Abstract
The effects of stress on the atherosclerosis are complex. Here we discuss how the chronic complex stress (CCS), when combined with an atherogenic diet (AD), leads to the development of atherosclerosis in mice. The CCS mouse model consisted of physical and psychosocial stressors of different types and intensities presented in a random order. Eighty-seven mice were divided into a standard chow diet (n=44) and AD (n=43) groups and bred for 4 months. Both groups were subdivided into groups without and with CCS treatment. The CCS was performed during the last month of the study. Ultrasound bio microscopy, histopathological and fluorescence immunohistochemical examinations, ELISA, PCR, and flow cytometry were used. We showed that CCS, when combined with atherogenic diet, resulted in accelerated process of the atherosclerosis, as measured by the maximum intima media thickness and hypoechoic plaque formation in the ultrasound bio microscopy, mean aortic well area in the hematoxylin and eosin staining, max Oil-Red-O content in the Oil Red O staining, and the aortic triglyceride level. These changes were accompanied by endothelial dysfunction and excessive inflammation based on down-regulation of aortic Nos3 mRNA expression, up-regulation of aortic syndecan-1 (Sdc1) and thrombomodulin (Thbd) mRNA expressions, and an increased percentage of CD45-positive cells in the aorta. Stressed mice had up-regulation of aortic tumor necrosis factor-alpha (Tnf-α) gene expression, co-up-regulation of aortic nuclear receptor subfamily 3, group C, member 1 (Nr3c1) and Nfkb1 mRNA expressions, and hyperactivity of adrenal gland function based on increased phenyl ethanolamine-N-methyltransferase (PNMT) and NR3C1-positive cells, up-regulation of Nr3c1 and Pnmt mRNA expressions in the adrenal gland. These observations may demonstrate that CCS, via hyperactivity of the adrenal gland and aortic proinflammatory cytokine (Tnf-α)-related co-up-regulation of aortic Nr3c1 and Nfkb1 gene expressions, accelerated the atherosclerosis development in mice, especially when combined with atherogenic diet.
Key words
adrenal gland function, aortic proinflammatory cytokine, atherogenic diet, atherosclerosis, chronic complex stress
Introduction
The mortality rate due to cardiovascular disease is decreasing gradually; however, cardiovascular disease is still a major cause of death [1]. Atherosclerosis is a chronic inflammatory disease affecting large and medium arteries and is considered to be a major underlying cause of cardiovascular disease [2]. In addition to the traditional risk factors (hypertension, hyperlipidemia, diabetes), stress has been implicated in the pathogenesis of atherosclerosis [3-6]. Certainly, an association between stress and cardiovascular disease exists; however, the underlying mechanisms are complex and animal models of stress that similar to those that develop in humans still need development.
Activation of the autonomic nervous system and hypothalamic-pituitary-adrenal (HPA) axis are principle features of the stress response that affects the functions of other organs including the cardiovascular system [6]. When the HPA axis is activated, glucocorticoids (GCs) are released from the adrenal cortex. GCs are essential steroid hormones that regulate a variety of physiological processes including development, apoptosis, metabolism, and homeostasis. These biological actions of GCs are mediated by the glucocorticoid receptor (GR), a transcription factor that belongs to the nuclear receptor superfamily (nuclear receptor subfamily 3, group C, member 1; NR3C1). Activated GR translocates to the nucleus to regulate transcription of its target genes including nuclear factor (NF)-κB [7-9]. NF-κB, a transcription regulator, impacts many biological processes such as cell growth, proliferation, development, and inflammatory and immune responses [10,11].
Inflammation is believed to be the driving force behind the progression of atherosclerosis [6]. Tumor necrosis factor-alpha (TNF-α) activated NF-κB plays an important role in regulating the expression of proinflammatory cytokines and endothelial cell junctional molecules such as platelet endothelial cell adhesion molecule (PECAM)-1. Excessive or inappropriate leukocyte accumulation in tissues contributes to a number of pathologic conditions such as atherosclerosis and myocardial infarction [6]. The process of leukocyte emigration begins with the generation of inflammatory mediators such as chemokines and cytokines and the upregulation of endothelial cell adhesion molecules including PECAM-1, which plays an important role in the process of leukocyte emigration.
Development of atherosclerosis is preceded by endothelial dysfunction. Nitric oxide (NO), a potential vasodilator produced by endothelial cells, can inhibit monocyte adhesion, platelet aggregation, and vascular smooth muscle cell proliferation resulting in an inhibitory effect on the atherosclerotic process. Impaired endothelial barrier function and the subsequent inflammatory response induces production of adhesion molecules and recruitment of immune cells, which in turn perpetuate the vicious cycle of macrophage infiltration, atherosclerotic plaque formation, and adverse thrombotic events resulting in the acceleration of atherosclerosis [12].
Previously, the atherogenic diet (AD) was shown to accelerate the process of atherosclerosis in an ApoE-/- mouse model of atherosclerosis when compared to either the standard chow diet or Western diet [13]. These results suggest that atherosclerosis can be modulated through alterations in nutrition.
In this study, we discussed how the chronic complex stress (CCS) leaded to the development of atherosclerosis in mice, especially when combined with the atherogenic diet. This mouse model consisted of 5 stressors (physical and psychosocial stressors) of different intensities presented in a random order, similar to the stressors that develop in human lives. We hypothesized that the hyperactivity of the adrenal gland and the co-activation of aortic proinflammatory cytokines and NF-κB mediated endothelial dysfunction, and that an excessive aortic inflammatory response is the underlying mechanism of the CCS-accelerated process of atherosclerosis in mice, especially when combined with the atherogenic diet.
Methods
Animal preparation
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Yamaguchi University (Permit Number: 13-013). Ultrasound bio microscopy was performed under isoflurane in oxygen anesthesia, and all efforts were made to minimize suffering.
Adult (12-week) male C57BL/6j mice were from Charles River Japan, Inc. (Tokyo, Japan). The mice were housed individually on a 12-hour light/12-hour dark cycle and fed with standard chow diet (SCD, a diet of 56% carbohydrate, 23% protein, 5% fat) or the atherogenic diet (AD, a diet of 12% carbohydrate, 52% protein, 21% fat, #7685-130119, Oriental Bio Service Inc. Kyoto, JAPAN, referring to the previous study for 16 weeks after baseline abdominal aorta ultrasound imaging recording [13]. Mice were weighed at study entry and weekly thereafter. Food intake was calculated weekly. The study population started out with 87 mice. The mice were randomly divided into SCD (n =44) and AD (n=43) groups; then those groups were randomly subdivided into groups without and with CCS treatment; finally, 23 SCD mice without CCS, 21 SCD mice with CCS, 22 AD mice without CCS, and 21 AD mice with CCS were used in the present study.
CCS protocol in conscious mice
CCS was performed during the last month of the study. It consisted of 5 stressors (physical and psychosocial stressors) at different stress exposure levels presented in random order, similar to stressors that develop in humans, was produced with reference to previous studies [14,15]. In brief, these included restraint stress (physical stressor): mice were placed for a certain period of time in individual vented plexiglass restrainers without tail movement limitation; conspecific exposure (psychosocial stressor): two mice (each from a different cage) were placed together in a new cage with clean bedding devoid of signals (i.e. urine scent) of previously established dominance; water avoidance (unpredictable mild stressor): a clean empty cage was drenched with 0.3-cm depth of cold water (20–22°C), and a 1-cm-high, 3-cm-diameter platform was provided as a dry environment; damp bedding (unpredictable mild stressor): a clean empty cage was drenched with 0.3-cm depth of cold water (20–22°C), where mice stayed during the stress; cage tilting (unpredictable mild stressor): cage inclination at 20 degrees, where mice stayed during the stress. To avoid habituation, stressors were assigned in random order with one stressor per day for a duration of 6-h each from a.m. 08:30 (-10:00) to p.m. 14:30 (-16:00) for 5 consecutive days (from Monday to Friday) followed by 2 days of rest (Saturday and Sunday) during the last month of the study. With the exception of the CCS, identical procedures including water and food deprivation were performed on the mice without CCS during the same time period.
Blood and tissue collection
At the end of the study, mice were anesthetized with 0.5 to 1% isoflurane in oxygen, and then blood samples were collected from the left ventricle; after systemic perfusion with isotonic saline via the left ventricle, the adrenal gland and aorta (from the aortic arch to the iliac bifurcation) were harvested. One side of the adrenal gland and a part of abdominal aorta tissue pieces were completely submerged in the collection vessel containing RNA later RNA stabilization reagent (Qiagen, Tokyo, Japan); this tissue was incubated overnight at 4°C and then removed from the reagent and transferred to –80°C for storage until the real-time reverse transcriptase polymerase chain reaction (RT-PCR) analysis, and aortic protein concentration and triglyceride level measurements. The remaining adrenal gland and aorta tissue were fixed in 4% paraformaldehyde in 0.1-M phosphate-buffered saline (PBS), then washed in 15% sucrose, and finally stored at 4 °C until further analysis. The blood sample was collected in a tube with heparin and centrifuged at 15,000 × g for 20 minutes at 4°C immediately after collection to obtain the plasma, then was stored at -80 °C until further analysis.
Aortic protein and triglyceride level measurements
The aortic samples were homogenized with lysis buffer (Cell Signal Technology Inc., Danvers, MA) and centrifuged at 10,000 rpm (9,170 × g) for 10 minutes at 4°C. The resultant supernatants were used as the total aortic protein for the aortic protein concentration and aortic triglyceride level measurements. The protein concentrations were determined by Bradford’s method, and the aortic triglyceride level were measured with an automatic analyzer (FDC4000; Fuji Medical Systems, Tokyo, Japan).
