In the past few decades, the emergence of novel information from rigorous clinical trials and observational studies has reset the priorities regarding the management of type 2 diabetes [T2D]. Most recent results have focused on therapeutic strategies that, while promoting adequate glycemic control, are also capable of reducing cardiovascular [CV] and renal morbidity and mortality. Consequently, glucose-specific microvascular complications as well as macro-vascular diseases are prevented/minimized and, the quality of life of patients with T2D improves significantly. One important aspect of diabetes treatment, however, which has not been sufficiently emphasized, is the fact that in order to acquire long-term meaningful benefits, early and aggressive initiation with multi-drug therapy is necessary.
In this review article, we propose a sound and practical approach to the management of T2D patients based on data extracted from various studies addressing therapy with anti-hyperglycemic agents that also affect CV and renal outcome. To enhance the chances of risk reduction, drug treatment must be started early in ‘pre-diabetes’ subjects and with the addition of multiple medications. Therapeutic choices are guided by disease progression and the risk for developing CV/renal diseases over time, while keeping glycemic parameters at targeted goals. The primary goals of aggressive therapy are to achieve/maintain near-normoglycemia concomitant with preservation of beta cell function and improvement in insulin resistance. A feasible and reasonable diet with calories/nutrient manipulations that induce sustainable weight loss and fat re-distribution should be implemented together with agents that correct the underlying metabolic and hormonal defects in diabetes. Dietary adjustments, regular physical activity and early treatment with anti-diabetic drugs that provide CV/renal benefits represent the mainstay of this new management plan.
The events that lead to the appearance and progression of type 2 diabetes [T2D] have been well-described [1-3]. We have learned that most individuals at risk for developing T2D are in a metabolic state of insulin resistance. There is a limited response to the biological action of insulin in various organs and tissues. This condition is partially inherited and tends to worsen with exposure to the environment. Subsequently, the demand on beta cells for the secretion of the insulin is increased. The resultant hyperinsulinemia is able to, initially, overcome the tissue resistance to the action of insulin and, normoglycemia is temporarily maintained. Continued contact with adverse environmental factors further accentuates the degree of tissue insulin resistance, which eventually reaches a maximum. Following a persistent stimulation, the pancreatic hypersecretion of insulin begins to recede and hyperglycemia ensues. The “timing” and the “rate” of decline in beta-cell function depends upon several elements [1,2]. Regardless, a state of relative insulin deficiency emerges and the disease advances. Combined with extreme tissue insulin resistance and the presence of hyperglycemia, there is a rapid deterioration in the beta-cell secretory capacity. Postprandial first followed by fasting hyperglycemia become apparent and, both of which meet the criteria for the diagnosis of diabetes [4]. Therefore, the combination of tissue insulin resistance and beta-cell failure are two fundamental disturbances that lead to the development of T2D.
The state of insulin resistance is inherited in patients with T2D and, some specific cellular and molecular abnormalities that affect the biologic action of insulin in tissues have been identified [5,6]. The responsible genes are known to be linked to some common gene variations, known as single nucleotide polymorphisms [SNPs]. Although the actual genetic defects underlying the risk for T2D have not yet been fully identified. It is believed that susceptible individuals carry genotypes that predispose the development of tissue insulin resistance at the cellular level. Whether there are additional genes that account for defective islet-cell responses to glucose and other stimuli is not entirely clear. In any case, this genetic material has very high penetrance and tends to occur frequently in first-degree relatives. In addition to the genetic imprint, numerous environmental factors, most importantly body weight and fat excess, sedentary lifestyle and lack of exercise, play a critical role and aggravate the inherited resistance to insulin action [7]. It is now recognized that in susceptible individuals, the presence of one or many of these conditions contributes to worsening of the metabolic state of tissue insulin resistance. In the long-term, the invariable outcome is the acceleration of beta-cell failure evolving to complete exhaustion manifested with overt type 2 diabetes [8].
Under normal circumstances, insulin secretion by pancreatic beta cells follows a pattern of continuous release alternating with quick bursts of transient elevations in circulating insulin levels. These oscillations occur both in the portal system and in peripheral blood. They are primarily a function of the changes in glucose concentration in the extra-cellular space, particularly the one surrounding the pancreatic islet cells. In a typical 24-hour circadian rhythm, the magnitude and the rapidity of the changes in glucose concentration after meals are critical elements that determine the rates of insulin secretion [9]. In healthy people without diabetes, the postprandial increments in insulin secretion also are influenced by the release of gastro-intestinal peptides (“Incretins”) that act on pancreatic islet cells and further potentiate the insulin secretory response [10,11]. In T2D patients, on the other hand, increments in the rates of insulin secretion often are not aligned with the postprandial elevations in circulating glucose levels. The insulin secretory rates are considerably diminished and, higher glucose concentrations are necessary to elicit a response [12]. Although not proven, it is conceivable that a simultaneous defect in molecular mechanisms and/or biosynthetic pathways in beta cells is also inherited, which would further affect the pattern of insulin secretion. The fact is that the presence of these two essential disturbances, i.e., tissue insulin resistance and inadequate insulin secretion, represents the phenotypic manifestation of the diabetes genotype [5]. It seems that unfavorable environmental conditions may be necessary for the full development of T2D. Since there is no effective gene therapy yet, all strategies designed to delay the initiation and progression of diabetes rely heavily on modifications of environmental factors. Thus, the primary goals of preventing diabetes are to reduce the degree of tissue insulin resistance and concomitantly improve the beta cell capacity to secrete insulin. Better tissue response to insulin action together with protection of the beta cells can delay the appearance of diabetes and prevent its complications [13-17].
The known factors and conditions that contribute to the development of T2D are summarized in (Figure 1). Obesity, sedentary lifestyle and the accumulation of excess body fat are prevalent among T2D patients and, together with ectopic and visceral fat deposition are major factors in worsening tissue insulin resistance and beta-cell dysfunction [18,19]. Specifically, accumulation of fat in skeletal muscle fibers deteriorates insulin action in muscle tissue, while in the endocrine pancreas it impairs insulin secretion. Elevated circulating fatty acids [lipotoxicity] and chronic hyperglycemia [glucotoxicity] both aggravate tissue insulin resistance and inhibit beta-cell insulin secretion [20,21]. The ability to discharge insulin adequately and proportionately is also affected by derangements in the “entero-pancreatic axis”, i.e., dysregulated release of gut hormones (“incretins”). [11].
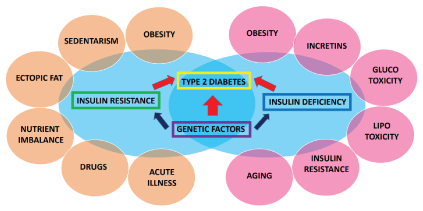
Figure 1. The diagram shows the many conditions that contribute to the development of type 2 diabetes. Individuals at risk carry genotypes responsible for insulin resistance at the molecular level, which result in increased demand for pancreatic insulin hypersecretion. Hyperinsulinemia precedes insulin deficiency and, eventually, complete beta-cell failure develops. Among environmental factors, obesity, sedentarism and ectopic fat deposition are highly prevalent in the population at large and play an important role in both worsening insulin resistance and in accelerating beta cell degradation. Moreover, inadequate release and inappropriate action of gut hormones (“Incretins”) also contribute to limit the ability of the beta cells to secrete insulin. In addition to the functional loss of the endocrine pancreas with aging, the presence of chronic hyperglycemia [“Glucotoxicity”], elevation of circulating fatty acids [“Lipotoxicity”] and tissue insulin resistance itself further impair insulin secretion and beta cell survival. Nutrient imbalance, particularly the over-consumption of simple carbohydrates, “refined sugars”, can potentially hamper sensitivity to insulin action and may affect insulin secretion by beta cells. The use of steroid-based drugs and adrenergic agents transiently inhibit tissue insulin action and, if used repeatedly, can provoke irreversible damage. Likewise, episodes of acute illness, trauma or surgery, by generating an inflammatory response, will further affect cellular action of insulin.
Tissue insulin action can be affected by other factors such as an imbalance in nutrient intake, as for instance the exaggerated consumption of “refined sugars”, i.e., simple carbohydrates. Moreover, frequent use of steroid-based drugs or active adrenergic agents, also can transiently deteriorate tissue response to insulin action and harm the ability to secrete insulin [22-24]. Likewise, episodes of acute illnesses, trauma or surgery, by generating an inflammatory response, can block insulin activity at the cellular level [25,26]. Finally, the loss of islet cell function with aging may be somewhat accelerated because of the accompanying state of insulin resistance [8].
