Abstract
Atherosclerosis is one of the most investigated disorders among cardiovascular anomalies and its clinical manifestations are responsible for the deaths of over 17 million patients worldwide. Atheromatous plaques block the blood flux and bring about long-term serious consequences. Although the precise cause is still unknown, atherosclerosis onset is related to risk factors such as high cholesterol levels, high blood pressure, obesity, diabetes, smoking, imbalanced diet, family history and genetic predisposition. The inherited risk of atherosclerosis is increased in families with multiple affected members. Therefore, family history of atherosclerosis account for genetic counseling and genetic testing. In the present review, we explore the influence of genetic polymorphism of target genes (eNOS, GSTM1, GSTT1 and TP53) and on atherosclerosis. Here we show the possible influence of genetic polymorphisms and atherosclerosis. Knowledge of genetic variants that increase susceptibility of atherosclerosis may launch more efficient methods of prevention, diagnostic, treatment, and genetic counseling in the important field of cardiology. Although discrepancies are seen, each target gene show promisor activity as atherosclerosis markers that could be used in diagnostic and treatment.
Keywords
atherosclerosis, eNOS, GSTM1, GSTT1, TP53
Introduction
Atherosclerosis is the most common disorder among cardiovascular anomalies and its clinical manifestations are responsible for the high rate of mortality (17 million deaths) observed worldwide [1-3]. The main characteristic of the disease is the progressive narrowing of artery lumen due to the development of atheromatous plaques, which limits the flow of oxygen-rich blood to organs and other parts of the body. The blood flux blockage performed by atheromatous plaques can affect large and medium-size arteries leading to severe consequences. Although the precise cause is still unknown, atherosclerosis onset is related to risk factors such as high cholesterol levels [4], high blood pressure [5], obesity [6,7], diabetes [8,9], smoking [10,11], imbalanced diet [7], family history and genetic predisposition [12,13].
Risk factors act synergistically and stimulate the synthesis and release of proinflammatory cytokines, oxidative degradation of lipids and oxidative stress. The outcome is increased cell adhesion [14] and endothelium-derived relaxing factors and anticoagulants a reduction [15,16]. Atherosclerosis is asymptomatic at first [17], but it might evolve to peripheral [18] or coronary [19] artery disease, ischemic stroke [20], myocardial infarction [21], severe thrombosis [22], artery stenosis [23], kidney [24,25], liver [26,27], lung [28] and intestines [29] injuries. Atherosclerosis is a complex condition, usually starts early in life and progresses as people age [26]. In addition, several works have shown the influence of DNA damage to atherosclerosis [30-32].
Inflammation is intimately linked to atherosclerosis lesion formation and progression [33]. The endothelial dysfunction observed in atherosclerosis is responsible for the proin?ammatory and prothrombotic environment generated by atheromatous plaques, lesions and inflammation, which might lead to severe clinical manifestations of the disease [16]. Atheromatous plaques comprises macrophages, monocytes, low-density lipoproteins (LDL), cholesterol crystals, smooth muscle cells, cellular debris and extracellular matrix. Atherosclerosis initiates as monocytes from blood adhere to the endothelium and then diapedeses between endothelial cells to enter the sub-endothelial region, differentiate into macrophage and accumulate lipoprotein [34]. LDL is then oxidized leading to oxygen reactive species formation (ROS), lesion and consequently inflammation response driven by cytokine production. Next, macrophages are recruited, they phagocyte oxidized LDL turning into foam cells. The inflammatory estate is aggravated by death of foam, a process which will recruit more macrophages and lead to the production of more foam cells [35,36].
Atherosclerosis has a complex genetic trait. Hundreds of genes have been reported to be related to atherosclerosis and they regulated a myriad of physiological processes, including macromolecules metabolism, coagulation, detoxification, endothelial function and coagulation. Several of those genes are polymorphic and variations in their sequence contribute to the molecular pathogenesis of the disease [37–41]. The inherited risk of atherosclerosis is increased in families with multiple affected members [42]. Therefore, family history of atherosclerosis account for genetic counseling and genetic testing. In the present review, we explore the influence of genetic polymorphism of target genes (eNOS, GSTM1, GSTT1 and TP53) and on atherosclerosis.