Histopathological and fluorescence immunohistochemical examination
Three-μm-thick sections of mice aortae (2 fractions of descending aorta and 2 fractions of abdominal aorta) were processed for hematoxylin and eosin (HE) staining, and 8-μm-thick frozen sections of mice aortae (2 fractions of descending aorta and 2 fractions of abdominal aorta) were processed for Oil Red O staining to quantitatively evaluate the mean aortic area in HE and the max Oil-Red-O content in the Oil Red O staining using the computerized microscope system (BZ-II VIEWER and BZ-II ANALYZER 2007; Keyence Corporation, Osaka, Japan) at 10 × magnification.
For immunofluorescence staining, 8-μm-thick frozen sections of mouse adrenal glands embedded in 4% paraformaldehyde in 0.1-M PBS were cut by cryostat (Leica CM1850), then incubated with the primary antibodies of NR3C1 (H-300, rabbit polyclonal; Santa Cruz Biotechnology) and phenyl ethanolamine-N-methyltransferase (PNMT, ab167427, rabbit monoclonal; Abcam, Cambridge, UK), and further incubated with a Cy3-labeled anti-rabbit IgG (AP182C, Chemicon) diluted at 1:1,000. Nuclei were stained with TO-PRO®-3 iodide (Molecular Probes, Carlsbad, CA). Finally, sections were observed under confocal laser scanning microscopy (LSM5 Pascal/Version 3.2; Carl Zeiss Microlmaging Co. Ltd., Zeiss, Germany). The total number of cells (number of nuclei) and the total number of immuno-positive cells (immunofluorescence-expressing cells which include nuclei) were each counted in 10 consecutive regions under a confocal microscope at 20 × magnification. The ratio of each value was calculated as a percentage of immune-reaction-positive cells.
Ultrasound bio microscopy protocol
Ultrasound bio microscopy was performed on a VisualSonics Vevo 660TM Micro-Ultrasound system (Toronto, Canada) with a transducer frequency of 55 MHz using isoflurane in oxygen anesthesia at the baseline and the end of the study. Following the procedure in a previous study in mice, the maximum intima media thickness (max IMT) and plaque were evaluated [16]. The abdominal aorta lesions were visualized in both long-axis and short-axis views and recorded by B-mode and M-mode ultrasound. Plaques were defined as hyperechoic or hypoechoic plaque that showed higher or lower echogenicity when compared to their adventitia, according to a previous study in humans [17]. Hypoechoic plaques are unstable with pronounced inflammatory infiltration, high lipid content, and prone to rupture while hyperechoic plaques are stable with an abundance of fibrous tissue and calcium [see review in reference 17]. The cine loops of B-mode and M-mode images were saved and max IMT was measured off-line using the built-in software of the Visual Sonics Vevo 660TM Micro-Ultrasound system.
Plasma analysis
Plasma was analyzed with an automatic analyzer (FDC4000i; Fuji Medical Systems, Tokyo, Japan) for blood total cholesterol (TCHO) level, with noradrenaline research ELISA kit (BA E-5200, LDN, Nordhorn, Germany) for blood noradrenaline level according to the manufacturer’s instructions.
Real-time reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from the abdominal aorta tissue using the RNeasy® Fibrous Tissue Mini Kit (Qiagen, Tokyo, Japan) and from the adrenal gland using RNeasy Mini Kit (Qiagen, Tokyo, Japan). ReverTra Ace® qPCR RT Master Mix with gDNA Remover was used to remove gDNA and to synthesize cDNA according to the manufacturer’s instructions. RT-PCR was performed for the quantitative assessment of mRNA expression using an Applied Biosystems StepOne™ system (Applied Biosystems, Foster City, CA, USA). Assays for gene expression included oxidized low density lipoprotein (lectin-like) receptor 1 (ID: Mm00454588_g1, NCBI Reference Sequence: NM_138648.2, NM_001301094.1, and NM_001301096.1; gene symbol: Olr1), nitric oxide synthase 3, endothelial cell (ID: Mm00435204_m1, NCBI Reference Sequence: NM_008713.4; gene symbol: Nos3), syndecan1 (ID: Mm00448920_g1, NCBI Reference Sequence: NM_011519.2; gene symbol: Sdc1), thrombomodulin (ID: Mm00437014_s1, NCBI Reference Sequence: NM_009378.3, gene symbol: Thbd), tumor necrosis factor (ID: Mm99999068_m1, NCBI Reference Sequence: NM_013693.3; gene symbol: Tnf-α), nuclear factor of kappa light polypeptide gene enhancer in B cells 1, p105 (ID: Mm00476361_m1, NCBI Reference Sequence: NM_008689.2; gene symbol: Nfkb1), nuclear receptor subfamily 3, group C, member 1 (ID: Mm00433832_m1, NCBI Reference Sequence: NM_008173.3; gene symbol: Nr3c1), phenylethanolamine-N-methyltransferase (ID: Mm00476993_m1, NCBI Reference Sequence: NM_008890.1, gene symbol: Pnmt), and platelet/endothelial cell adhesion molecule 1 (ID: Mm01246167_m1, GenBank: BC085502.1 and BC008519.1; gene symbol: Pecam1); all were purchased from Applied Biosystems. The relative expressions of the Olr1, Sdc1, Thbd, Nos3, Nr3c1, Pnmt, Nfkb1, Tnf-α, and Pecam1 mRNAs were normalized to the amount of glyceraldehyde-3-phosphate dehydrogenase (ID: Mm99999915_g1, NCBI Reference Sequence: NM_001289726.1 and NM_008084.3; Gene symbol: Gapdh) mRNA in an identical cDNA sample, using the comparative quantitative method recommended by the manufacturer; the relative expression was expressed as the fold change from the values of SCD without CCS. Nos3, Sdc1, and Thbd were used as markers of endothelial damage with reference to methods in previous studies [18,19].
Flow cytometry analysis of leukocyte accumulation within mice aortae isolation of mice aorta
At the end of the study a part of mice were anesthetized with 0.5 to 1% isoflurane in oxygen, and the whole aorta (from the aortic arch to the iliac bifurcation) was harvested after systemic perfusion with Hanks’ balanced salt solution (HBSS) via the left ventricle. Adipose tissues and para-aortic lymph nodes were carefully dissected away from the aorta. The isolated aorta was placed in a collection tube with HBSS.
Preparation of single cell suspensions
An aorta dissociation enzyme stock solution (ADESS) was created using a tumor dissociation kit for mice (130-096-730, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany). In brief, 100 μL Enzyme D, 50 μL Enzyme R, and 12.5 μL Enzyme A were set to 1.5 mL with 1 % BSA-10 mM HEPES-HBSS buffer in gentleMACSTM C tube (130-093-237, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) and the aorta was incubated at 37°C. The whole aorta was removed from the HBSS to the gentleMACSTM C tube containing the prepared ADESS. The aortae were cut into small pieces and homogenized by crushing using the gentleMACS™ Dissociator (Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) after being incubated with the ADESS for 30 minutes at 37°C. This procedure was repeated a total of three times; then, 10 mL autoMACSTM running buffer (130-091-221, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) was added, and the homogenate was passed through a pre-separation filter, 70 μm (130-095-823, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) into 15 mL tubes. The cells were pelleted by centrifugation (300 × g, 10 minutes, and 4°C) to prepare single-cell suspensions from the total aorta.
Stain aortic cell suspensions with CD45-PerCP antibody
Up to 106 nucleated cells were suspended with 45 μL of autoMACSTM Running Buffer, and 5 μL of CD45-PerCP antibody (common leukocyte antigen marker) was added, followed by thorough mixing and incubation for 10 minutes at 4°C in the dark. Cells were washed by adding 1 mL of autoMACSTM Running Buffer and centrifuged at 300 × g for 10 minutes. Supernatant was aspirated completely. The cell pellet was resuspended in a suitable amount (0.5 mL) of autoMACSTM Running Buffer and 5 μL PI (Live/Dead cell viability staining). Samples were run on a flow cytometer (Cytomics FC500, Beckman Coulter, CA).
Statistical analysis
Continuous data are expressed as means ± standard deviations (SDs). Extreme values were excluded by the Smirnov-Grubbs test. Sample size (numbers of mice) for group each had been determined in the experimental design before the study. However, the numbers of mice introduced to each analysis in group each were also include the excluded numbers with Smirnov-Grubbs test, the discarded numbers due to sudden death during the CCS, and the number used for reproducibility experiment resulted in the numbers of mice in each group were not the same finally. The plaque formation in the abdominal aorta was evaluated by the contingency table (m x n) chi-square test followed Bonferroni post hoc test among 4 groups, and deference of hypoechoic plaque formation in the abdominal aorta between without and with CCS in mice fed AD was evaluated by contingency table (2x2) chi-square test only. Continuous-variable comparisons between 2 groups (before and during the CCS) were tested by paired t test, while those continuous-variable comparisons among 4 groups were tested by 1-way analysis of variance (ANOVA). When an F-value was found to be significant by ANOVA, a Bonferroni/Dunn post hoc test was used for multiple comparisons. Statistical analysis was performed using Statcel2 for Windows software (OMS Publishing, Inc., Saitama, Japan). Value of p < 0.05 was considered statistically significant.