Based on our current knowledge of the natural history of T2D and, with the understanding that most environmental aspects involved in the disease process are either preventable or reversible, a series of interventions that reduce the risk of the disease, its progression and complications have been proposed. First, in order to prevent the initial surge of hyperglycemia, which is the landmark manifestation and diagnostic criteria of T2D, it is necessary to halt the progression of both tissue insulin resistance and the rate of loss of beta-cell function. Secondly, once hyperglycemia is established, in addition to maintaining all other preventive measures, therapies that help mitigate potential deleterious effects due to “glucotoxicity” must be implemented. Chronic hyperglycemia results from the combined inability of insulin to promote glucose active transport and intra-cellular metabolism [insulin resistance]. Uninhibited release of endogenous glucose into the circulation due to the relative insulin deficiency that accompanies beta cell failure further contributes to hyperglycemia. The excess glucose that accumulates in the extra-cellular space penetrates into tissues/organs and, by undergoing alternate metabolic processes, causes oxidative stress that leads to significant end-organ damage. Therefore, the management of T2D patients in the initial phase requires aggressive pharmacotherapy combined with intensive nutritional and lifestyle changes. These are aimed essentially at improving insulin resistance and enhancing insulin secretion in order to restore near-normoglycemia. Suggested lifestyle changes, nutritional and the aforementioned drug therapy must be complementary and, altogether, they should help to facilitate tissue insulin action, stimulate insulin release and promote beta-cell preservation. In other words, protection of the endocrine pancreas by slowing the rapid deterioration of beta cells is a critical component in diabetes prevention and early treatment. In this way, near-normalization of plasma glucose is likely to be more durable and accompanied by a significant delay in the development of retinopathy, nephropathy and neuropathies. Following these general principles, once plasma glucose decreases and, a proportionate reduction in the appearance and progression of glucose-specific micro-vascular complications follows [27-29].
In T2D patients substantial morbidity is associated with micro-vascular and cardio-vascular [CV] complications, even though CV and renal diseases account for the high mortality rates [30,31]. Thus, achieving and maintaining near-normoglycemia are critical steps that mitigate diabetes morbidity in the mid to long-term. In order to decrease mortality rates and substantially improve the quality of life, however, preventive measures with an aggressive plan that addresses CV risks and renal complications is required. Decreasing the chances for developing atherosclerotic cardiovascular diseases [ASCVD], such as coronary heart disease, stroke and peripheral vascular diseases, in addition to congestive heart failure [CHF] and chronic kidney diseases [CKD] must take priority. In fact, therapies designed to halt the progression of macro-vascular complications have now become an essential part of any diabetes management plan [32-39]. Currently, these goals can be accomplished with a multitude of available anti-hyperglycemic pharmacologic agents, used either alone or in combination. All of which have been shown to effectively reduce blood glucose levels while simultaneously providing cardiac and renal protection [34-39]. Considering that, the majority of patients with T2D are likely to experience some form of cardiac or renal complication late in the course of the disease, treatment with drugs that decelerate the processes of ASCVD, CHF and CKD are clearly indicate early on to reduce clinical events and mortality. Hence, in addition to addressing hormonal and metabolic disturbances characteristic of T2D patients, a strategic plan that covers hemodynamic abnormalities, reverses the pro-inflammatory state and neutralizes the atherogenic factors is indispensable. Obviously, there are risk factors that co-exist in all patients with macro-vascular diseases that are not exclusive to diabetes. These include dyslipidemias, arterial hypertension, and smoking, among others, which should be dealt with appropriately. The combination of these conditions is referred to as the “cardio-renal metabolic syndrome”, a condition that is common in T2D patients [40-42].
Even though it is recognized that there are marked diversities in the metabolic profile, drug responses, clinical evolution and other aspects of obesity and diabetes among the various ethnic groups, between male and female individuals and, during the different stages of physical development in life, these are not covered in the current review.
Obesity and body fat excess are the most obvious targets when designing a nutritional program aimed at the prevention and delaying the progression of diabetes. Body weight and fat reduction, particularly the elimination and/or redistribution of visceral ectopic fat, are the cornerstone of the nutritional therapy that is known to retard the appearance of metabolic disturbances, including hyperglycemia [13-15]. Regardless of the source of calorie restriction or nutrient manipulation in the diet, it has been repeatedly shown that achieving ~5% of body weight loss is accompanied by a significant decrease in the conversion rate from pre-diabetes to diabetes [13,14]. Based on several clinical studies, specific recommendations have been proposed to counsel obese subjects. These are primarily aimed at delaying the development of overt diabetes and its complications, in addition to reducing many risks associated with body fat excess accumulation. Lifestyle changes and dietary adjustments tend to be more efficient when implemented together with regular physical activity. Exercise repetition must include aerobic (“cardiac fitness”) alternating with anaerobic (“muscle building”) activities for at least 30-45 minutes, 3 to 5 times every week.
It must be emphasized, however, that most clinical trials have included a predominant number of white Caucasian male and female subjects and, thus, results may not apply equally to all ethnic groups. On the other hand, the few studies addressing ethnic and gender inequalities, in general, tend to support these principles in lifestyle modifications and nutritional recommendations for all. This is not true for people at different stages of physical development, including young adolescents, pregnant/lactating women, elderly individuals and others.
Dietary restriction of ~500 calories per day, i.e., a reduction in either carbohydrate, fat intake or a combination of both usually represents a good start. An effort to limit the consumption of simple carbohydrates (“refined sugars”), with less saturated animal fat (red meat and dairy products) and no added sodium, i.e., salt intake, are also important steps. These guidelines encourage the consumption of natural unprocessed foods, such as fruits, vegetables and fiber-rich products containing complex carbohydrates. Vegetable oils, particularly olive oil, should substitute for animal fat and, people must give preference to the consumption of nuts, an excellent source of vegetable oil. The main objective of lifestyle changes and dietary modifications are to establish healthier body composition. Consequently, there will be adequate metabolic balance and cardiovascular fitness. Once a reduction of ~5% in body weight is attained, there is shrinkage in total body fat excess, especially the fat deposited in ectopic areas such as in the muscle and liver. The latter is sufficient to alleviate some of the tissue insulin resistance and enable restoration closer to normal hepatic glucose and fat metabolism. Of greater importance, perhaps, because of the weight loss, the demand on pancreatic insulin secretion diminishes. If these outlined lifestyle and nutritional modifications can be safely implemented and adhered to for a long period, there is solid evidence that the rate of conversion from pre-diabetes to diabetes is reduced and the time for the development of complications is extended [43].
When overt hyperglycemia arises and one converts from the state of pre-diabetes to diabetes, a greater emphasis on the reduction in the total daily consumption of carbohydrates, especially “refined sugar”, to a maximum of 50 grams/day (200 calories/day) becomes necessary. The intake of saturated fat of animal origin no greater than 7% of the total daily calories must be enforced. The avoidance of simple sugars in the diet can be compensated by a higher ingestion of fresh fruits, vegetables, nuts and vegetable oil [44]. Implementation of the Mediterranean or DASH diets may help to “package” these changes for most patients. In addition to promoting body weight/fat loss, these nutrient manipulations induce sustained decreases in plasma glucose and circulating lipids in T2D patients [45]. Despite the fact that dietary adjustments and lifestyle changes are critical in all comprehensive diabetes management plans, it should be recognized that adequate glycemic control is inevitably lost. Most patients will require adjunct drug therapy to further delay the progression of the disease and early drug therapy may have its advantages.