Endothelial nitric oxide synthase
Endothelial dysfunction is characterized by apoptosis, down regulation of the endothelial nitric oxide synthase (eNOS), a reduction of nitric oxide levels and an increase in ROS formation. These features lead to inflammation and are highly related to atherosclerosis onset and progression. The most important eNOS function is the regulation of vascular tonus and integrity [43], producing nitric oxide (NO), an important relaxing factor that acts on vasodilation [44,45], leukocyte adhesion [46], antithrombotic [47] and antioxidant processes [48], and platelet aggregation [49]. NO diffuses freely across vascular smooth muscle cell membranes and activates a cascade pathway resulting in vasodilation [50]. eNOS is highly polymorphic and studies have showed that variations in eNOS gene might increase the susceptibility to vascular [37,38] and other diseases such as diabetes [51], retinopathies [52] and erectile dysfunction [51,53].
Among the most frequent polymorphisms of the eNOS gene are on exon 7 (894 G/T), intron 11(-30 A/G), intron 18(27A/C), intron 23(10G/T) within the coding region and T786C, A922G and T1468A in the flanking region of the gene [54]. G894T and T786C are the most investigated eNOS polymorphisms.
The relation between the T786C eNOS polymorphism and risk of atherosclerosis has been contradictory. Ethnic groups may respond differently regarding susceptibility to disease [55]. T786C polymorphism features the substitution of thymine (TT genotype) to cytosine (CC or TC genotypes) at the 786 locus of the gene. Carriers of the mutant allele (homozygotes or heterozygotes) might have increased susceptibility to vascular diseases. Higher frequencies of the wild type has been identified in the Brazilian [37,56], Greek [57], Chilean [58] and Italian [59] population with no statistically significant result for the influence of the polymorphism on cardiovascular diseases. Conversely, the mutant allele was more frequent in the Turkish [60] and Ukrainian [61] populations, for which the polymorphism may be an increased risk factor for atherosclerosis.
The replacement of guanine for a thymine at position 894 of the eNOS gene characterizes the G894T polymorphism. G894T carriers show less NO production and it may be a factor that increases patients’ susceptibility to atherosclerotic disease. The frequency of the mutant allele was more frequent in the Brazilian [62], Mexican [63] and Italian [64] populations. The wild type allele is more frequent in England [54], Germany [65], Turkey [66], South Africa [67] and in the African-American population [68]. The wild type is also predominant in the Asia population such as Korea [69], Japan [70] and India [71]; but interestingly, TT the genotype is practically absent. Although, the distribution of these polymorphisms vary among populations, eNOS gene seems to be related to the pathogeny of artherosclerosis. Future studies are required to shed some light on observed discrepancies and bring a better understanding of the relation between polymorphisms and disease.
Glutathione S-transferase
Glutathione S-transferase (GST) belong to a family of genes and proteins from phase II metabolic isozymes. They catalyze the reduced form of glutathione into xenobiotic substrates, turning such compounds more water-soluble in order to fulfill their function of detoxification [72]. GSTs polymorphisms are linked to innumerous diseases and anomalies such cancer [73], drug resistance [74], diabetes [75], atherosclerosis [39,40], and other inflammatory [76] and autoimmune diseases [77,78].
Glutathione S-transferase Mu 1 (GSTM1) and glutathione S-transferase theta-1 (GSTT1) take part in the detoxification of carcinogens, drugs, xenobiotics and products generated by oxidative stress. GSTs from the mu and theta classes are particularly polymorphic. Null mutations in GSTM1 have been associated with a large variety of diseases and an increased susceptibility to toxins [39,73]. Null mutations in GSTM1 increases the susceptibility diseases as well [40,75]. The GSTM1 present genotype was identified in a higher frequency in the Brazilian [39], Turkish [79] and British [80] population. They found statistically significant difference comparing the null and present genotype in healthy and atherosclerotic patients, suggesting that the null genotype is a risk factor for the disease.
GSTT1 present genotype has been found to be more frequent in certain populations from Brazil [40,81], India [82] and Serbia [83]. Interistingly, a study conducted in a Turkish population has shown that GSTT1 null genotype was nine-fold more frequent in atherosclerotic patients compared to healthy ones [79]. Another interesting fact is the difference between frequencies of null and present GSTT1 genotypes in males and females, it has been found that the present genotype is more frequent in males [40,84–86].
Understanding the polymorphisms that affect the GST family, especially GSTM1 and GSTT1, is important in order to elucidate their connections with onset and progression of atherosclerosis. Moreover, the results from worldwide indicates that having GSTM1 and GSTT1 as molecular markers of atherosclerosis in order to establish better prevention, diagnostic, treatment, and genetic counseling in cardiovascular diseases.