Results
Plasma TCHO, body weight, and consumption of nutrients and calories
Mice were discarded due to sudden death (3 of SCD and 2 of AD) and movement disorder (1 of SCD) during CCS; eventually, 40 SCD mice (23 without and 17 with CCS) and 41 AD mice (22 without and 19 with CCS) were analyzed in the present study. We used an atherogenic diet (AD) of 12% carbohydrate, 52% protein, 21% fat (445 kcal/100g diet) or a standard chow diet (SCD) of 56% carbohydrate, 23% protein, 5% fat (362 kcal/100g diet).
The plasma TCHO (Figure 1A) was significantly increased in mice fed the AD without CCS, while CCS suppressed this increase in mice fed the same AD (Figure 1A) at the end of the study.
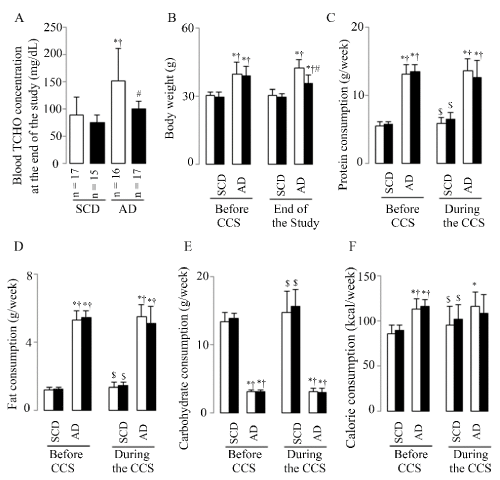
Figure 1. Animal basic characteristics
Representative results of plasma TCHO (A), BW (B), and protein (C), fat (D), carbohydrate (E), calorie (F) consumptions. White bars represent the mice without CCS and black bars represent mice with CCS; 40 SCD mice (23 without and 17 with CCS) and 41 AD mice (22 without and 19 with CCS) were analyzed from B to F; Results are shown as means ± SDs. *p <0.05 vs. SCD without CCS, †p<0.05 vs. SCD with CCS, #p<0.05 vs. AD without CCS in the ANOVA followed the Bonferroni/Dunn post hoc test statistical analysis, $p<0.05 vs. Before CCS of them in the paired t test.
Before CCS (after 3 months SCD or AD breeding) BW (Figure 1B) showed significantly increases in mice fed the AD (both of without and with CCS mice) as compared to mice fed the SCD (both of without and with CCS mice). At the end of the study, BW (Figure 1B) also showed significantly increases in mice fed the AD (both of without and with CCS mice) as compared to mice fed the SCD (both of without and with CCS mice), however CCS suppressed this AD-induced BW increment in mice fed AD with CCS. In contrast, CCS showed no significant effects on BW in mice fed SCD. These results were accompanied with the high consumption of protein (Figure 1C), fat (Figure 1D), and calorie (Figure 1F), while the low carbohydrate consumption (Figure 1E) both before and during the CCS in mice fed the AD (both of without and with CCS mice) as compared to mice fed the SCD (both of without and with CCS mice). But compared to them of before CCS, mice fed AD with CCS showed the suppressed trend of protein, fat, carbohydrate, and calorie consumption (the statistical analysis found no difference, p = 0.0567 in all); in contrast to these, the mice fed SCD with CCS showed the increased consumption of them (Figure 1C-1F, p < 0.05 in all).
The results indicated that AD diet produced hyperlipidemia and greater body weight increase; however, AD mice undergoing CCS lost body weight and suppressed the elevation in blood TCHO at the end of the study due to the comprehensive suppressed consumption of nutrients and calories during the stress treatment.
Max IMT and plaque formation in the abdominal aorta
Increased max IMT (Figure 2A and 2C) and plaque formation (Figure 2B) were shown in SCD with CCS, AD without CCS, and AD with CCS group mice as compared to SCD without CCS mice, and the highest max IMT and hyperechoic plaque formation were shown in stressed mice fed AD. Hyperechoic formation were 4.5% (SCD without CCS), 73% (SCD with CCS), 38% (AD without CCS), and 100% (AD with CCS) respectively. Moreover, hypoechoic plaques, the unstable plaques, only appeared in the mice fed the AD diet, and CCS increased this hypoechoic plaque formation (8 of 14 (57%) in mice with CCS vs. 2 of 21 (10%) in mice without CCS, χ2 = 9.3, p < 0.05).
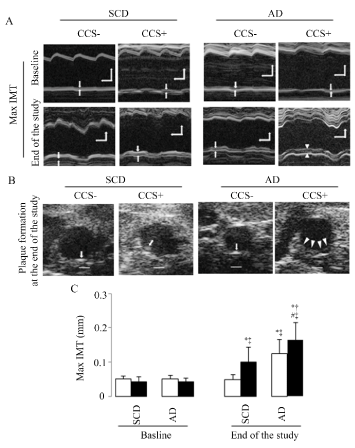
Figure 2. Max IMT and plaque formation assessed by ultrasound bio microscopy imaging
(A) Representative examples of the abdominal aortic max IMT at baseline and at the end of the study, the space between the two arrows indicate max IMT, horizontal scale bar = 100ms, vertical scale bar = 300μm. (B) Representative examples of the formation of hard (arrows) and soft (arrowheads) plaques in the abdominal aorta at the end of the study; these can be defined by higher and lower echogenicity compared to the adventitia, respectively; scale bar = 300μm. (C) Representative results in max IMT. White bars represent the mice without CCS and black bars represent mice with CCS; 37 SCD mice (22 without and 15 with CCS) and 35 AD mice (21 without and 14 with CCS) were analyzed in C; Results are shown as means ± SDs. *p<0.05 vs. SCD without CCS, †p<0.05 vs. SCD with CCS, #p<0.05 vs. AD without CCS in the ANOVA followed the Bonferroni/Dunn post hoc test statistical analysis, ‡p<0.05 vs. baseline of them in the paired t test.
Pathology phenotype of atherosclerosis based on mean aortic wall area, max aortic Oil-Red-O content and aortic triglyceride level, and Pecam1- and Olr1-mediated leukocyte accumulation in the whole aortic cell suspension
Five mice were processed for HE is staining each group, however, 1 of SCD without CCS and 1 of AD without CCS were discarded due to the unsuccessful tissue slice preparation; Both AD and CCS induced an increase in the mean aortic wall area, evaluated by HE is staining, compared to the SCD mice without CCS (Figure 3A and 3B).
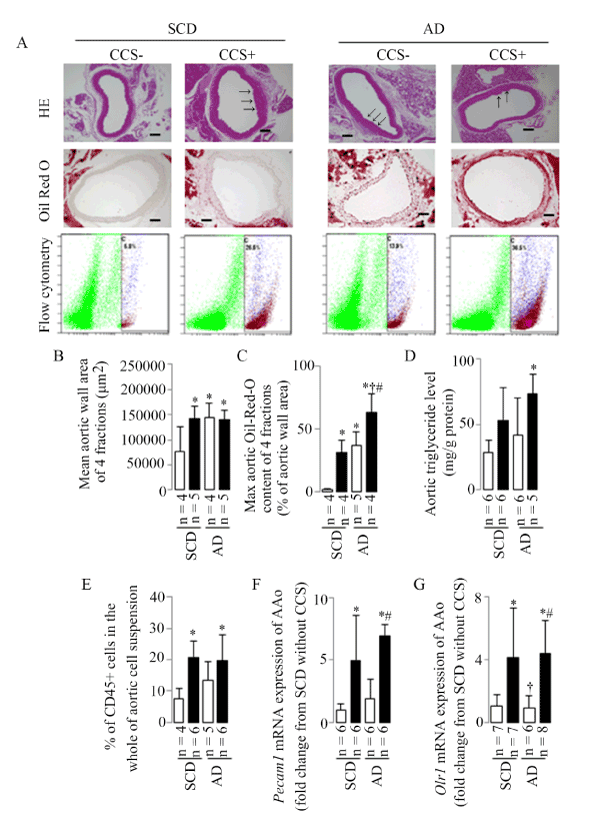
Figure 3. Aortic mean wall area, max Oil-Red-O content, triglyceride level, leukocyte accumulation, and Pecam1 and Orl1 mRNA expressions
(A) Representative examples of the aortic wall area (HE is staining), Oil-Red-O content (Oil Red O staining), and CD45+ cells in the whole of aortic cell suspension (flow cytometry), arrows indicate the regional lesions, scale bar = 100μm. (B) Representative results in aortic mean area. (C) Representative results in aortic max Oil-Red-O content. (D) Representative results in aortic triglyceride level. (E) Representative results in aortic leukocyte accumulation identified as CD45+ cells in the whole of the aortic cell suspension using flow cytometry, the numbers represent the percentages of live aortic CD45+ leukocytes in C gate (purple cell population). (F) Representative results in aortic Pecam1 mRNA expression. (G) Representative results in aortic Orl1 mRNA expression. Results are shown as means ± SDs; white bars represent mice without CCS and black bars represent mice with CCS. *p<0.05 vs. SCD without CCS, †p<0.05 vs. SCD with CCS, #p<0.05 vs. AD without CCS in the ANOVA followed the Bonferroni/Dunn post hoc test statistical analysis.
Five mice were processed for Oil Red O staining each group, however, 1 of SCD without CCS, 1 of SCD with CCS, and 1 of AD with CCS were discarded due to the unsuccessful tissue slice preparation; Both AD and CCS induced an increase in the max aortic Oil-Red-O content, evaluated by Oil Red O staining compared to the SCD mice without CCS, especially in AD mice with CCS (Figure 3A and 3C).