The best strategy to delay the development and progression of diabetes and thus, minimize the appearance of serious complications is to complement lifestyle and nutritional interventions with anti-hyperglycemic agents. Early treatment of high-risk subjects diagnosed with pre-diabetes (also considered “prevention” of diabetes), plays a crucial role in an effort to reduce morbidity and mortality related to T2D. In addition to the lifestyle changes and nutritional manipulations, the drug metformin, of the class biguanides, is the preferred choice as the initial pharmacological agent in the treatment of pre-diabetes and recently diagnosed diabetes. Metformin is a time-honored, extensively studied medication that has been demonstrated to significantly decrease the rate of conversion from pre-diabetes to diabetes [46,47]. It is usually started at the dose of 500-1000 mg once daily prior to the morning meal to eventually reach a maximum effective dose of 1000 mg twice daily. The primary action of metformin is to inhibit the rate of endogenous, essentially hepatic glucose production. As a result, in the fasting state, but also in the postprandial period, abnormally elevated rates of endogenous glucose production are lowered. This leads to a decrease in fasting glucose concentration and attenuates post-meal hyperglycemic excursions [46]. Most pre-diabetic subjects will have fasting plasma glucose concentration between 100-125 mg/dl, hemoglobin A1C values between 5.8% and 6.5% and/or 2-hour post-oral glucose tolerance test [standard challenge with 75 grams of dextrose] between 140-199 mg/dl [48]. Furthermore, metformin is weight-neutral and sometimes, it can induce weight loss but does not usually induce body weight gain. Following the addition of metformin, periodic [for example, every 3-6 months] re-assessment of clinical status, adherence to dietary regimens, medication compliance, reports of adverse effects, etc. should be implemented. Changes in plasma glucose and/or hemoglobin A1C should be closely monitored and, the dose of metformin must be adjusted upwards, as tolerated. Metformin can be associated with diarrhea or gastrointestinal upset and, it is contra-indicated in individuals with glomerular filtration rate below 30 ml/min, because the risk of lactic acidosis becomes high. For the same reason, patients should discontinue the use of metformin a few days prior to elective surgery/procedures and immediately upon hospitalization. Metformin can be re-started when the patient is stable and adequate renal function is ascertained, usually at discharge from the hospital or on the day after the procedure. In the outpatient management, titration follows a good “rule-of-thumb” that calls for an increase the metformin dose when the average fasting plasma glucose and/or the hemoglobin A1C rise. In case these values remain stable or decrease, no dose modifications are needed. Metformin total daily dose above 2000 mg does not lead to enhanced glucose lowering and may carry a greater risk of adverse effects. If patients develop sustained hyperglycemia and the maximum tolerated dose of metformin has been achieved, the next best option is to add an insulin sensitizer as a second agent, such as pioglitazone.
The anti-diabetic class of insulin sensitizers, the PPAR-gamma agonists, also known as thiazolidinediones [TZDs] were introduced as an alternative to treat patients with T2D a few decades ago [49,50]. Several TZDs have been tested but today and, pioglitazone is the preferred TZD in the U.S. marketplace. These drugs bind to the peroxisome proliferator-activated receptor gamma that is located in the nucleus of the target cells. The binding, in turn, activates a series of molecular events that lead to the enhancement of transmembrane glucose transport and intra-cellular metabolism. This effect is most noticeable in insulin-dependent tissues, such as the skeletal muscle and the liver, although it has direct effects on adipose tissue biology [51]. A few weeks after pioglitazone intake, there is an increase in insulin-mediated glucose utilization in peripheral tissues simultaneous with inhibition of endogenous glucose production. Consequently, plasma glucose concentration decreases and hemoglobin A1C levels fall by approximately 0.5-1.0% [16,52]. One interesting additional action of pioglitazone is the mobilization of ectopic pro-inflammatory, mostly visceral, adipose tissue, to the subcutaneous fat. This is accomplished by stimulating adipocyte differentiation and formation of new adipose depot, located in eutopic areas. Moreover, atherogenic lipids particles in plasma tend to decrease and, there is a modest increase in circulating adiponectin, an anti-inflammatory adipocytokine [53,54]. Because of the decline in leptin levels, in conjunction with a possible surge in some orexigenic peptides, patients using pioglitazone tend to have an increase in appetite. Mild fluid retention often develops, which contributes to weight gain during treatment with pioglitazone. The initial recommended dose is of 15 mg once daily and, after 4-6 weeks, can be increased to 30 mg daily dose. The maximum dose of pioglitazone 45 mg daily is rarely prescribed, presumably because of the small increase in the overall anti-hyperglycemic benefit that is associated with a higher frequency of adverse effects, such as edema and weight gain. In patients with ventricular dysfunction who are at risk for pulmonary edema and, in those with more advanced congestive heart failure, pioglitazone is contra-indicated [52]. Of interest, however, several clinical trials [55-57] and observational studies [58,59] have shown clinically significant anti-atherosclerotic effects of pioglitazone. These include carotid and coronary artery atheromatous plaque regression [60,61] and improved CV outcomes [55-58], noticed even in non-diabetic individuals [59]. Some of these beneficial effects are likely indirect, related perhaps, to the reduction in CV risk factors and to the release of the anti-inflammatory cytokine adiponectin. The possibility that pioglitazone may also have direct cardio-protective effects is suggested by some in vitro experiments indicating that it interferes with a hyperactive insulin mitogenic signaling in coronary vascular smooth muscle cells [62,63]. It has been speculated that by attenuating the arterial smooth muscle cell migration and proliferation, vascular wall plaque formation and growth can be retarded. The use of pioglitazone is safe in patients with reduced glomerular filtration rates, even those in dialysis treatment. This is important because patients with end-stage renal disease cannot use metformin. Pioglitazone is also a good choice for the treatment of pre-diabetes, as it has been demonstrated to be very effective in preventing the conversion to diabetes [17].
The same principles that guide the dose titration of metformin also apply to the use of pioglitazone. Therefore, if the average fasting plasma glucose and/or the hemoglobin A1C values rise, the initial dose of 15 mg of pioglitazone should be increased to 30 mg, daily. An individual with pre-diabetes may then end up in dual therapy, i.e. metformin and pioglitazone. Keep in mind that, even though the diagnosis of diabetes has not yet been established, this regimen represents an early and aggressive form of therapy. In these circumstances, plasma glucose concentration and hemoglobin A1C values may well still be below 126 mg/dl and 6.5%, respectively. In those rare, exceptional patients in whom neither metformin nor pioglitazone can be utilized for some reasons outline above, the choice of different drugs can be entertained, which we will address later. The theoretical basis for the use of these two anti-diabetic agents together rests on their reported ability to reduce the conversion from the state of pre-diabetes to diabetes and, the fact that they are effective in halting the progression of the disease [14,17]. Via different and additive mechanisms, metformin and pioglitazone both decrease the rate of endogenous glucose production, which is primarily a manifestation of hepatic insulin resistance. In addition, pioglitazone improves insulin resistance in peripheral tissues and, as a result, decreases the demand for insulin secretion, thus preserving beta-cell function [16,17]. Of note, if these agents are started too late in the disease process, the opportunity to gain this beneficial action might be lost. For instance, tissue insulin resistance may have already reached a very high level and the beta-cell reserve may be near-exhaustion, even at the point of irreversibility [64]. Based on the natural history of the disease and following these management principles, by the time the diagnosis of diabetes is confirmed, all subjects should be already receiving adequate nutritional therapy with maximally tolerated doses of metformin and pioglitazone in combination.
It is worth mentioning that, although sulfonylureas [SU] are a common first, sometimes second therapeutic choice in the treatment of recently diagnosed T2D patients at large, these agents are not listed in most current recommendations. SU are a well-known class of anti-hyperglycemic insulin secretagogues commonly used early in the management of diabetes, especially at a time when other oral agents were not available. All SU, short and long acting, stimulate the secretion of insulin after binding to specific receptors in the beta-cell membrane. In response, insulin is released into the portal circulation, the magnitude of which is solely dependent upon the dose of SU taken. The anti-hyperglycemic effect following a dose of SU is completely independent of changes in plasma glucose levels and of incretins [65,66]. The activation of SU receptors triggers the exocytosis of insulin molecules from pancreatic beta cells and circumvents the normal required cellular pathways that involve glucose uptake/oxidation [67]. Thus, the risk of hypoglycemia is high, as there is frequently a transient hyperinsulinemia. There is also a tendency for body weight gain and fat accumulation due to the chronic hyperinsulinemia generated with long-term use of SU. There have been no studies indicating that SU therapy is associated with preservation of beta-cell function over time, whereas some have suggested that it may actually accelerate its destruction [65-67]. To date, no clinical studies have demonstrated prolonged, sustained anti-hyperglycemic effects extending beyond 1-2 years, with the use of SU in patients with T2D. The short durability of the SU anti-hyperglycemic action represents further evidence that the insulin secretory capacity from beta cells is not protected, which represents an additional important negative aspect of the SU in the treatment of diabetes [67]. In other words, once started, all SU agents are accompanied by a glucose lowering effect that is short-lived, although suitable in the beginning. Furthermore, whether or not SU have deleterious CV effects remains in question, whereas clear-cut improvements in CV outcomes have never been reported [68]. In the past few decades, numerous alternate therapeutic agents for patients with T2D have emerged, rendering the choice of SU rather obsolete. In general, patients treated with SU, who are enjoying adequate glycemic control need not to make changes concerning glycemic control. On the other hand, inclusion of anti-diabetic agents with known cardio-renal protective effects is now strongly recommended.