Tumor protein 53
Tumor protein 53 (TP53) gene is classified as an oncogene (tumor suppressor gene). The protein coded by TP53 (p53) exerts a function of protector of the genome, in a way to maintain the genetic material stability [87]. The protein p53 regulates cell cycle and cellular growth [88], apoptosis [87], transcription [89] and angiogenesis [90]. The TP53 location within the human genome is 17p13.1. The gene is proximately 20 kb and comprises 11 exons. The nucleotide sequence is highly conserved and structurally homologous across species from different kingdoms [91]. DNA damage induces TP53 expression which consists of cell cycle arrest, repair or apoptosis [87].
Mutations on p53 alters the capacity of the cell to control mitosis [92]. This single nucleotide polymorphism changes de structure of the protein, losing its capacity to bind to DNA [93] and consequently increasing the risk of disease. The most frequent p53 polymorphism is characterized by a substitution of the base guanine by a cytosine at codon 72 and consequently within the protein the amino acid arginine is substituted for proline. There are three possible genotypes for the TP53 (arg/arg, arg/prol, prol/prol).
Polymorphic studies about p53 polymorphisms and atherosclerosis show some discrepancies. Studies performed with Italian [94,95], Kwait [96], Brazilian [41,97] did not find any relation between the polymorphic p53 and occurrence of atherosclerosis. Conversely, studies performed in different population from Italy [98,99], Chile [100] and Japan [101] have shown an intrinsic influence of p53 codon 72 polymorphism on atherosclerosis. Clearly, the large number of function exerted by TP53 and its protein is somehow related to a series of inflammation disorders such as atherosclerosis, however, more studies should be performed in order to identify what causes lie underneath this influence. Large samples studies and wide genome association studies may shed some light on answered questions.
Concluding remarks
Knowledge of genetic variants that increase susceptibility of atherosclerosis may launch more efficient methods of prevention, diagnostic, treatment, and genetic counseling in the important field of cardiology. Molecular biology and molecular genetics have contributed to our understanding of the complex and multifactorial diseases such as cardiovascular disorders. The most important polymorphic genes related to atherosclerosis, eNOS, GSTM1, GSTT1 and TP53 (Figure 1) may show some conflicting results but they have definitely shown a relationship with onset and progression of atherosclerosis.
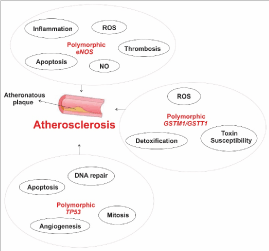
Figure 1. A schematic view of important polymorphic genes related to atherosclerosis. The eNOS gene produces nitric oxide (NO), important to prevent reactive oxygen species formation. Polymorphic eNOS induces inflammation, ROS and thrombosis and reduces apoptosis rate and NO production. GSTM1 and GSTT1 are related to detoxification processes and mutations in their sequence lead to increased susceptibility to toxins and carcinogens. Polymorphic TP53 promotes cell division control loss, reduced apoptosis and angiogenesis rate and affects DNA repair.
References
- Zamani P, Jacobs DR Jr, Segers P, Duprez DA, Brumback L, et al. (2014) Reflection magnitude as a predictor of mortality: the Multi-Ethnic Study of Atherosclerosis. Hypertension 64: 958-964. [Crossref]
- Yilmaz H, Çelik HT, Gurel OM, Bilgic MA, Namuslu M, et al. (2015) Increased serum levels of GDF-15 associated with mortality and subclinical atherosclerosis in patients on maintenance hemodialysis. Herz 40: 305-312. [Crossref]
- Maeda Y, Inoguchi T, Etoh E, Kodama Y, Sasaki S, et al. (2014) Brachial-ankle pulse wave velocity predicts all-cause mortality and cardiovascular events in patients with diabetes: the Kyushu Prevention Study of Atherosclerosis. Diabetes Care 37: 2383-2390. [Crossref]
- Pan X, Jiang XC, Hussain MM (2013) Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation 128: 1758-1769. [Crossref]
- Miedema MD, Lopez FL, Blaha MJ, Virani SS, Coresh J, et al. (2015) Implications of the Eighth Joint National Committee Guidelines for the Management of High Blood Pressure for Aging Adults: Atherosclerosis Risk in Communities Study. Hypertension 66: 474-480. [Crossref]
- Lovren F, Teoh H, Verma S (2015) Obesity and atherosclerosis: mechanistic insights. Can J Cardiol 31: 177-183. [Crossref]
- Nguyen HN, Ahn YJ, Medina EA, Asmis R (2018) Dietary 23-hydroxy ursolic acid protects against atherosclerosis and obesity by preventing dyslipidemia-induced monocyte priming and dysfunction. Atherosclerosis 275: 333-341. [Crossref]
- Singh R, Devi S, Gollen R (2015) Role of free radical in atherosclerosis, diabetes and dyslipidaemia: larger-than-life. Diabetes Metab Res Rev 31: 113-126. [Crossref]
- Zeadin MG, Petlura CI, Werstuck GH (2013) Molecular mechanisms linking diabetes to the accelerated development of atherosclerosis. Can J Diabetes 37: 345-350. [Crossref]
- McEvoy JW, Blaha MJ, DeFilippis AP, Lima JA, Bluemke DA, et al. (2015) Cigarette smoking and cardiovascular events: role of inflammation and subclinical atherosclerosis from the MultiEthnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol 35: 700-709. [Crossref]
- Siasos G, Tsigkou V, Kokkou E, Oikonomou E, Vavuranakis M, et al. (2014) Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Curr Med Chem 21: 3936-3948.