The elevated aortic triglyceride level was shown only in the AD mice with CCS compared to the SCD mice without CCS (Figure 3D), upward trend (without significant statistical difference) showed in mice fed SCD with CCS and in mice AD without CCS.
Flow cytometry was performed to analyze leukocytes within the mice aorta using CD45 staining in conjunction with Live/Dead viability dye (propidium iodide staining) in the present study. Six mice were processed for flow cytometry study each group, however, 2 of SCD without CCS and 1 of AD without CCS were discarded due to the extreme values during the statistical analysis. The percentage of aortic leukocytes in the whole aortic cell suspension (infiltration of CD45+leukocytes) was increased only in mice with CCS both the SCD- and AD-fed mice (Figure 3A and 3E).
PECAM-1 contributes to the pathogenesis of atherosclerosis through its ability to mediate leukocyte infiltration, and oxidized LDL has been shown to promote monocyte migration through cytokine-stimulated endothelial cells in vitro by a mechanism involving upregulation of endothelial cell PECAM-1 [see review in in reference [20]. In the present study, CCS, but not AD-induced hyperlipidemia, induced a significant up-regulation of Pecam1 and Orl1 (the receptor of oxidized LDL) mRNA expressions (Figure 3F and 3G).
Endothelial dysfunction
In the present study, the CCS-induced leukocyte infiltration was accompanied by endothelial dysfunction based on changed aortic gene expressions of Nos3 (Figure 4A), Thbd (Figure 4B), and Sdc1 (Figure 4C) in stressed mice, as evidenced by down-regulated Nos3 mRNA expression and the up-regulated Thbd and/or Sdc1 mRNA expressions in mice with CCS as compared to the mice fed SCD without CCS.
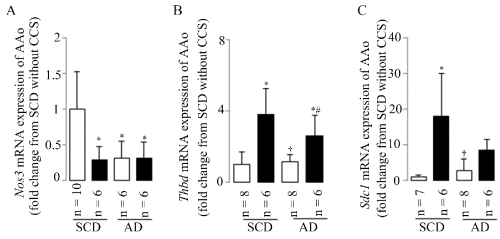
Figure 4. Endothelial dysfunction
Endothelial dysfunction was assessed by aortic Nos3 (A), Thbd (B), and Sdc1 (C) mRNA expressions in the present study. Results are shown as means±SDs; white bars represent mice without CCS and black bars represent mice with CCS. *p<0.05 vs. SCD without CCS, †p<0.05 vs. SCD with CCS, #p<0.05 vs. AD without CCS using the ANOVA followed the Bonferroni/Dunn post hoc test statistical analysis.
Tnf-α-related co-up-regulation of aortic Nr3c1 and Nfkb1 mRNA expressions was accompanied with leukocyte infiltration and endothelial dysfunction
CCS may induce leukocyte infiltration and endothelial dysfunction especially when combined with atherogenic diet at least in partly. These changes were accompanied by the significant up-regulation or upward trends (without statistical differences) of aortic Nr3c1 (Figure 5A), Nfkb1 (Figure 5B), and Tnf-α (Figure 5C) mRNA expressions in mice with CCS. The results indicated that Tnf-α related co-up-regulation of aortic Nr3c1 and Nfkb1 mRNA expressions may in part mediate the aortic inflammatory response and endothelial dysfunction during CCS, especially in AD mice with CCS.
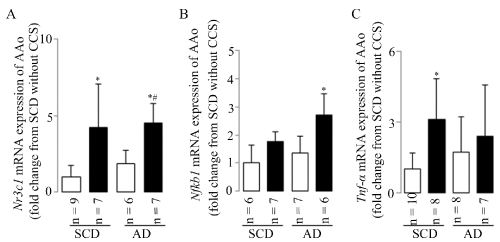
Figure 5. Tnf-α-related co-up-regulation of aortic Nr3c1 and Nfkb1 mRNA expression
CCS up-regulated aortic mRNA expressions of the Nr3c1 (A), Nfkb1 (B) and Tnf-α (C). Results are shown as means ± SDs; white bars represent mice without CCS and black bars represent mice with CCS. *p<0.05 vs. SCD without CCS, #p<0.05 vs. AD without CCS in the ANOVA followed the Bonferroni/Dunn post hoc test statistical analysis.
Stress-induced adrenal hyperactivation
Stress leads to activation of stress-responsive organs, beginning with cortisol and epinephrine release into circulation. CCS-induced changes in adrenal gland function were therefore determined. As shown in Figure 6, CCS, especially in mice combined with atherogenic diet, activated the adrenal gland function as indicated by increases in Nr3c1 and Pnmt mRNA expressions (Figure 6A and 6B), the percentage of NR3C1- and PNMT-positive cells (Figure 6C-6E). CSS induced the increase in the blood noradrenaline concentrations in mice fed standard chow diet (Figure 6F); however, only the upward trend (without significant statistical difference) in AD mice with CCS. These results showed that CCS may activate hormone epinephrine via the Pnmt gene, which is glucocorticoid-regulated and similar to results of the repeated immobilization stress at least in partly [21,22].
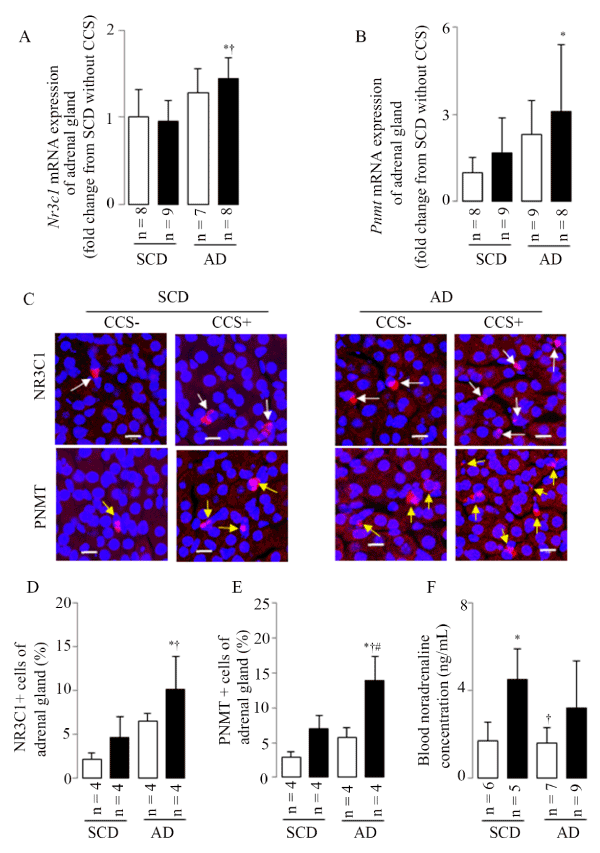
Figure 6. CCS-induced adrenal hyperactivation
(A) Representative results in adrenal gland Nr3c1 mRNA expression. (B) Representative results in adrenal gland Pnmt mRNA expression. (C) Representative examples of adrenal gland NR3C1- (white arrows) and PNMT- (yellow arrows) positive cells as assessed by LSM; scale bar=10μm. (D) Representative results in the percentage of NR3C1-positive cells. (E) Representative results in the percentage of PNMT-positive cells. (F) Representative results in the blood noradrenaline concentration. Results are shown as means ± SDs; white bars represent mice without CCS and black bars represent mice with CCS. *p<0.05 vs. SCD without CCS, † p<0.05 vs. SCD with CCS, #p<0.05 vs. AD without CCS in the ANOVA followed the Bonferroni/Dunn post hoc test statistical analysis.
Discussion
A new complex chronic mental stress (CCS) mouse model was used in the present study. This mouse model consisted of physical and psychosocial stressors of different types and intensities, presented in a random order similar to the stressors that develop in humans. Our data demonstrate that the process of atherosclerosis can be controlled by CCS in mic fed standard chow diet and the atherogenic diet. The synergy between CCS and atherogenic diet may also accelerate the atherosclerosis development in mice. CCS could induce endothelial dysfunction and excessive leukocyte accumulation at the aortic wall, leading to the process of atherosclerosis in mice. These effects may be modulated in part by CCS-induced hyperactivity of the adrenal gland and the proinflammatory cytokine (Tnf-α) related co-up-regulation of aortic Nr3c1 and Nfkb1 gene expressions.
Animal stress models for atherosclerosis study
Insight into the link between stress and atherosclerosis disease can be gained from studies in animals. Animal stress models are typically divided into physical stress models and psychological stress models. It is important for animal models to mimic the natural courses of disease and stress responses found in humans; unfortunately, stress in humans is complex, and the animal models of stress used in previous studies are very limited in complexity [see review in reference 6]. Referring to methods in previous studies, we used a chronic complex stress mouse model in the present study [6,14]. This mouse model consisted of the physical and psychosocial stressors of different types and intensities, presented in a random order, similar to what human’s experience. Restraint stress is a physical method of stress induction. A previous study reported that a restraint stress (120 min/day, 14 days) induced endothelial dysfunction by oxidative stress [23]. Conspecific exposure is a psychosocial stress that can abate social interaction and increase anxiety-derived behaviors similar to stress-induced psychopathology in humans. The social defeat test is associated with sympathetic hyperactivity and tachyarrhythmia in rat studies [see review in reference 24]. In a mouse study, the volume fraction of fibrosis was six folds larger than in control animals due to the appearance of more microscopic scarring [25]. Unpredictable chronic mild stresses: In a study with ApoE -/- mice, unpredictable chronic mild stress promoted plaque features associated with vulnerable lesions that cause myocardial infarction and stroke in humans [26]. Three kinds of unpredictable chronic mild stresses (water avoidance, damp bedding, and cage tilting) were used in the present study. Our chronic complex stress mouse model applied various stress modules in a random order, with one stressor per day for a duration of 6-h each for 5 consecutive days (from Monday to Friday) followed by 2 days of rest (Saturday and Sunday) during the last month of the study. CCS were shown to accelerate the process of atherosclerosis, suggesting that this chronic complex stress mouse model may be useful in gaining insight into the link between stress and atherosclerosis in humans.