In patients who are at high risk and in those who have documented CV and/or renal disease, despite adequate glycemic control, the addition of a Glucagon-Like Peptide 1 – Receptor Agonist [GLP-1RA] or a Sodium-Glucose Co-Transporter-2 [SGLT-2] inhibitor must be considered at this stage. Either is obviously also necessary in those subjects who are receiving maximum tolerated doses of metformin and pioglitazone in combination and demonstrate deterioration in glycemic control. It is always a good practice to review aspects of lifestyle changes, regular exercises plans and nutritional therapy prior to advancing to triple-therapy. GLP-1RA and SGLT-2 inhibitors are drugs that are able to correct hyperglycemia and concomitantly delay the progression of the disease. These agents act through different and independent mechanisms to lower plasma glucose and to preserve the beta-cell secretory capacity. Both agents are capable of reducing body weight/fat by diverse actions that can create negative energy balance [69,70]. Triple-therapy combination certainly delays and, in some cases actually prevents, microvascular complications, such as retinopathy, nephropathy and neuropathies [27-29]. Moreover, in the case of GLP-1RA agents, near-normalization of blood glucose levels is often associated with preservation of the insulin secretory capacity, maybe even with some restoration of the beta-cell function [71-73]. The same also occurs with the use of SGLT-2i agents, although to a lesser extent [74]. The GLP-1RA agents tend to slow gastric emptying and create an immediate sensation of fullness, translated into lack of appetite. In the mid- to long-term, hunger can be further suppressed mediated by the action on appetite and satiety centers in the hypothalamus. Therefore, as blood glucose falls over time with the use of GLP-1 RA, there is also consistent body weight loss [69]. SGLT-2i agents promote the loss of glucose in the urine, thus, producing a negative calorie balance. As a result, in the absence of a compensatory increase in caloric intake, a reduction in body weight ensues [70]. Considering that agents in both classes offer comparable benefits regarding glycemic control and body weight reduction, albeit by different means, the decision to choose a GLP-1RA or SGLT-2 inhibitors depends essentially on individual clinical conditions and risks, medication tolerance and affordability.
There is a variety of choices in the class of GLP-1-RA and most have shown equivalent efficacy and safety profiles in patients with T2D. Exenatide as single injections given twice, prior to the morning and evening meals, was the first GLP-1 RA agent introduced. It is a synthetic derivative of a chemical found in the Hela monster [“desert lizard”] saliva with strong insulinotropic and glucagonostatic properties [75]. At the initial dose of 5 micrograms, which can be increased to a subcutaneous dose injection of 10 micrograms, twice daily, exenatide can reach pharmacological levels capable of stimulating both the secretion of insulin and the inhibition of glucagon release in humans [76,77]. It also leads to slowing of the gastric emptying, which produces a sensation of early satiety. There is sufficient drug penetration of the drug into the central nervous system to bind hypothalamic receptors to induce appetite suppression. Nausea and vomiting occur in up to 40%, initially, but tends to wane over time. There have been rare cases of acute pancreatitis during the use of exenatide and thus, it should be used with caution in patients at risk. Most GLP-1RA agents are contra-indicated in patients with personal or family history of thyroid medullary cancer. Exenatide treatment is followed by a significant decrease in postprandial plasma glucose, which reflects in an average decline in HbA1C of 1.0-1.2% after 6-12 months of therapy. There is usually a concomitant body weight loss in the range of 4-6 kilograms. Liraglutide administered at the dose of 0.6, 1.2, up to 1.6 mg once daily produces similar blood glucose lowering and body weight reduction with a convenient once daily subcutaneous injection. The same gastro-intestinal adverse events and contra-indications reported with exenatide are expected. Either one of these GLP-1 RA agents can be safely added to metformin and pioglitazone to configure a “triple-therapy”. The latter regimen was shown to be safe and highly effective in the initial management of T2D patients [78].
Long-acting GLP-1 RA formulations have been introduced more recently and, tend to be more effective for glycemic control compared to the short-form counter-parts. In addition to the reduction in postprandial plasma glucose, long-acting GLP-1 RA agents may also induce lowering in fasting glucose. Although, there is no clear-cut explanation for the effect on morning blood glucose levels, this has been consistently observed. Exenatide 2 mg given once weekly is associated with an average decrease in HbA1C of up to ~2.0% and with weight reduction in the range of 4-6 lbs. over a 6-month period [77]. A mean decline in HbA1C of ~0.9% has also been documented with dulaglutide, starting at 0.75 and up to 1.5 mg weekly injections. Similarly, there is a body weight decrease of approximately 3-4 lbs. over 6 months. A most recent addition to the long-acting class of GLP-1 RA is semaglutide. Weekly injections at the dose of 0.25, 0.5 and up to 1.0 mg are reported to induce a fall in HbA1C in the range of 1.0-1.5% and weight reduction up to 6-8 lbs. after 6 months of therapy [79]. An oral version of semaglutide is now commercially available, which must be given daily at a starting dose of 3 mg for the first month, ten may be titrated up to the maintenance doses of 7 and 14 mg daily. These doses have been reported to significantly improve glycemic control and reduce body weight [80]. Higher doses than with subcutaneous injection are required because gastro-intestinal absorption is limited.
Most adverse effects of the long-acting and oral GLP-1 RA agents are very similar, albeit milder and less frequent than the short-acting preparations. These include nausea, vomiting and diarrhea and similarly, they are contra-indicated in patients with medullary thyroid cancer and recurrent pancreatitis. There is ample evidence that all GLP-1 RA agents are safe when used in combination with a variety of other drugs in the treatment of T2D [81]. In practice, the preference of patients for the use of either short or long-acting GLP-1 RA, injectable or oral, is personal. This decision is essentially dependent upon cost and convenience, unless the patient has or is at high risk of CVD or renal insufficiency, in which case only longer acting GLP-1 RA’s have reported CV benefit. At times, the substitution of one drug for another in the same GLP-1 RA class may have transient benefits, but long-term data are lacking. (Table 1) summarizes clinical pharmacological and adverse effects, as well as contra-indications of GLP-1 RA agents and SGLT-2 inhibitors currently available in the US market.
Table 1. GLP-1RA and SGLT-2 Inhibitor Agents in the Treatment of Type 2 Diabetes
GLP-1RA = glucagon-like peptide-1 receptor agonist; SGLT-2 = sodium-glucose co-transporter-2; exenatide twice daily; liraglutide once daily, semaglutide oral daily; DKA = diabetes keto-acidosis; LDL = low density lipoprotein cholesterol.
* = major CV events include non-fatal myocardial infarction, stroke, CV mortality and heart failure in patients with diabetes; ** = major CV events include non-fatal myocardial infarction, stroke, CV mortality and heart failure in patients with diabetes and non-diabetes (dapagliflozin) *** = renal benefits include reduction in proteinuria, slowing the progression to end-stage renal disease, requiring dialysis/renal transplantation and less renal-related deaths.
It is worth mentioning that, despite the enthusiasm among primary care providers treatment of diabetes that includes Dipeptidyl-Peptidase-4 [DPP-4] inhibitor agents has not shown to have a persistent and robust anti-hyperglycemic effect. It is especially disappointing, that these drugs do not usually attain a reasonable degree of attenuation in postprandial hyperglycemia. This is possibly related to the fact that simple inhibition of the endogenously derived DPP-4 enzyme activity is not potent enough to generate measurable pharmacological and clinical actions.
There is general agreement that the total amount of DPP-4 enzyme present in the microcirculation in individuals who develop T2D is severely diminished. Thus, preserving its activity, even if for a long period, is not capable of enhancing insulin secretion and suppressing the release of glucagon by the endocrine pancreas. As a result, there is little impact on chronic glycemic control in the majority of patients. Although in some cases transient improvements in post-meal glycemic elevations has been reported, this is not sustained in mid to long-term therapy [82]. Furthermore, the utilization of DPP-4 inhibitors combined with GLP-1 RA agents appears futile. Since potentiation of beta cell insulin secretion with simultaneous suppression of glucagon release from the alpha-cells induced by GLP-1-RA is overwhelming, similar action by DPP-4 inhibitors on islet-cells becomes mute. Whether there are additional, unrelated benefits derived from the inhibition of the endogenous DPP-4 enzyme activity in such as combination regimen are yet to be proven. The use of DPP-4 inhibitors is further discouraged because large clinical trials have not demonstrated clear-cut reductions in cardio-vascular events [82,83] and, actually, CV outcomes trial using sitagliptin and alogliptin reported higher risk of hospitalization for heart failure [83]. Therefore, in those patients with T2D who are under current treatment with DPP-4 inhibitors, the most appropriate recommendation is to maintain a close follow-up and always to re-evaluate whether substitution for other anti-diabetic agents might be worthwhile.