- Ye Z, Bailey KR, Austin E, Kullo IJ (2016) Family history of atherosclerotic vascular disease is associated with the presence of abdominal aortic aneurysm. Vasc Med 21: 41-46. [Crossref]
- Van daele CM, De Meyer T, De Buyzere ML, Gillebert TC, Denil SL, et al. (2013) Addition of a novel, protective family history category allows better profiling of cardiovascular risk and atherosclerotic burden in the general population. The Asklepios Study. PLoS One 8: e63185. [Crossref]
- Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, et al. (2014) Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation 129: 66-76. [Crossref]
- MacKenzie A (2011) Endothelium-derived vasoactive agents, AT1 receptors and inflammation. Pharmacol Ther 131: 187-203. [Crossref]
- Mudau M, Genis A, Lochner A, Strijdom H (2012) Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr 23: 222–231. [Crossref]
- Niu L, Zhang Y, Meng L, Xiao Y, Wong KK, et al. (2014) Detection of subclinical atherosclerosis in asymptomatic subjects using ultrasound radiofrequency-tracking technology. PLoS ONE. 9: e111926. [Crossref]
- Zimmermann A, Senner S, Eckstein HH, Pelisek J (2015) Histomorphological evaluation of atherosclerotic lesions in patients with peripheral artery occlusive disease. Adv Med Sci 60: 236-239. [Crossref]
- Zabalza M, Subirana I, Lluis-Ganella C, Sayols-Baixeras S, de Groot E (2015) Association Between Coronary Artery Disease Genetic Variants and Subclinical Atherosclerosis: An Association Study and Meta-analysis. Rev Esp Cardiol 68: 869-877. [Crossref]
- Reina SA, Llabre MM, Allison MA, Wilkins JT, Mendez AJ (2015) HDL cholesterol and stroke risk: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 243: 314-319. [Crossref]
- De Jesus NM, Wang L, Herren AW, Wang J, Shenasa F (2015) Atherosclerosis exacerbates arrhythmia following myocardial infarction: Role of myocardial inflammation. Heart Rhythm 12: 169-178. [Crossref]
- Badimon L, Vilahur G (2014) Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med 276: 618–632. [Crossref]
- Horie N, Tateishi Y, Morikawa M, Morofuji Y, Hayashi K, et al. (2016) Acute stroke with major intracranial vessel occlusion: Characteristics of cardioembolism and atherosclerosis-related in situ stenosis/occlusion. J Clin Neurosci 32: 24-29. [Crossref]
- Roos CJ, Delgado V, de Koning EJ, Rabelink TJ, Jukema JW, et al. (2014) Associations of atherosclerosis in the descending thoracic aorta on CTA with arterial stiffness and chronic kidney disease in asymptomatic patients with diabetes mellitus. Int J Cardiovasc Imaging 30: 1151-1159. [Crossref]
- Palanca A, Castelblanco E, Perpiñán H, Betriu A, Soldevila B, et al. (2018) Prevalence and progression of subclinical atherosclerosis in patients with chronic kidney disease and diabetes. Atherosclerosis 276: 50-57. Crossref
- Puig J, Blasco G, Daunis-I-Estadella J, Loshuertos E, Codina J, et al. (2015) Nonalcoholic fatty liver disease and age are strong indicators for atherosclerosis in morbid obesity. Clin Endocrinol (Oxf) 83: 180-186. Crossref
- Peters KM, Wilson RB, Borradaile NM (2018) Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr Opin Lipidol. [Crossref]