CCS accelerated the development of atherosclerosis
The link between stress and cardiovascular diseases has not been clarified completely. Overreaction of the autonomic nervous system to a chronic or unexpected stressor can produce cardiovascular dysfunction. Previous studies have shown that acute restraint stress caused lethal bradycardia associated with the shift of cardiac sympathovagal balance toward sympathetic predominance in rats, and patients with major depressive disorder were shown to have extraordinarily high sympathetic nervous activity, including the sympathetic outflow to the heart [14,27]. In the present study, 5 kinds of stressful situations were used in the CCS project, and the effects of CCS on the progress of atherosclerosis, a major underlying cause of cardiovascular disease, were assessed. Our data suggest that CCS may have accelerated the progress of atherosclerosis (Figure 2) along with the leukocyte overresponses (Figure 3A and 3E) and aortic endothelial dysfunction (Figure 4) in mice. These results were associated with overreaction of the autonomic nervous system based on the increased secretion of adrenal PNMT and plasma noradrenaline concentration (Figure 6C, 6E, and 6F), and activation of the HPA axis as evidenced by increases in secretion of adrenal NR3C1 (Figure 6C and 6D). Our results indicate that the HPA axis and the autonomic nervous system, the first two responders to stressful situations, play a role in CCS-accelerated development of atherosclerosis.
CCS may via Tnf-α–related co-up-regulation of Nr3c1 and Nfkb1 mRNA expressions induced endothelial dysfunction and excessive leukocyte accumulation in aortic wall especially when combined with atherogenic diet
The vascular endothelium plays an important role in the initiation and development of atherosclerotic diseases. The primary role of the endothelium is to adjust vascular function by producing NO, which decreases vascular resistance, inhibits platelet adhesion, and decreases vascular smooth muscle cell proliferation [28]. Stress activates the HPA axis, leading to increased secretion of adrenal GCs [see review in reference 12], and the actions of GCs are mediated through the glucocorticoid receptor (GR), a transcription factor that belongs to the nuclear receptor superfamily (NR3C1). Incubation of human endothelial cells with cortisol reduced NOS3 mRNA and protein to 60–70% of control expression levels, and these responses were prevented by the glucocorticoid receptor antagonist mifepristone [29]. The sugar-protein glycocalyx coats the vascular endothelium. Damage to the glycocalyx in the vascular endothelium causes impaired endothelial barrier function and an increase in platelet and leucocyte adhesion and shear stress in the vessel [30]. Higher levels of syndecan-1 (Sdc-1, a biomarker of endothelial glycocalyx damage) were observed during acute coronary syndrome, suggesting that damage to endothelial glycocalyx may participate in the atherosclerotic plaque vulnerability process in patients [31]. Thrombomodulin, an endothelial membrane glycoprotein, is an essential part of the protein C anti-coagulant pathway and has a role in the regulation of fibrinolysis [32]. Our results are consistent with previous studies showing that CCS-induced up-regulations of Nr3c1 mRNA both in the adrenal gland and aorta were accompanied by the down-regulation of aortic Nos3 mRNA expression. Furthermore, CCS-induced up-regulation of Nr3c1 mRNA was also accompanied with the up-regulation of Sdc1 and Thbd mRNA expressions, suggesting impaired endothelial barrier function and Thbd-mediated fibrinolysis regulation. In addition, binding of activated NR3C1 on glucocorticoid-responsive elements and interaction with NF-КB are the main mechanisms of regulation associated with glucocorticoid-mediated transactivation and trans repression [33].
NF-КB is a family of constitutively expressed transcription factors that impact cell growth, proliferation, and development, and inflammatory and immune responses [see review in reference 34]. The NF-КB pathway has been considered as a prototypical proinflammatory signaling pathway due to the role of NF-КB in the expression of proinflammatory genes including cytokines, chemokines, and adhesion molecules. In response to diverse internal and external stressors, NF-КB is response to inflammatory stimuli, such as the proinflammatory cytokine TNF-α signaling, and the activated NF-КB also drives proinflammatory cytokines, chemokines, receptors, and adhesion molecules productions including TNF-α [34,35]. Previous studies reported that chronic mild stress increased the circulating level of TNF-α in rats, and a higher level of TNF-α occurs in school teachers who are under a greater degree of psychological stress [36,37]. In addition to this study, several findings have demonstrated that anti-TNF-α therapies may improve endothelial dysfunction in various pathophysiological conditions [38,39]. These findings are consistent with our results. We found that CCS-induced Nos3 mRNA down-regulation was accompanied by up-regulation of aortic Tnf-α, Nr3c1, and Nfkb1 mRNA expressions. Thus, CCS activates the HPA axis leading to increased production of NR3C1 both in the adrenal gland and aorta, increases aortic proinflammatory mediators (TNF-α in the present study), and results in the endothelial dysfunction observed in CCS.
Excessive leukocyte accumulation in the aortic wall contributes to the progress of atherosclerosis. Our results showed that CCS-induced endothelial dysfunction (Figure 4) was associated with excessive leukocyte accumulation (Figure 3A and 3E) in the aortic wall. Endothelial dysfunction and the subsequent inflammatory response induce production of adhesion molecules such as PECAM-1 and recruitment of immune cells such as leukocytes, which in turn perpetuate the vicious cycle of leukocyte infiltration, atherosclerotic plaque formation, and adverse thrombotic events. The process of leukocyte emigration begins with the generation of inflammatory mediators (such as TNF-α) and upregulation of endothelial cell adhesion molecules. These support the initial attachment of leukocytes to endothelial cells and their migration through junctions between adjacent endothelial cells in vessel walls, and the changes in levels of numerous endothelial cell junctional molecules including PECAM-1 [see review in reference 20]. The NF-КB signal transduction pathway in artery endothelial cells is primed for atherosclerotic lesion formation, and PECAM-1-deficient mice showed no detectable activation of NF-КB or evidence of downstream inflammatory genes in regions of disturbed flow [40,41]. Oxidized LDL upregulated PECAM-1 and downregulated VE-cadherin at the endothelial junctions, and then promoted monocyte entry by enhanced homophilic binding to monocyte PECAM-1, and by a disrupted junctional barrier, respectively [42]. Furthermore, previous studies showed that hyperinsulinemia accelerated atherosclerosis by directly enhancing neutrophil trans-endothelial migration via increased endothelial PECAM-1 expression [43,44]. An in vitro study showed that rapamycin, which can inhibit oxidized LDL accumulation in human umbilical vein endothelial cells, reduced mechanistic target of its phosphorylation and subsequently inhibit NF-κB activation and suppresses LOX-1 resulted in a reduction in oxidized LDL uptake in human umbilical vein endothelial cells [45]. The present study showed that the CCS-induced Pecam1 mRNA up-regulation was accompanied with Olr1 (the receptor of oxidized LDL) mRNA up-regulation in the mice aorta. Together with the previous studies, up-regulated aortic Pecam1 and Olr1 mRNA expressions during the progress of atherosclerosis may at least in part be responsible for the leukocyte accumulation in the aortic wall observed in CCS.
Atherogenic diet is also responsible for the development of atherosclerosis
In a study with male ApoE-/- mice, mice fed the atherogenic diet of 12% carbohydrate, 43% fat, 45% protein, and 0.15% cholesterol showed the more extensive progress in atherosclerosis than mice fed Western’ diet (43% carbohydrate, 42% fat, 15% protein, and 0.15% cholesterol), despite of the similar dietary fat and cholesterol content in the diet, and reduced weight gain [13]. This conclusion was evidenced by the significantly more atheroma and the larger cross-sectional plaque area at the level of the aortic valve. In the present study, wild type mice (C57BL/6j) fed AD of 12% carbohydrate, 52% protein, 21% fat also showed the more extensive progress in atherosclerosis, as evidenced by the increased max IMT and hypoechoic plaque formation, aortic mean area, aortic max Oil-Red-O content, and aortic triglyceride level (Figure 2 and 3), furthermore, CCS in mice fed the AD showed the highest max IMT, hypoechoic plaque formation, Oil-Red-O content, and triglyceride level in the four groups. Our results indicated that the synergy between CCS and AD may responsible for the atherosclerosis development in mice. However, the changes of the aortic Pecam1, Olr1, Nos3, Thbd, Sdc1, and Tnf-α at the message level are insufficient to address the synergy (between CCS and AD)-induced the atherosclerosis development completely, further study for the underlying mechanisms is necessary.
