, Margarito MA, Quaranta N (2004) The effects of 'supra-physiological' vitamin B12 administration on temporary threshold shift. Int J Audiol. 43: 162-165. [Crossref]
- Tavanai E, Mohammadkhani G (2016) Role of antioxidants in prevention of age-related hearing loss: a review of literature. Eur Arch Otorhinolaryngol