- Donohue KM, Hoffman EA, Baumhauer H, Guo J, Ahmed FS (2013) Asthma and lung structure on computed tomography: the Multi-Ethnic Study of Atherosclerosis Lung Study. J Allergy Clin Immunol 131: 361-368. [Crossref]
- Schramm R, Appel F, Reinacher M, Schäfers HJ, Bierbach B, et al. (2011) Atherosclerosis aggravates ischemia/reperfusion injury in the gut and remote damage in the liver and the lung. Inflamm Res 60: 555-567. [Crossref]
- Wu H, Roks AJM (2014) Genomic instability and vascular aging: a focus on nucleotide excision repair. Trends Cardiovasc Med 24: 61-68. [Crossref]
- Bautista-Niño PK, Portilla-Fernandez E, Vaughan DE, Danser AH, Roks AJ (2016) DNA Damage: A Main Determinant of Vascular Aging. Int J Mol Sci 17: 5. [Crossref]
- Shah AV, Bennett MR (2017) DNA damage-dependent mechanisms of ageing and disease in the macro- and microvasculature. Eur J Pharmacol 816: 116-128. [Crossref]
- Pant S, Deshmukh A, Gurumurthy GS, Pothineni NV, Watts TE (2014) Inflammation and atherosclerosis-revisited. J Cardiovasc Pharmacol Ther 19: 170-178. [Crossref]
- Schwartz CJ , Valente AJ , Sprague EA , Kelley JL , Cayatte AJ, et al. (1992) Atherosclerosis. Potential targets for stabilization and regression. Circulation 86: 117-123.
- Raghavan S, Singh NK, Gali S, Mani AM, Rao GN (2018) PKCθ via Activating Transcription Factor 2-Mediated CD36 Expression and Foam Cell Formation of Ly6Chi Cells Contributes to Atherosclerosis. Circulation. [Crossref]
- Yu XH, Fu YC, Zhang DW, Yin K, Tang CK (2013) Foam cells in atherosclerosis. Clin Chim Acta 424: 245-252. [Crossref]
- Barbosa AM, Silva KSF, Lagares MH, Rodrigues DA, da Costa IR, et al (2017) Atherosclerosis: analysis of the eNOS (T786C) gene polymorphism. Genet Mol Res 16: 3. [Crossref]
- Campedelli FL, E Silva KSF, Rodrigues DA, Martins JVM, Costa IR, et al. (2017) Polymorphism of the gene eNOS G894T (Glu298Asp) in symptomatic patients with aterosclerosis. Genet Mol Res 16: 2 [Crossref]
- Rodrigues DA, Martins JV, E Silva KS, Costa IR, Lagares MH, et al (2017) GSTM1 polymorphism in patients with clinical manifestations of atherosclerosis. Genet Mol Res 16: 1. [Crossref]
- Martins JVM, Rodrigues DA, Silva KSF, Costa IR, Lagares MH, et al. (2017) Molecular analysis of the GSTT1 gene polymorphism in patients with clinical manifestation of atherosclerosis. Genet Mol Res 16: 3. [Crossref]
- Lagares MH, Silva KSF, Barbosa AM, Rodrigues DA, Costa IR, et al. (2017) Analysis of p53 gene polymorphism (codon 72) in symptomatic patients with atherosclerosis. Genet Mol Res 16: 3. [Crossref]
- Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, et al. (2013) Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 504: 432–436. [Crossref]
- Palmer RM, Ferrige AG, Moncada S (1987) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524-526. [Crossref]
- Cockcroft JR (2005) Exploring vascular benefits of endothelium-derived nitric oxide. Am J Hypertens 18: 177S-183S. [Crossref]
- Villanueva C, Giulivi C (2010) Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med 49: 307-316. [Crossref]
- Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, et al. (2011) Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J Clin Invest 121: 120-131. [Crossref]
- Walford G, Loscalzo J (2003) Nitric oxide in vascular biology. J Thromb Haemost 1: 2112–2118.