Limitations
The chronic complex stress (CCS) mice model used in the present study only induced mild atherosclerotic lesions in the wild type mice. In a recent study, a chronic immobilization stress (4 hours in an immobilization stress tube daily for 12 weeks) enhanced vascular endothelial senescence and atherosclerotic plaque growth in ApoE-/- mice [46]. To further investigate the impact of CCS and AD on atherosclerosis, an established atherosclerosis mice model regarding CCS and AD in an ApoE-/- and Ldlr-/- mice is necessary.
In the present study, descending and abdominal aorta were used; however, arteriosclerosis is relatively hard to develop in descending and abdominal aorta compared to aortic root. This limitation may be responsible at least in partly for the not evident changes of mean aorta wall areas evaluated by HE is staining images in mice with CCS, in contrast with other results, such as the max IMT (Figure 2A and 2C) and % of CD45+ cells in the whole aorta (Figure 3A and 3E). However, to investigate the full particulars of CCS and AD on atherosclerosis, all of the aortic fractions including the aortic root, outflow tract are essential, even the generic noninvasive ultrasound bio microscopy and flow cytometry methods were used.
In the present study, we evaluated the relationship between gene phenotypes and CCS-accelerated atherosclerosis development. However, CCS accelerated the process of atherosclerosis in mice are not only account for the gene phenotypes. The further molecular and biology study for the atherosclerosis-related cells in the aorta are necessary.
Conclusions
Psychological stress may lead to adverse atherosclerosis; however, the mechanisms have not been well understood. Nervous and humoral systems are activated with variations in the type and intensity of stressors, suggesting the importance of creating chronic complex stress animal models. The CCS used in the present study may constitute a better model of the kind of unexpected stressful stimuli that develop in human lives. CCS, especially when combined with an atherogenic diet, accelerated the process of atherosclerosis via endothelial dysfunction and excessive leukocyte accumulation despite a decrease in blood TCHO in mice. The hyperactivity of adrenal gland function, the co-up-regulation of aortic proinflammatory genes (Tnf-α, Pecam1, and Olr1) and Nfkb1 mRNA expressions may be involved in endothelial dysfunction and the activation of the aortic inflammatory response.
Acknowledgement
The authors would like to thank Kenichi Sasamoto (Miltenyibiotec Tokyo), Ihoko Fujimoto (Yamaguchi University), Yuki Tanaka (Yamaguchi University), and Fuka Haruse (Yamaguchi University) for technical assistance.
Funding and grants
This study was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (no. 25460869 to Jinyao Liu and no. 17K09265 to Jinyao Liu) for data collection and analysis.
Conflict of interest
Not declaredAbstract
The effects of stress on the atherosclerosis are complex. Here we discuss how the chronic complex stress (CCS), when combined with an atherogenic diet (AD), leads to the development of atherosclerosis in mice. The CCS mouse model consisted of physical and psychosocial stressors of different types and intensities presented in a random order. Eighty-seven mice were divided into a standard chow diet (n=44) and AD (n=43) groups and bred for 4 months. Both groups were subdivided into groups without and with CCS treatment. The CCS was performed during the last month of the study. Ultrasound bio microscopy, histopathological and fluorescence immunohistochemical examinations, ELISA, PCR, and flow cytometry were used. We showed that CCS, when combined with atherogenic diet, resulted in accelerated process of the atherosclerosis, as measured by the maximum intima media thickness and hypoechoic plaque formation in the ultrasound bio microscopy, mean aortic well area in the hematoxylin and eosin staining, max Oil-Red-O content in the Oil Red O staining, and the aortic triglyceride level. These changes were accompanied by endothelial dysfunction and excessive inflammation based on down-regulation of aortic Nos3 mRNA expression, up-regulation of aortic syndecan-1 (Sdc1) and thrombomodulin (Thbd) mRNA expressions, and an increased percentage of CD45-positive cells in the aorta. Stressed mice had up-regulation of aortic tumor necrosis factor-alpha (Tnf-α) gene expression, co-up-regulation of aortic nuclear receptor subfamily 3, group C, member 1 (Nr3c1) and Nfkb1 mRNA expressions, and hyperactivity of adrenal gland function based on increased phenyl ethanolamine-N-methyltransferase (PNMT) and NR3C1-positive cells, up-regulation of Nr3c1 and Pnmt mRNA expressions in the adrenal gland. These observations may demonstrate that CCS, via hyperactivity of the adrenal gland and aortic proinflammatory cytokine (Tnf-α)-related co-up-regulation of aortic Nr3c1 and Nfkb1 gene expressions, accelerated the atherosclerosis development in mice, especially when combined with atherogenic diet.
Key words
adrenal gland function, aortic proinflammatory cytokine, atherogenic diet, atherosclerosis, chronic complex stress
Introduction
The mortality rate due to cardiovascular disease is decreasing gradually; however, cardiovascular disease is still a major cause of death [1]. Atherosclerosis is a chronic inflammatory disease affecting large and medium arteries and is considered to be a major underlying cause of cardiovascular disease [2]. In addition to the traditional risk factors (hypertension, hyperlipidemia, diabetes), stress has been implicated in the pathogenesis of atherosclerosis [3-6]. Certainly, an association between stress and cardiovascular disease exists; however, the underlying mechanisms are complex and animal models of stress that similar to those that develop in humans still need development.
Activation of the autonomic nervous system and hypothalamic-pituitary-adrenal (HPA) axis are principle features of the stress response that affects the functions of other organs including the cardiovascular system [6]. When the HPA axis is activated, glucocorticoids (GCs) are released from the adrenal cortex. GCs are essential steroid hormones that regulate a variety of physiological processes including development, apoptosis, metabolism, and homeostasis. These biological actions of GCs are mediated by the glucocorticoid receptor (GR), a transcription factor that belongs to the nuclear receptor superfamily (nuclear receptor subfamily 3, group C, member 1; NR3C1). Activated GR translocates to the nucleus to regulate transcription of its target genes including nuclear factor (NF)-κB [7-9]. NF-κB, a transcription regulator, impacts many biological processes such as cell growth, proliferation, development, and inflammatory and immune responses [10,11].
Inflammation is believed to be the driving force behind the progression of atherosclerosis [6]. Tumor necrosis factor-alpha (TNF-α) activated NF-κB plays an important role in regulating the expression of proinflammatory cytokines and endothelial cell junctional molecules such as platelet endothelial cell adhesion molecule (PECAM)-1. Excessive or inappropriate leukocyte accumulation in tissues contributes to a number of pathologic conditions such as atherosclerosis and myocardial infarction [6]. The process of leukocyte emigration begins with the generation of inflammatory mediators such as chemokines and cytokines and the upregulation of endothelial cell adhesion molecules including PECAM-1, which plays an important role in the process of leukocyte emigration.
Development of atherosclerosis is preceded by endothelial dysfunction. Nitric oxide (NO), a potential vasodilator produced by endothelial cells, can inhibit monocyte adhesion, platelet aggregation, and vascular smooth muscle cell proliferation resulting in an inhibitory effect on the atherosclerotic process. Impaired endothelial barrier function and the subsequent inflammatory response induces production of adhesion molecules and recruitment of immune cells, which in turn perpetuate the vicious cycle of macrophage infiltration, atherosclerotic plaque formation, and adverse thrombotic events resulting in the acceleration of atherosclerosis [12].
Previously, the atherogenic diet (AD) was shown to accelerate the process of atherosclerosis in an ApoE-/- mouse model of atherosclerosis when compared to either the standard chow diet or Western diet [13]. These results suggest that atherosclerosis can be modulated through alterations in nutrition.
In this study, we discussed how the chronic complex stress (CCS) leaded to the development of atherosclerosis in mice, especially when combined with the atherogenic diet. This mouse model consisted of 5 stressors (physical and psychosocial stressors) of different intensities presented in a random order, similar to the stressors that develop in human lives. We hypothesized that the hyperactivity of the adrenal gland and the co-activation of aortic proinflammatory cytokines and NF-κB mediated endothelial dysfunction, and that an excessive aortic inflammatory response is the underlying mechanism of the CCS-accelerated process of atherosclerosis in mice, especially when combined with the atherogenic diet.
Methods
Animal preparation
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Yamaguchi University (Permit Number: 13-013). Ultrasound bio microscopy was performed under isoflurane in oxygen anesthesia, and all efforts were made to minimize suffering.
Adult (12-week) male C57BL/6j mice were from Charles River Japan, Inc. (Tokyo, Japan). The mice were housed individually on a 12-hour light/12-hour dark cycle and fed with standard chow diet (SCD, a diet of 56% carbohydrate, 23% protein, 5% fat) or the atherogenic diet (AD, a diet of 12% carbohydrate, 52% protein, 21% fat, #7685-130119, Oriental Bio Service Inc. Kyoto, JAPAN, referring to the previous study for 16 weeks after baseline abdominal aorta ultrasound imaging recording [13]. Mice were weighed at study entry and weekly thereafter. Food intake was calculated weekly. The study population started out with 87 mice. The mice were randomly divided into SCD (n =44) and AD (n=43) groups; then those groups were randomly subdivided into groups without and with CCS treatment; finally, 23 SCD mice without CCS, 21 SCD mice with CCS, 22 AD mice without CCS, and 21 AD mice with CCS were used in the present study.