The addition of SGLT-2 inhibitors as an alternate third choice to treat individuals with pre-diabetes, whose response to the therapeutic combination of metformin and pioglitazone is poor, has recently gained some traction [84]. SGLT-2 inhibitors are capable of inducing a decrease in plasma glucose by blocking the renal tubular reabsorption of glucose and producing glycosuria. It seems intuitive that with higher plasma glucose concentrations greater glucose lowering effects are expected. This derives from the fact that the renal glucose load dictates the total amount of urinary glucose excretion in face of SGLT-2 inhibition. Even in normoglycemic conditions glucosuria has been shown, despite minimal decreases in plasma glucose levels. The latter is primarily due to the compensatory elevation in endogenous glucose production, which is proportional to the rate of glucose excretion in the urine [70,85]. Thus, hypoglycemia is avoided and rarely occurs, except when sulfonylureas or insulin is added to the regimen. Moreover, because of the negative energy balance secondary to the urinary loss of ~200-400 calories daily in the form of glucose, SGLT-2 inhibitors often promote body weight loss. As a rule, patients taking SGLT-2 inhibitors are unable to match the loss of calories in the urine with equal intake. Careful analyses have concluded that, initially, there is no sufficient compensatory increase in appetite or any specific desire for carbohydrate-rich food intake. There are occasional reports of individuals on SGLT-2 inhibitors who specifically seek carbohydrate consumption, the so-called “sugar-cravings”. This has been observed following a few months on the drug and may help to explain why the rate of loss in body weight declines [70]. In most cases, however, the initial decrease in body weight tends to reach a nadir and stabilize over 3-6 months. Surprisingly, there is no evidence that the body weight returns to baseline values, even after long-term therapy [85]. Therefore, the addition of SGLT-2 inhibitors to triple-therapy regimens provides glucose lowering combined with body weight reduction safely. This triple-therapy regimen benefits individuals with pre-diabetes and those with recent onset diabetes. It reduces glucotoxicity, which also enables improvements both in insulin secretion and in tissue sensitivity to the action of insulin and, better CV and renal outcomes have been reported [85]. In summary, early aggressive triple-therapy regimens have been associated with superior, sustained glycemic control and a delay in the appearance of diabetes complications.
There is good evidence that triple-therapy combination with metformin, pioglitazone and GLP-1 RA slows the progression of diabetes and reduces micro-vascular complications [78]. Although not extensively evaluated, the inclusion of SGLT-2 inhibitors, instead of a GLP-1 RA, in a triple-therapy regimen, is anticipated to provide similar, or even better, outcomes. Currently, there is a tendency to implement these three (or four) agents sequentially, primarily based on elevations in HbA1C. These drugs are also added when sustained increments in fasting plasma glucose concentration occur. This aggressive multi-drug regimen is often recommended to maintain near-normoglycemia, prevent progressive loss of beta-cell function and decrease the risk for CV events [78,84,85]. Based on the abundant evidence published more recently, strategies to introduce three or four agents together in patients with documented CV and/or renal disease, independent of glycemic parameters, has gained some traction [86]. It is worth noting that early treatment with multiple agents replaces the traditional practice of “treat-to-failure”, a term frequently used in insulin therapy regimens. Essentially, anti-hyperglycemic medications are only added when glycemic control is already lost. We concur with the proposal that, although, an important goal of therapy, there is no need to wait for increments in plasma glucose or HbA1C above target levels to initiate multiple drug therapy. This novel approach, referred to as “treat-to-target”, is primarily aimed at mitigating deleterious effects of hyperglycemia, while simultaneously preventing CV and renal complications, which are responsible for substantial morbidity and mortality in diabetes. Following this general principle, the addition of SGLT-2 inhibitors in combination with GLP-1 RA on the background of metformin plus pioglitazone, has become a most effective therapy choice for T2D [87,88].
Numerous studies have shown that in patients with T2D combining SGLT-2 inhibitors with GLP-1 RA agents provides additive beneficial effects regarding in glycemic control, weight loss and other metabolic and clinical parameters [89-91]. There is convincing evidence to support the utilization of GLP-1RA and SGLT-2 inhibitors in T2D patients, particularly in those at risk or with known CV disease and/or mild-to-moderate chronic renal insufficiency [84-87]. In fact, some SGLT-2 inhibitors are effective agents in primary prevention of CV disease with reduced mortality [84,86]. Thus, a new revised algorithm has been suggested for the management of T2D to maintain adequate glycemic control and simultaneously protect against CV and renal disease, as summarized in (Figure 2).
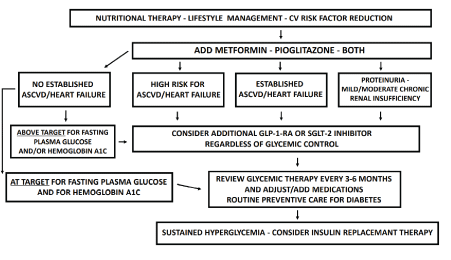
Figure 2. Novel Treatment Approach for Pre-Diabetes and Type 2 Diabetes Patients.
New recommendations for the treatment of pre-diabetes and type 2 diabetes [T2D] extends beyond glucose lowering. These are based on recent introduction of anti-hyperglycemic agents that can protect insulin secretory capacity with demonstrated cardiovascular [CV] and renal benefits. Subjects at risk for developing T2D, for instance, obese, sedentary individuals with positive family history of type 2 diabetes in first-degree relatives qualify for initial implementation of nutritional therapy, lifestyle management and CV risk factor reduction strategies. Thereafter, clinical and laboratory data are gathered and subjects characterized as pre-diabetes are started on metformin (500 mg twice daily) or pioglitazone 15mg/day. Within 3-6 months, the metformin or pioglitazone dose must be doubled and, a thorough re-assessment of the nutritional goals and the adherence to lifestyle modifications must be undertaken. Recommendations should be personalized and guided by competent professionals in each area of expertise. Patients with no risks for atherosclerotic cardiovascular diseases [ASCVD] or heart failure and, who are within target for fasting plasma glucose [i.e., 70-110 mg/dl] and HbA1C ≤ 6.5%, should follow up every 3- 6 months. The overall risks are then reviewed and routine preventive care for diabetes is provided. In contrast, those who are at high risk or have established CV disease and, those who have mild-to-moderate renal insufficiency i.e., estimated GFR ≤ 60 ml/min.1.73m2 with proteinuria, must be started on GLP-1 RA or SGLT-2 inhibitor therapy. The latter is also suggested for patients who are above target for glycemic indices, although they might not have evidence of CV or renal disease. The choice of the GLP-1-RA or SGLT-2 inhibitor depends upon the overall clinical characteristics, drug tolerance and affordability for each patient. At this stage, all that meet the criteria outlined above should be given full dose triple therapy with metformin, pioglitazone and either a GLP-1-RA or a SGLT-2 inhibitor. Many other alternative anti-diabetic agents might be considered, but when hyperglycemia is sustained, insulin replacement therapy must be started. At this stage, basal insulin therapy is initiated, as a single daily dose of a long-acting preparation. Pre-prandial doses of short- or rapid-acting insulin formulae should follow. All anti-diabetic drugs, oral and injectable, except for sulfonylureas should be maintained.
Based on rigorous clinical trials, it is now imperative to offer treatment with one or more of these agents to all T2D. Nonetheless, despite all efforts to attain near-normoglycemia using available anti-diabetic agents, loss of glycemic control eventually occurs. According to the natural history of the disease, the fact that multi-drug therapy fails to sustain glycemic targets is an indication that patients have entered into a later stage of the disease. This signal near-complete exhaustion of the beta-cell insulin reserve and, replacement therapy with the administration of exogenous insulin becomes necessary.