- Ma L, Chen S, Li S, Deng L, Li Y, et al. (2018) Effect of Allicin against Ischemia/Hypoxia-Induced H9c2 Myoblast Apoptosis via eNOS/NO Pathway-Mediated Antioxidant Activity. Evid Based Complement Alternat Med 1: 3207973.
- Radziwon-Balicka A, Lesyk G, Back V, Fong T, Loredo-Calderon EL, et al. (2017) Differential eNOS-signalling by platelet subpopulations regulates adhesion and aggregation. Cardiovasc Res 113: 1719-1731. [Crossref]
- Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, et al. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286: 1583-1587.
- Hermans MP1, Ahn SA, Rousseau MF (2012) eNOS [Glu298Asp] polymorphism, erectile function and ocular pressure in type 2 diabetes. Eur J Clin Invest 42: 729-737. [Crossref]
2021 Copyright OAT. All rights reserv
- Taverna MJ, Elgrably F, Selmi H, Selam JL, Slama G (2005) The T-786C and C774T endothelial nitric oxide synthase gene polymorphisms independently affect the onset pattern of severe diabetic retinopathy. Nitric Oxide 13: 88-92. [Crossref]
- Lee YC, Huang SP, Liu CC, Yang YH, Yeh HC, et al. (2012) The association of eNOS G894T polymorphism with metabolic syndrome and erectile dysfunction. J Sex Med 9: 837-843. [Crossref]
- Hingorani AD, Liang CF, Fatibene J, Lyon A, Monteith S, et al. (1999) A common variant of the endothelial nitric oxide synthase (Glu298-->Asp) is a major risk factor for coronary artery disease in the UK. Circulation 100: 1515-1520.
- Luo JQ, Wen JG, Zhou HH, Chen XP, Zhang W (2014) Endothelial nitric oxide synthase gene G894T polymorphism and myocardial infarction: a meta-analysis of 34 studies involving 21,068 subjects. PLoS ONE 9: e87196. [Crossref]
- da Costa Escobar Piccoli J1, Manfredini V, Hamester FI, Bandinelli JB, Turkienicz IM, et al. (2012) Interaction between endothelial nitric oxide synthase gene polymorphisms (-86T>C, 894G>T and intron 4 a/b) and cardiovascular risk factors in acute coronary syndromes. Arch Med Res 43: 205-211. [Crossref]
- Ragia G, Nikolaidis E, Tavridou A, Arvanitidis KI, Kanoni S, et al. (2010) Endothelial nitric oxide synthase gene polymorphisms -786T >C and 894G >T in coronary artery bypass graft surgery patients. Hum Genomics 4: 375-383. [Crossref]
- Jaramillo PC, Lanas C, Lanas F, Salazar LA (2010) Polymorphisms of the NOS3 gene in Southern Chilean subjects with coronary artery disease and controls. Clin Chim Acta 411: 258-262. [Crossref]
- Ghilardi G, Biondi ML, DeMonti M, Bernini M, Turri O, et al. (2002) Independent risk factor for moderate to severe internal carotid artery stenosis: T786C mutation of the endothelial nitric oxide synthase gene. Clin Chem 48: 989-993.
- Ciftçi C, Melil S, Cebi Y, Ersöz M, Cagatay P, et al. (2008) Association of endothelial nitric oxide synthase promoter region (T-786C) gene polymorphism with acute coronary syndrome and coronary heart disease. Lipids Health Dis 7: 5. [Crossref]
- Dosenko VIe, Zahorii VIu, Lutai IaM, Parkhomenko OM, Moibenko OO (2005) [Allelic polymorphism of endothelial NO-synthase (T(-786)-->C) promoter gene as risk factor of acute coronary syndrome]. Fiziol Zh 51: 72-76.
- Tardin OMA, Pereira SB, Velloso SMW, Balieiro HM, Costa B, et al. (2013) Polimorfismo G894T da óxido nítrico-sintetase endotelial e o prognóstico na insuficiência cardíaca. Arquivos Brasileiros de Cardiologia 101: 352-358. [Crossref]
- García-González I1, Solís-Cárdenas Ade J2, Flores-Ocampo JA2, Alejos-Mex R3, Herrera-Sánchez LF, et al. (2015) [G894T (NOS3) and G1958A (MTHFD1) gene polymorphisms and risk of ischemic heart disease in Yucatan, Mexico]. Clin Investig Arterioscler 27: 64-73. [Crossref]
- Fatini C, Sofi F, Sticchi E, Bolli P, Sestini I, et al. (2005) eNOS G894T polymorphism as a mild predisposing factor for abdominal aortic aneurysm. J Vasc Surg 42: 415-419. [Crossref]
- Krex D, Fortun S, Kuhlisch E, Schackert HK, Schackert G (2006) The role of endothelial nitric oxide synthase (eNOS) genetic variants in European patients with intracranial aneurysms. J Cereb Blood Flow Metab 26: 1250-1255. [Crossref]
- Afrasyap L, Ozturk G (2004) NO level and endothelial NO synthase gene polymorphism (Glu298Asp) in the patients with coronary artery disease from the Turkish population. Acta Biochim Biophys Sin (Shanghai) 36: 661-666.