CCS protocol in conscious mice
CCS was performed during the last month of the study. It consisted of 5 stressors (physical and psychosocial stressors) at different stress exposure levels presented in random order, similar to stressors that develop in humans, was produced with reference to previous studies [14,15]. In brief, these included restraint stress (physical stressor): mice were placed for a certain period of time in individual vented plexiglass restrainers without tail movement limitation; conspecific exposure (psychosocial stressor): two mice (each from a different cage) were placed together in a new cage with clean bedding devoid of signals (i.e. urine scent) of previously established dominance; water avoidance (unpredictable mild stressor): a clean empty cage was drenched with 0.3-cm depth of cold water (20–22°C), and a 1-cm-high, 3-cm-diameter platform was provided as a dry environment; damp bedding (unpredictable mild stressor): a clean empty cage was drenched with 0.3-cm depth of cold water (20–22°C), where mice stayed during the stress; cage tilting (unpredictable mild stressor): cage inclination at 20 degrees, where mice stayed during the stress. To avoid habituation, stressors were assigned in random order with one stressor per day for a duration of 6-h each from a.m. 08:30 (-10:00) to p.m. 14:30 (-16:00) for 5 consecutive days (from Monday to Friday) followed by 2 days of rest (Saturday and Sunday) during the last month of the study. With the exception of the CCS, identical procedures including water and food deprivation were performed on the mice without CCS during the same time period.
Blood and tissue collection
At the end of the study, mice were anesthetized with 0.5 to 1% isoflurane in oxygen, and then blood samples were collected from the left ventricle; after systemic perfusion with isotonic saline via the left ventricle, the adrenal gland and aorta (from the aortic arch to the iliac bifurcation) were harvested. One side of the adrenal gland and a part of abdominal aorta tissue pieces were completely submerged in the collection vessel containing RNA later RNA stabilization reagent (Qiagen, Tokyo, Japan); this tissue was incubated overnight at 4°C and then removed from the reagent and transferred to –80°C for storage until the real-time reverse transcriptase polymerase chain reaction (RT-PCR) analysis, and aortic protein concentration and triglyceride level measurements. The remaining adrenal gland and aorta tissue were fixed in 4% paraformaldehyde in 0.1-M phosphate-buffered saline (PBS), then washed in 15% sucrose, and finally stored at 4 °C until further analysis. The blood sample was collected in a tube with heparin and centrifuged at 15,000 × g for 20 minutes at 4°C immediately after collection to obtain the plasma, then was stored at -80 °C until further analysis.
Aortic protein and triglyceride level measurements
The aortic samples were homogenized with lysis buffer (Cell Signal Technology Inc., Danvers, MA) and centrifuged at 10,000 rpm (9,170 × g) for 10 minutes at 4°C. The resultant supernatants were used as the total aortic protein for the aortic protein concentration and aortic triglyceride level measurements. The protein concentrations were determined by Bradford’s method, and the aortic triglyceride level were measured with an automatic analyzer (FDC4000; Fuji Medical Systems, Tokyo, Japan).
Histopathological and fluorescence immunohistochemical examination
Three-μm-thick sections of mice aortae (2 fractions of descending aorta and 2 fractions of abdominal aorta) were processed for hematoxylin and eosin (HE) staining, and 8-μm-thick frozen sections of mice aortae (2 fractions of descending aorta and 2 fractions of abdominal aorta) were processed for Oil Red O staining to quantitatively evaluate the mean aortic area in HE and the max Oil-Red-O content in the Oil Red O staining using the computerized microscope system (BZ-II VIEWER and BZ-II ANALYZER 2007; Keyence Corporation, Osaka, Japan) at 10 × magnification.
For immunofluorescence staining, 8-μm-thick frozen sections of mouse adrenal glands embedded in 4% paraformaldehyde in 0.1-M PBS were cut by cryostat (Leica CM1850), then incubated with the primary antibodies of NR3C1 (H-300, rabbit polyclonal; Santa Cruz Biotechnology) and phenyl ethanolamine-N-methyltransferase (PNMT, ab167427, rabbit monoclonal; Abcam, Cambridge, UK), and further incubated with a Cy3-labeled anti-rabbit IgG (AP182C, Chemicon) diluted at 1:1,000. Nuclei were stained with TO-PRO®-3 iodide (Molecular Probes, Carlsbad, CA). Finally, sections were observed under confocal laser scanning microscopy (LSM5 Pascal/Version 3.2; Carl Zeiss Microlmaging Co. Ltd., Zeiss, Germany). The total number of cells (number of nuclei) and the total number of immuno-positive cells (immunofluorescence-expressing cells which include nuclei) were each counted in 10 consecutive regions under a confocal microscope at 20 × magnification. The ratio of each value was calculated as a percentage of immune-reaction-positive cells.
Ultrasound bio microscopy protocol
Ultrasound bio microscopy was performed on a VisualSonics Vevo 660TM Micro-Ultrasound system (Toronto, Canada) with a transducer frequency of 55 MHz using isoflurane in oxygen anesthesia at the baseline and the end of the study. Following the procedure in a previous study in mice, the maximum intima media thickness (max IMT) and plaque were evaluated [16]. The abdominal aorta lesions were visualized in both long-axis and short-axis views and recorded by B-mode and M-mode ultrasound. Plaques were defined as hyperechoic or hypoechoic plaque that showed higher or lower echogenicity when compared to their adventitia, according to a previous study in humans [17]. Hypoechoic plaques are unstable with pronounced inflammatory infiltration, high lipid content, and prone to rupture while hyperechoic plaques are stable with an abundance of fibrous tissue and calcium [see review in reference 17]. The cine loops of B-mode and M-mode images were saved and max IMT was measured off-line using the built-in software of the Visual Sonics Vevo 660TM Micro-Ultrasound system.
Plasma analysis
Plasma was analyzed with an automatic analyzer (FDC4000i; Fuji Medical Systems, Tokyo, Japan) for blood total cholesterol (TCHO) level, with noradrenaline research ELISA kit (BA E-5200, LDN, Nordhorn, Germany) for blood noradrenaline level according to the manufacturer’s instructions.
Real-time reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from the abdominal aorta tissue using the RNeasy® Fibrous Tissue Mini Kit (Qiagen, Tokyo, Japan) and from the adrenal gland using RNeasy Mini Kit (Qiagen, Tokyo, Japan). ReverTra Ace® qPCR RT Master Mix with gDNA Remover was used to remove gDNA and to synthesize cDNA according to the manufacturer’s instructions. RT-PCR was performed for the quantitative assessment of mRNA expression using an Applied Biosystems StepOne™ system (Applied Biosystems, Foster City, CA, USA). Assays for gene expression included oxidized low density lipoprotein (lectin-like) receptor 1 (ID: Mm00454588_g1, NCBI Reference Sequence: NM_138648.2, NM_001301094.1, and NM_001301096.1; gene symbol: Olr1), nitric oxide synthase 3, endothelial cell (ID: Mm00435204_m1, NCBI Reference Sequence: NM_008713.4; gene symbol: Nos3), syndecan1 (ID: Mm00448920_g1, NCBI Reference Sequence: NM_011519.2; gene symbol: Sdc1), thrombomodulin (ID: Mm00437014_s1, NCBI Reference Sequence: NM_009378.3, gene symbol: Thbd), tumor necrosis factor (ID: Mm99999068_m1, NCBI Reference Sequence: NM_013693.3; gene symbol: Tnf-α), nuclear factor of kappa light polypeptide gene enhancer in B cells 1, p105 (ID: Mm00476361_m1, NCBI Reference Sequence: NM_008689.2; gene symbol: Nfkb1), nuclear receptor subfamily 3, group C, member 1 (ID: Mm00433832_m1, NCBI Reference Sequence: NM_008173.3; gene symbol: Nr3c1), phenylethanolamine-N-methyltransferase (ID: Mm00476993_m1, NCBI Reference Sequence: NM_008890.1, gene symbol: Pnmt), and platelet/endothelial cell adhesion molecule 1 (ID: Mm01246167_m1, GenBank: BC085502.1 and BC008519.1; gene symbol: Pecam1); all were purchased from Applied Biosystems. The relative expressions of the Olr1, Sdc1, Thbd, Nos3, Nr3c1, Pnmt, Nfkb1, Tnf-α, and Pecam1 mRNAs were normalized to the amount of glyceraldehyde-3-phosphate dehydrogenase (ID: Mm99999915_g1, NCBI Reference Sequence: NM_001289726.1 and NM_008084.3; Gene symbol: Gapdh) mRNA in an identical cDNA sample, using the comparative quantitative method recommended by the manufacturer; the relative expression was expressed as the fold change from the values of SCD without CCS. Nos3, Sdc1, and Thbd were used as markers of endothelial damage with reference to methods in previous studies [18,19].
Flow cytometry analysis of leukocyte accumulation within mice aortae isolation of mice aorta
At the end of the study a part of mice were anesthetized with 0.5 to 1% isoflurane in oxygen, and the whole aorta (from the aortic arch to the iliac bifurcation) was harvested after systemic perfusion with Hanks’ balanced salt solution (HBSS) via the left ventricle. Adipose tissues and para-aortic lymph nodes were carefully dissected away from the aorta. The isolated aorta was placed in a collection tube with HBSS.
Preparation of single cell suspensions
An aorta dissociation enzyme stock solution (ADESS) was created using a tumor dissociation kit for mice (130-096-730, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany). In brief, 100 μL Enzyme D, 50 μL Enzyme R, and 12.5 μL Enzyme A were set to 1.5 mL with 1 % BSA-10 mM HEPES-HBSS buffer in gentleMACSTM C tube (130-093-237, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) and the aorta was incubated at 37°C. The whole aorta was removed from the HBSS to the gentleMACSTM C tube containing the prepared ADESS. The aortae were cut into small pieces and homogenized by crushing using the gentleMACS™ Dissociator (Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) after being incubated with the ADESS for 30 minutes at 37°C. This procedure was repeated a total of three times; then, 10 mL autoMACSTM running buffer (130-091-221, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) was added, and the homogenate was passed through a pre-separation filter, 70 μm (130-095-823, Miltenyi Biotec Gmbh Inc. Bergisch Glabdach, Germany) into 15 mL tubes. The cells were pelleted by centrifugation (300 × g, 10 minutes, and 4°C) to prepare single-cell suspensions from the total aorta.