Patients with worsening glycemic control, despite efforts with quadruple-therapy, are candidates for treatment with subcutaneous insulin injections [92]. The first step, once the decision is made, is to start with a single daily dose of a long-acting basal insulin preparation. To calculate the individual starting dose one can utilize the ratio of 0.2 units per kilogram body weight. This initial dose is then adjusted up or down, every 5-7 days. When capillary fasting glucose reads consistently above 120 mg/dl, the insulin dose should be increased by ~10%. However, if morning blood glucose is below 80 mg/dl, the basal insulin dose must be decreased by ~10-20% the next day. As a rule, there is no need to discontinue metformin, pioglitazone, GLP-1 RA or SGLT-2 inhibitors. In patients receiving insulin therapy in combination with other agents, changes in blood glucose levels should be first corrected by adjusting the insulin dose. Unless patients describe or specific adverse events are noticed during the course of a multi-agent therapy, the insulin dose is always the first to be altered. In patients in whom the basal insulin daily requirements are too high, such as above 1.5 to 2.0 units per kilogram body weight, twice-daily injections of a long-acting insulin preparation may be more adequate. In hospitalized patients, frequent injections of either intermediary or short-acting insulin preparations are safer [93]. The same is true for patients with advanced stages of renal insufficiency, whereby insulin dosing every 12 hours reduces the risk of hypoglycemia [94]. Ideally, in the latter patients, the total dose of long-acting insulin must be decreased, if possible, and pre-meal short-acting insulin injections should be dosed with caution. Meal consumption and physical activity in the hospital are often unpredictable, thus the risk of hypoglycemia is enhanced. Poor glycemic control with persistent elevations in hemoglobin A1C over time is a good justification for an evaluation with a continuous subcutaneous glucose monitoring [CGM] device. Results derived from the CGM analysis could help to guide dietary recommendations, lifestyle changes and, determine whether pre-prandial insulin injections are indicated and/or require adjustments.
The addition of short-acting insulin injections should be considered in those patients with exaggerated post-meal glycemic elevations and are unresponsive to dietary manipulations. The administration and dose-adjustment of pre-prandial insulin is independent of the overall diabetes management, including anti-diabetic drugs and basal insulin injections. Using CGM devise, the magnitude of the increase in glucose concentration after the ingestion of specific meals can be analyzed. This can also be undertaken with a series of 2-hour postprandial capillary glucose measurements. A correction with appropriate dose adjustment of the rapid-acting insulin must be sought. Currently, preference is given to the addition of a single pre-prandial injection to the basal insulin after the largest meal of the day. This is referred to as the “basal-plus” insulin regimen, whereby one pre-meal insulin dose is added at a time [95]. Conversely, some prefer the initiation of multiple daily pre-meal insulin injections in an ‘intensive therapy”, also known as the basal-bolus regimen. The “basal-plus” regimen provides more flexibility, is convenient and enables self-adjustment of the insulin doses prior to every meal. If the meal is not consumed, the patient need not to inject rapid-acting insulin. At every meal, patients can supplement the standard dose of rapid-acting insulin, in case pre-meal hyperglycemia is detected. In this way, near-normoglycemia is restored in the postprandial period. While long-acting basal insulin injections are intended to cover the underlying body insulin requirements, rapid-acting insulin administered prior to each meal cover for the hyperglycemic effect of any given meal. There are numerous modalities available for the administration of exogenous insulin [94-97]. In addition to traditional vials, insulin can be injected subcutaneously using pens with accurate and easy-to-handle dosing titration dial buttons. A new inhaled insulin preparation designed only as “prandial insulin” has been used successfully in both in type 1 and 2 diabetes. These inhaled insulin doses must always be utilized in combination with either oral or injectable medications, including long-acting insulin [97]. Rapid or short-acting Insulin can also be administered subcutaneously with the assistance of electro-mechanical pumps, either alone or paired with subcutaneous glucose sensors [94-96]. Insulin pumps can perform with remarkable accuracy and reproducibility and, some are actually disposable. Most common site for the injection of subcutaneous insulin and for the insertions of tubing/sensors is the abdomen. Some patients, including pregnant women, feel more comfortable using the thigh or the upper arms. In all cases, however, despite insulin therapy, diabetes management using all four above-mentioned anti-diabetic agents must be continued. These agents are capable of restoring near-normoglycemia while protecting beta cells and, they can prevent against the CV and renal complications in T2D patients.
It is of paramount importance to highlight that the majority of clinical trials and pharmacologic studies cited in this review do not address specific ethnic disparities and, provide little information regarding drug metabolism and therapeutic responses in individuals who are at different stages of development. Numerous publications are available that provide a better understanding of these critical aspects of pre-diabetes and diabetes treatment.
In summary, based on our current understanding of the natural history of T2D, implementation of an early and aggressive multi-drug treatment to delay the appearance of hyperglycemia and avoid its consequences has become necessary. A sustained glucose-lowering effect indicates better tissue insulin resistance and beta-cell secretory capacity, thus delaying the progression of disease. Furthermore, to decrease the morbidity and mortality in T2D patients, therapy must be aimed at preventing CV and renal complications. The latter can be accomplished with utilization of drugs known to reduce CV and renal outcomes, in addition to appropriate nutritional manipulations and lifestyle changes [98]. As a rule, pre-diabetes subjects must be started on low-carbohydrate, low-calorie diets with an incentive to consume fresh fruits, vegetables, oils, nuts and, to limit intake of animal fat and processed food. Regular physical activity and smoking cessation should also be encouraged. The preferred initial therapeutic agents are metformin and pioglitazone, used either alone or in combination. In combination with measures and agents that help to normalize arterial blood pressure and circulating lipid particles, medications that belong to the GLP- 1RA and SGLT-2 inhibitor classes should be considered early in the management of T2D. Once this multi-drug therapeutic program is established and tolerated, the evidence suggests that T2D patients are enjoying the most effective treatment currently available. Nonetheless, the development of persistent hyperglycemia, despite quadruple-therapy, is an indication that patients have reached an advanced stage of the disease, with near-complete failure of the beta-cell reserve [99]. At this point, basal insulin replacement followed by pre-prandial insulin doses is needed, although all other agents should be adjusted and maintained. This early aggressive multi-drug therapy approach together with an adequate nutritional and exercise plan will provide the best chance for a healthier outcome in T2D patients.
- Eriksson J, Franssila-Kallunki A, Ekstrand A, Saloranta C, Widen E, et al. (1989) Early metabolic defects in persons at increased risk for non-insulin dependent diabetes mellitus. N Engl J Med 321: 337-343. [Crossref]
- Leahy JL (1990) Natural history of beta cell dysfunction in NIDDM. Diabetes Care 13: 992-1010. [Crossref]
- Harris MI, Flegal KM, Cowie CC, Eberhardt MS, Goldstein DE, et al. (1998) Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. Diabetes Care 21: 518-524. [Crossref]
- Standards of Medical Care in Diabetes (2021) Classification and Diagnosis of Diabetes. Diabetes Care 44: S8-S16. [Crossref]
- Dean L, McEntyre J (2004) The Genetic Landscape of Diabetes. Chapter 3, Genetic Factors in Type 2 Diabetes. National Center for Biotechnology Information (US), Bethesda (MD).
- Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, et al. (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38: 320-323. [Crossref]OpenUrl
- Murea M, Ma L, Freedman BI (2012) Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications. Rev Diabet Stud 9: 6-22.[Crossref]
- DeFronzo RA, Bonadonna RC, Ferrannini E (1992) Pathogenesis of NIDDM. A balanced overview. Diabetes Care 15: 318-368. [Crossref]
- Polonsky KS, Given BD, Hirsch L, Shapiro ET, Tillil H, et al. (1998) Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 81: 435-441. [Crossref]
- Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132: 2131-2157. [Crossref]
- Nauck MA, Meier JJ (2018) Incretin hormones: Their role in health and disease. Diabetes Obes Metab 20: 5-21. [Crossref]
- Jones CNO, Pei D, Staris P, Polonsky KS, Chen YD, Reaven GM (1997) Alterations in the glucose-stimulated insulin secretory dose-response curve and in insulin clearance in nondiabetic insulin-resistant individuals. J Clin Endocrinol Metab 82: 1834–1838. [Crossref]
- Tuomilehto J, Lindström J, Eriksson JG, Valle TT, Hämäläinen H, et al. (2001) Prevention of Type 2 Diabetes Mellitus by Changes in Lifestyle among Subjects with Impaired Glucose Tolerance. New Engl J Med 344: 1343-1350. [Crossref]
- Knowler WC, Connor EB, Fowler SE, Hamman RF, Lachin JM, et al. (2002) Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. New Engl J Med 346: 393-403. [Crossref]
- Shai I, Schwarzfuchs D, Henkin Y, Shahar DR, Witkow S, et al. (2008) Weight Loss with a Low-Carbohydrate, Mediterranean or Low-Fat Diet. New Engl J Med 359: 229-241. [Crossref]
- Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, Mari A, et al. (2007) Thiazolidinediones improve beta-cell function in type 2 diabetic patients. Am J Physiol Endocrinol Metab 292: E871-E883. [Crossref]
- DeFronzo RA, Tripathy D, Schwenke DC, Banerji MA, Bray GA, et al. (2011) Pioglitazone for Diabetes Prevention in Impaired Glucose Tolerance. N Engl J Med 364: 12: 1104-1115.