- Hillermann R, Carelse K, Gebhardt GS (2005) The Glu298Asp variant of the endothelial nitric oxide synthase gene is associated with an increased risk for abruptio placentae in pre-eclampsia. J Hum Genet 50: 415-419. [Crossref]
- Li R, Lyn D, Lapu-Bula R, Oduwole A, Igho-Pemu P, et al. (2004) Relation of endothelial nitric oxide synthase gene to plasma nitric oxide level, endothelial function, and blood pressure in African Americans. Am J Hypertens 17: 560-567. [Crossref]
- Moon J, Yoon S, Kim E, Shin C, Jo SA, et al. (2002) Lack of evidence for contribution of Glu298Asp (G894T) polymorphism of endothelial nitric oxide synthase gene to plasma nitric oxide levels. Thromb Res 107: 129-134.
- Kato N, Sugiyama T, Morita H, Nabika T, Kurihara H, et al. (1999) Lack of evidence for association between the endothelial nitric oxide synthase gene and hypertension. Hypertension 33: 933-936.
- Nishevitha NS, Angeline T, Jeyaraj N (2009) Endothelial nitric oxide synthase (eNOS) Glu298-->Asp polymorphism (G894T) among south Indians. Indian J Med Res 129: 68-71.
- Oakley A (2011) Glutathione transferases: a structural perspective. Drug Metab Rev 43: 138-151. [Crossref]
- Geng M, Wang L, Chen X, Cao R, Li P (2013) The association between chemosensitivity and Pgp, GST-π and Topo II expression in gastric cancer. Diagn Pathol 8: 198. [Crossref]
- Zeng X, Morgenstern R, Nyström AM (2013) Nanoparticle-directed sub-cellular localization of doxorubicin and the sensitization breast cancer cells by circumventing GST-mediated drug resistance. Biomaterials 35: 1227-1239. [Crossref]
- S Nath S, Das S, Bhowmik A, Ghosh SK, Choudhury Y (2017) The GSTM1 and GSTT1 null genotypes increase the risk for Type 2 diabetes mellitus and the subsequent development of diabetic complications: A meta-analysis. Curr Diabetes Rev. [Crossref]
- Stocco G, Pelin M, Franca R, De Iudicibus S, et al. (2014) Cuzzoni E, Pharmacogenetics of azathioprine in inflammatory bowel disease: a role for glutathione-S-transferase?. World J Gastroenterol 20: 3534-3541. [Crossref]
- Lundström E, Hartshorne T, Li K, Lindblad S, Wick MC, et al. (2011) Effects of GSTM1 in rheumatoid arthritis; results from the Swedish EIRA study. PLoS ONE 6: e17880. [Crossref]
- Glesse N, Rohr P, Monticielo OA, Rech TF, Brenol JC, et al. (2014) Genetic polymorphisms of glutathione S-transferases and cytochrome P450 enzymes as susceptibility factors to systemic lupus erythematosus in southern Brazilian patients. Mol Biol Rep 41: 6167-6179. [Crossref]
- Taspinar M, Aydos S, Sakiragaoglu O, Duzen IV, Yalcinkaya A, et al. (2012) Impact of genetic variations of the CYP1A1, GSTT1, and GSTM1 genes on the risk of coronary artery disease. DNA Cell Biol 31: 211-218. [Crossref]
- Wilson MH1, Grant PJ, Hardie LJ, Wild CP (2000) Glutathione S-transferase M1 null genotype is associated with a decreased risk of myocardial infarction. FASEB J 14: 791-796.
- Bazo AP, Salvadori D Jr, Salvadori RA, Sodré LP, da Silva GN, et al. (2011) DNA repair gene polymorphism is associated with the genetic basis of atherosclerotic coronary artery disease. Cardiovasc Pathol 20: e9-15. [Crossref]
- Girisha KM, Gilmour A, Mastana S, Singh VP, Sinha N, et al. (2004) T1 and M1 polymorphism in glutathione S-transferase gene and coronary artery disease in North Indian population. Indian J Med Sci 58: 520-526.
- Zivkovic M, Stankovic A, Djuric T, Koncar I, Kolakovic A, et al. (2014) Effects of glutathione S-transferase T1 and M1 deletions on advanced carotid atherosclerosis, oxidative, lipid and inflammatory parameters. Mol Biol Rep 41: 1157-1164. [Crossref]
- Maciel SS, Pereira Ada C, Silva GJ, Rodrigues MV, Mill JG, et al. (2009) Association between glutathione S-transferase polymorphisms and triglycerides and HDL-cholesterol. Atherosclerosis 206: 204-208. [Crossref]
- Schreiber R, Mill JG, Krieger JE, Pereira AC, Nadruz W (2013) Association between glutathione S-transferase M1 polymorphism and urinary sodium excretion in a Brazilian population. Am J Hypertens 26: 1024-1029. [Crossref]
- Wang R, Wang Y, Wang J, Yang K (2012) Association of glutathione S-transferase T1 and M1 gene polymorphisms with ischemic stroke risk in the Chinese Han population. Neural Regen Res 7: 1420-1427. [Crossref]
- Wawryk-Gawda E, Chylinska-Wrzos P, Lis-Sochocka M, Chlapek K, Bulak K, et al. (2014) P53 protein in proliferation, repair and apoptosis of cells. Protoplasma 251: 525-533. [Crossref]
- Surget S, Khoury MP, Bourdon JC (2014) Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther 7: 57-68. [Crossref]
- Walker KK, Levine AJ (1996) Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci U.S.A 93: 15335-15340.
- Song R, Tian K, Wang W, Wang L (2015) P53 suppresses cell proliferation, metastasis, and angiogenesis of osteosarcoma through inhibition of the PI3K/AKT/mTOR pathway. Int J Surg 20: 80-87. [Crossref]
- Soussi T, May P (1990) Structural aspects of the p53 protein in relation to gene evolution. Oncogene 5: 945-952.
- Syvänen AC (2001) Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet 2: 930-942. [Crossref]
- Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS, Kinzler KW, et al. (1994) Vogelstein. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc Natl Acad Sci U S A 91: 1998-2002.
- D'Agostini F, Fronza G, Campomenosi P, Izzotti A, Petrilli GL, et al. (1995) Cancer biomarkers in human atherosclerotic lesions: no evidence of p53 involvement. Cancer Epidemiol Biomarkers Prev 4: 111–115.
- Manfredi S, Masetti S, Botto N, Colombo MG, Terrazzi M, et al. (2002) P53 codon 72 polymorphism in coronary artery disease: no evidence for association with increased risk or micronucleus frequency. Environ Mol Mutagen 40: 110-115. [Crossref]
- Alkhalaf M, Al-Bustan S, Hamoda H, Abdella N (2007) Polymorphism of p53 gene codon 72 in Kuwaiti with coronary artery disease and diabetes. Int J Cardiol 115: 1-6. [Crossref]
- Smith MA, Silva MD, Cendoroglo MS, Ramos LR, Araujo LM, et al. (2007) TP53 codon 72 polymorphism as a risk factor for cardiovascular disease in a Brazilian population. Braz J Med Biol Res 40: 1465-1472.
- Gloria-Bottini F, Saccucci P, Magrini A, Bottini E, Banci M, et al. (2012) p53 codon 72 polymorphism and coronary artery disease: evidence of association with left ventricular ejection fraction. Am J Med Sci 343: 127-130. [Crossref]
- Bonafè M, Olivieri F, Mari D, Baggio G, Mattace R, et al. (1999) P53 codon 72 polymorphism and longevity: additional data on centenarians from continental Italy and Sardinia. Am J Hum Genet 65: 1782-1785. [Crossref]
- Caamaño JL, Saavedra N, Jaramillo PC, Lanas C, Lanas F, et al. (2009) Polimorfismo Pro72Arg del gen TP53 se asocia a enfermedad coronaria en individuos chilenos. Rev chil cardiol 28: 151-157.
- Kojima S, Goto Y, Nonogi H, Horie H, Kinoshita M, et al. (2000) Role of a p53 polymorphism in luminal narrowing after balloon coronary angioplasty. Atherosclerosis 151: 585–586.