- Rasouli N, Molavi B, Elbein SC, Kern PA (2006) Ectopic fat accumulation and metabolic syndrome. Diabetes Obes Metab 9: 1-10.[Crossref]
- Yki-Jarvinen H (2002) Ectopic fat accumulation: an important cause of insulin resistance in humans. J R Soc Med 95: 39-45. [Crossref]
- McGarry JD, Dobbins RL (1999) Fatty acids, lipotoxicity and insulin secretion. Diabetologia 42: 128-38. [Crossref]
- Kaiser NN, Leibowitz G, Nesher R (2003) Glucotoxicity and beta cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab 16: 5-22. [Crossref]
- Macdonald IA (2016) A review of recent evidence relating to sugars, insulin resistance and diabetes. Eur J Nutr 55: 17-23. [Crossref]
- Hwang JL, Weiss RE (2014) Steroid-induced diabetes: a clinical and molecular approach to understanding and treatment. Diabetes Metab Res Rev 30: 96-102. [Crossref]
- Deibert DC, DeFronzo RA (1980) Epinephrine-induced Insulin Resistance in Man. J Clin Invest 65: 717-721. [Crossref]
- Li L, Messina JL (2009) Acute Insulin Resistance Following Injury. Trends Endocrinol Metab 20: 429-435. [Crossref]
- Cahill, Jr GF (1984) Sir David Cuthbertson lecture: Insulin and trauma: Some thoughts. Clin Nutr 2: 169-172.
- Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, et al. (1993) The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N Engl J Med 329: 977-986. [Crossref]
- Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, et al. (1995) Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 28: 103-117. [Crossref]
- UK Prospective Diabetes Study Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 DM (UKPDS 33): UK Prospective Diabetes Study (UKPDS) Group. Lancet 352: 837-853. [Crossref]
- Garcia MJ, McNamara MP, Gordon T, Kannell WB (1974) Morbidity and Mortality in Diabetics In the Framingham Population: Sixteen-Year Follow-up Study. Diabetes 23: 105-111. [Crossref]
- Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, et al. (2014) Changes in Diabetes-Related Complications in the United States, 1990–2010. N Engl J Med 370: 1514-1523. [Crossref]
- Gerstein HC, Miller ME, Byington RP, Goff DC, Bigger JT, et al. (2008) Effects of intensive glucose lowering in Type 2 Diabetes. N Engl J Med 358: 2545-2559. [Crossref]
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, et al. (2009) Glucose control and vascular complications in veterans with Type 2 Diabetes. N Engl J Med 360: 129-139. [Crossref]
- Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, et al. (2015) Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med 373: 2117-2128. [Crossref]
- Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, et al. (2017) Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med 377: 644-657. [Crossref]
- Marso SP, Daniels GH, Frandsen KB, Kristensen P, Mann JFE, et al (2016) Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 375: 311-322.[Crossref]
- Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, et al. (2019) Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 380: 347-357. [Crossref]
- Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, et al (2019) Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 380: 2295-2306. [Crossref]
- Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. (2019) Dulaglutide and Cardiovascular Outcomes in Type 2 Diabetes (REWIND): a double-blind randomized placebo-controlled trial. Lancet 13: 394: 121-130. [Crossref]
- DeFronzo RA, Ferrannini E (1991) Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 14: 173-194. [Crossref]
- Alexander CM, Landsman PB, Teutsch SM, Haffner SM (2003) NCEP-Defined Metabolic Syndrome, Diabetes, and Prevalence of Coronary Heart Disease Among NHANES III Participants Age 50 Years and Older. Diabetes 52: 1210-1214. [Crossref]
- Grundy SM (2012) Pre-Diabetes, Metabolic Syndrome, and Cardiovascular Risk. J Am Coll Cardiol 59: 635-643. [Crossref]
- Schwarz P, Greaves C, Lindström J, Yates T, Davies MJ (2021) Non-pharmacological interventions for the prevention of type 2 diabetes mellitus. Nat Rev Endocrinol 8: 363-373. [Crossref]
- WHO Guideline (2015) Sugars intake for adults and children. World Health Organization, Geneva, 2021.
- Bazzano LA, Hu T, Reynolds K, Bunol C, Liu Y, et al. (2014) Effects of Low-Carbohydrate and Low-Fat Diets. Ann Intern Med 161: 309-318. [Crossref]
- Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE (1995) Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med 333: 550-554. [Crossref]
- Aroda VR, Ratner RE (2018) Metformin and Type 2 Diabetes Prevention. Diabetes Spectr 31: 336-342. [Crossref]
- Diabetes Care (2015) Standards of Medical Care in Diabetes. J Clin Applied Res Educ 38: S8-S16.
- Johnson MD, Campbell LK, Campbell RK (1998) Troglitazone: review and assessment of its role in the treatment of patients with impaired glucose tolerance and diabetes mellitus. Ann Pharmacother 32: 337-348. [Crossref]
- Bethge H, Haring HU (1998) The thiazolidinediones – a novel approach for the treatment of type 2 diabetes. Arzneim Forsch Drug Res 48: 97-119.
- Greenfield JR (2004) Thiazolidinediones: mechanisms of action. Aust Prescriber 2: 67-70.
- Alam F, Islam A, Mohamed M, Ahmed I, Kamal MA, et al. (2019) Efficacy and Safety of Pioglitazone Monotherapy in Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Sci Rep 9: 5389. [Crossref]
- Spanheimer R, Betteridge DJ, Tan MH, Ferrannini E, Charbonnel B (2009) Long-Term Lipid Effects of Pioglitazone by Baseline Anti-Hyperglycemia Medication Therapy and Statin Use from the PROactive Experience (PROactive 14). Am J Cardiol 104: 234-239. [Crossref]
- Betteridge JD (2007) Effects of pioglitazone on lipid and lipoprotein metabolism. Diabetes Obes Metab 9: 640-647. [Crossref]
- Charbonnel B, Dormandy J, Erdmann E, Massi-Benedetti M, Skene A (2004) The Prospective Pioglitazone Clinical Trial in Macrovascular Events (PROactive). Diabetes Care 27: 1647-1653. [Crossref]
- Wilcox R, Kupfer S, Erdmann E (2008) Effects of pioglitazone on major adverse cardiovascular events in high-risk patients with type 2 diabetes: results from PROspective pioglitAzone Clinical Trial In macro Vascular Events (PROactive 10). Am Heart J 155: 712-717. [Crossref]
- Asakura M, Kim J, Asanuma H, Nakama Y, Tsukahara K, et al. (2018) Cardiovascular Outcomes in Patients with Previous Myocardial Infarction and Mild Diabetes Mellitus Following Treatment with Pioglitazone - Reports of a Randomised Trial from The Japan Working Group for the Assessment Whether Pioglitazone Protects DM Patients Against Re-Infarction (PPAR Study). EclinicalMedicine 4: 10-24. [Crossref]
- Wilcox R, Bousser MG, Betteridge DJ, Schernthaner G, Pirags V, et al. (2007), for the PROactive Investigators. Effects of Pioglitazone in Patients with Type 2 Diabetes with or Without Previous Stroke. Stroke 38: 865-873. [Crossref]
- Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, et al. (2016) Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N Engl J Med 374:1321-1331.Lawrence M. Brass, M.D., is deceased. [Crossref]
- Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, et al. (2006) Effect of Pioglitazone Compared with Glimepiride on Carotid Intima-Media Thickness in Type 2 Diabetes: A Randomized Trial. JAMA 296: 2572-2581.
- Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, et al. (2008) Comparison of pioglitazone vs. glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA 299: 1561-1573. [Crossref]
- Cersosimo E, XiaoJing X, Musi N (2012) Role of insulin signaling in vascular smooth muscle cell migration, proliferation and inflammation. Am J Physiol Cell Physiol: C652-C657.
- Cersosimo E, XiaoJing X, Upala S, Triplitt C, Musi N (2014) Acute Insulin Resistance Stimulates and Insulin Sensitization Attenuates Vascular Smooth Muscle Cell Migration and Proliferation. Physiol Rep 2: e12123. [Crossref]
- DeFronzo RA (2009) From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 58: 773-795. [Crossref]
- Lebovitz HE, Feinglos MN (1983) Mechanism of action of the second-generation sulfonylurea glipizideMechanism of action of second-generation sulfonylurea. Am J Med 75: 46-54. [Crossref]
- Melander A (1987) Clinical pharmacology of sulfonylureas. Metabolism 36: 12-16. [Crossref]
- Proks P, Reimann F, Green N, Gribble F, Ashcroft F (2002) Sulfonylurea Stimulation of Insulin Secretion. Diabetes 51: S368-S376. [Crossref]
- Azoulay L, Suissa S (2017) Sulfonylureas and the Risks of Cardiovascular Events and Death: A Methodological Meta-Regression Analysis of the Observational Studies. Diabetes Care 40: 706-714. [Crossref]
- Hinnen D (2017) Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes. Diabetes Spectr 30: 202-210. [Crossref]
- Cersosimo E (2014) Renal Glucose Handling and the Kidney as a Target for Anti-Diabetic Medication. Curr Tren Endocrinol 7: 81-94.
- Retnakaran R, Kramer CK, Choi H, Swaminathan B, Zinman B (2014) Liraglutide and the Preservation of Pancreatic β-Cell Function in Early Type 2 Diabetes: The LIBRA Trial. Diabetes Care 37: 3270-3278. [Crossref]
- Zummo FP, Kirsty S, Cullen KS, Honkanen-Scott M, Shaw JAM, et al. (2017) Glucagon-Like Peptide 1 Protects Pancreatic β-Cells from Death by Increasing Autophagic Flux and Restoring Lysosomal Function. Diabetes 66: 1272-1285. [Crossref]
- Van Raalte DH, Verchere CB (2017) Improving glycaemic control in type 2 diabetes: Stimulate insulin secretion or provide beta‐cell rest? Diabetes Obes Metabol 19: 1205-1213. [Crossref]
- Asahara S, Ogawa W (2019) SGLT2 inhibitors and protection against pancreatic beta cell failure. Diabetol Int 10: 1-2. [Crossref]
- Furman BL (2012) The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon 59: 464-471.[Crossref]
- Knop FK, Brønden A, Vilsbøll T (2017) Exenatide: pharmacokinetics, clinical use, and future directions. Expert Opin Pharmacother 18: 555-571. [Crossref]
- Vega MM, Muñoz-Garach A, Tinahones FJ (2018) Pharmacokinetic drug evaluation of exenatide for the treatment of type 2 diabetes. Expert Opin Drug Metab Toxicol 14: 207-217. [Crossref]
- Abdul-Ghani MA, Puckett C, Triplitt C, Maggs D, Adams J, et al. (2015) Initial combination therapy with metformin, pioglitazone and exenatide is more effective than sequential add-on therapy in subjects with new-onset diabetes. Results from the Efficacy and Durability of Initial Combination Therapy for T2D (EDICT): a randomized trial. Diabetes Obes Metab 17: 268-275. [Crossref]
- Garber AJ (2011) Long-Acting Glucagon-Like Peptide 1 Receptor Agonists: A Review of Their Efficacy and Tolerability. Diabetes Care 34: S279-S284. [Crossref]
- Pratley R, Amod A, Hoff ST, Kadowaki T, Lingvay I, et al. (2019) Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomized, double blind, phase 3a trial. Lancet 394: 39-50.
- Htike ZZ, Zaccardi F, Papamargaritis D, Webb DR, Khunti K, et al. (2017) Efficacy and safety of glucagon‐like peptide‐1 receptor agonists in type 2 diabetes: A systematic review and mixed‐treatment comparison analysis. Diabetes Obes Metab 19: 524-536. [Crossref]
- Dicker D (2011) DPP-4 Inhibitors: Impact on Glycemic Control and Cardiovascular Risk Factors. Diabetes Care 34: S276-278. [Crossref]
- Toh S, Hampp C, Reichman ME, Graham DJ, Balakrishnan S, et al. (2016) Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other anti-hyperglycemic drugs: a retrospective cohort study. Ann Intern Med 164: 705-714. [Crossref]
- Van Baar MJB, Van Ruiten CC, Muskiet MHA, Van Bloemendaal L, Ijzerman GR, et al. (2018) SGLT2 Inhibitors in Combination Therapy: From Mechanisms to Clinical Considerations in Type 2 Diabetes Management. Diabetes Care 41: 1543-1556. [Crossref]
- Cersosimo E, Miles JM (2019) Hormonal, Metabolic and Hemodynamic Adaptations to Glycosuria in Type 2 Diabetes Patients Treated with Sodium-Glucose Co-Transporter Inhibitors. Curr Diabetes Rev 15: 314-327. [Crossref]
- Kalyani RR (2021) Glucose-Lowering Drugs to Reduce Cardiovascular Risk in Type 2 Diabetes. N Engl J Med 384: 1248-1260. [Crossref]
- Cahn A, Cefalu WT (2016) Clinical considerations for use of initial combination therapy in type 2 diabetes. Diabetes Care 39: S137-S145. [Crossref]
- Phung OJ, Sobieraj DM, Engel SS, Rajpathak SN (2014) Early combination therapy for the treatment of T2DM: systematic review and meta-analysis. Diabetes Obes Metab 16: 410-418. [Crossref]
- Fulcher G, Matthews DR, Perkovic V, de Zeeuw D, Mahaffey KW, et al. (2016) Efficacy and safety of canagliflozin when used in conjunction with incretin-mimetic therapy in patients with type 2 diabetes. Diabetes Obes Metab 18: 82-91. [Crossref]
- Frias JP, Guja C, Hardy E, Ahmed A, Dong F, et al. (2016) Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): a 28 week, multicentre, double-blind, phase 3, randomized controlled trial. Lancet Diabetes Endocrinol 4: 1004-1016. [Crossref]
- Martinez R, Al-Jobori H, Ali AM , Adams J, Abdul-Ghani M, et al.(2018) Endogenous Glucose Production and Hormonal Changes in Response to Canagliflozin and Liraglutide Combination Therapy. Diabetes 67: 1182-1189. [Crossref]
- Aschner P (2020) Insulin Therapy in Type 2 Diabetes. Am J Ther 27: e79-e90.
- Fischer KF, Lees JA, Newman JH (1986) Hypoglycemia in Hospitalized Patients. N Engl J Med 315: 1245-1250. [Crossref]
- Moen MF, Zhan M, Hsu VD, Walker LD, Einhorn LM, et al. (2009) Frequency of Hypoglycemia and Its Significance in Chronic Kidney Disease. Clin J Am Soc Nephrol 4: 1121-1127. [Crossref]
- Rodbard H, Visco V, Andersen H, Hiort L, Shu DW (2014) Treatment intensification with stepwise addition of prandial insulin aspart boluses compared with full basal-bolus therapy (Full Step study): a randomized, treat-to-target clinical trial. Lancet Diabetes Endocrinol 2: 30-37. [Crossref]
- Radermecker RP, Scheen AJ (2004) Continuous subcutaneous insulin infusion with short-acting insulin analogues or human regular insulin: Efficacy, safety, quality of life, and cost-effectiveness. Diabetes Metab Res Rev 20: 178-188. [Crossref]
- Mecklenburg RS, Benson EA, Benson JWJ, Blumenstein BA, Fredlund PN, Guinn TS, et al. (1985) Long-term metabolic control with insulin pump therapy. Report of experience with 127 patients. N Engl J Med 212: 465-468. [Crossref]
- Russell SJ, El-Khatib FH, Sinha M, Magyar KL, McKeon K, et al. (2014) Outpatient glycemic control with a bionic pancreas in type 1 diabetes. N Engl J Med 371: 313-325. [Crossref]
- Akturk HK, Snell-Bergeon JK, Rewers A, Klaff LJ, Bode BW, Peters AL, et al (2018) Improved postprandial glucose with inhaled technosphere insulin compared with insulin aspart in patients with type 1 diabetes on multiple daily injections: the STAT study. Diabetes Technol Ther 20: 639-645. [Crossref]