Stain aortic cell suspensions with CD45-PerCP antibody
Up to 106 nucleated cells were suspended with 45 μL of autoMACSTM Running Buffer, and 5 μL of CD45-PerCP antibody (common leukocyte antigen marker) was added, followed by thorough mixing and incubation for 10 minutes at 4°C in the dark. Cells were washed by adding 1 mL of autoMACSTM Running Buffer and centrifuged at 300 × g for 10 minutes. Supernatant was aspirated completely. The cell pellet was resuspended in a suitable amount (0.5 mL) of autoMACSTM Running Buffer and 5 μL PI (Live/Dead cell viability staining). Samples were run on a flow cytometer (Cytomics FC500, Beckman Coulter, CA).
Statistical analysis
Continuous data are expressed as means ± standard deviations (SDs). Extreme values were excluded by the Smirnov-Grubbs test. Sample size (numbers of mice) for group each had been determined in the experimental design before the study. However, the numbers of mice introduced to each analysis in group each were also include the excluded numbers with Smirnov-Grubbs test, the discarded numbers due to sudden death during the CCS, and the number used for reproducibility experiment resulted in the numbers of mice in each group were not the same finally. The plaque formation in the abdominal aorta was evaluated by the contingency table (m x n) chi-square test followed Bonferroni post hoc test among 4 groups, and deference of hypoechoic plaque formation in the abdominal aorta between without and with CCS in mice fed AD was evaluated by contingency table (2x2) chi-square test only. Continuous-variable comparisons between 2 groups (before and during the CCS) were tested by paired t test, while those continuous-variable comparisons among 4 groups were tested by 1-way analysis of variance (ANOVA). When an F-value was found to be significant by ANOVA, a Bonferroni/Dunn post hoc test was used for multiple comparisons. Statistical analysis was performed using Statcel2 for Windows software (OMS Publishing, Inc., Saitama, Japan). Value of p < 0.05 was considered statistically significant.
Results
Plasma TCHO, body weight, and consumption of nutrients and calories
Mice were discarded due to sudden death (3 of SCD and 2 of AD) and movement disorder (1 of SCD) during CCS; eventually, 40 SCD mice (23 without and 17 with CCS) and 41 AD mice (22 without and 19 with CCS) were analyzed in the present study. We used an atherogenic diet (AD) of 12% carbohydrate, 52% protein, 21% fat (445 kcal/100g diet) or a standard chow diet (SCD) of 56% carbohydrate, 23% protein, 5% fat (362 kcal/100g diet).
The plasma TCHO (Figure 1A) was significantly increased in mice fed the AD without CCS, while CCS suppressed this increase in mice fed the same AD (Figure 1A) at the end of the study.
Before CCS (after 3 months SCD or AD breeding) BW (Figure 1B) showed significantly increases in mice fed the AD (both of without and with CCS mice) as compared to mice fed the SCD (both of without and with CCS mice). At the end of the study, BW (Figure 1B) also showed significantly increases in mice fed the AD (both of without and with CCS mice) as compared to mice fed the SCD (both of without and with CCS mice), however CCS suppressed this AD-induced BW increment in mice fed AD with CCS. In contrast, CCS showed no significant effects on BW in mice fed SCD. These results were accompanied with the high consumption of protein (Figure 1C), fat (Figure 1D), and calorie (Figure 1F), while the low carbohydrate consumption (Figure 1E) both before and during the CCS in mice fed the AD (both of without and with CCS mice) as compared to mice fed the SCD (both of without and with CCS mice). But compared to them of before CCS, mice fed AD with CCS showed the suppressed trend of protein, fat, carbohydrate, and calorie consumption (the statistical analysis found no difference, p = 0.0567 in all); in contrast to these, the mice fed SCD with CCS showed the increased consumption of them (Figure 1C-1F, p < 0.05 in all).
The results indicated that AD diet produced hyperlipidemia and greater body weight increase; however, AD mice undergoing CCS lost body weight and suppressed the elevation in blood TCHO at the end of the study due to the comprehensive suppressed consumption of nutrients and calories during the stress treatment.
Max IMT and plaque formation in the abdominal aorta
Increased max IMT (Figure 2A and 2C) and plaque formation (Figure 2B) were shown in SCD with CCS, AD without CCS, and AD with CCS group mice as compared to SCD without CCS mice, and the highest max IMT and hyperechoic plaque formation were shown in stressed mice fed AD. Hyperechoic formation were 4.5% (SCD without CCS), 73% (SCD with CCS), 38% (AD without CCS), and 100% (AD with CCS) respectively. Moreover, hypoechoic plaques, the unstable plaques, only appeared in the mice fed the AD diet, and CCS increased this hypoechoic plaque formation (8 of 14 (57%) in mice with CCS vs. 2 of 21 (10%) in mice without CCS, χ2 = 9.3, p < 0.05).
Pathology phenotype of atherosclerosis based on mean aortic wall area, max aortic Oil-Red-O content and aortic triglyceride level, and Pecam1- and Olr1-mediated leukocyte accumulation in the whole aortic cell suspension
Five mice were processed for HE is staining each group, however, 1 of SCD without CCS and 1 of AD without CCS were discarded due to the unsuccessful tissue slice preparation; Both AD and CCS induced an increase in the mean aortic wall area, evaluated by HE is staining, compared to the SCD mice without CCS (Figure 3A and 3B).
Five mice were processed for Oil Red O staining each group, however, 1 of SCD without CCS, 1 of SCD with CCS, and 1 of AD with CCS were discarded due to the unsuccessful tissue slice preparation; Both AD and CCS induced an increase in the max aortic Oil-Red-O content, evaluated by Oil Red O staining compared to the SCD mice without CCS, especially in AD mice with CCS (Figure 3A and 3C).
The elevated aortic triglyceride level was shown only in the AD mice with CCS compared to the SCD mice without CCS (Figure 3D), upward trend (without significant statistical difference) showed in mice fed SCD with CCS and in mice AD without CCS.
Flow cytometry was performed to analyze leukocytes within the mice aorta using CD45 staining in conjunction with Live/Dead viability dye (propidium iodide staining) in the present study. Six mice were processed for flow cytometry study each group, however, 2 of SCD without CCS and 1 of AD without CCS were discarded due to the extreme values during the statistical analysis. The percentage of aortic leukocytes in the whole aortic cell suspension (infiltration of CD45+leukocytes) was increased only in mice with CCS both the SCD- and AD-fed mice (Figure 3A and 3E).
PECAM-1 contributes to the pathogenesis of atherosclerosis through its ability to mediate leukocyte infiltration, and oxidized LDL has been shown to promote monocyte migration through cytokine-stimulated endothelial cells in vitro by a mechanism involving upregulation of endothelial cell PECAM-1 [see review in in reference [20]. In the present study, CCS, but not AD-induced hyperlipidemia, induced a significant up-regulation of Pecam1 and Orl1 (the receptor of oxidized LDL) mRNA expressions (Figure 3F and 3G).
Endothelial dysfunction
In the present study, the CCS-induced leukocyte infiltration was accompanied by endothelial dysfunction based on changed aortic gene expressions of Nos3 (Figure 4A), Thbd (Figure 4B), and Sdc1 (Figure 4C) in stressed mice, as evidenced by down-regulated Nos3 mRNA expression and the up-regulated Thbd and/or Sdc1 mRNA expressions in mice with CCS as compared to the mice fed SCD without CCS.
Tnf-α-related co-up-regulation of aortic Nr3c1 and Nfkb1 mRNA expressions was accompanied with leukocyte infiltration and endothelial dysfunction
CCS may induce leukocyte infiltration and endothelial dysfunction especially when combined with atherogenic diet at least in partly. These changes were accompanied by the significant up-regulation or upward trends (without statistical differences) of aortic Nr3c1 (Figure 5A), Nfkb1 (Figure 5B), and Tnf-α (Figure 5C) mRNA expressions in mice with CCS. The results indicated that Tnf-α related co-up-regulation of aortic Nr3c1 and Nfkb1 mRNA expressions may in part mediate the aortic inflammatory response and endothelial dysfunction during CCS, especially in AD mice with CCS.
Stress-induced adrenal hyperactivation
Stress leads to activation of stress-responsive organs, beginning with cortisol and epinephrine release into circulation. CCS-induced changes in adrenal gland function were therefore determined. As shown in Figure 6, CCS, especially in mice combined with atherogenic diet, activated the adrenal gland function as indicated by increases in Nr3c1 and Pnmt mRNA expressions (Figure 6A and 6B), the percentage of NR3C1- and PNMT-positive cells (Figure 6C-6E). CSS induced the increase in the blood noradrenaline concentrations in mice fed standard chow diet (Figure 6F); however, only the upward trend (without significant statistical difference) in AD mice with CCS. These results showed that CCS may activate hormone epinephrine via the Pnmt gene, which is glucocorticoid-regulated and similar to results of the repeated immobilization stress at least in partly [21,22].
Discussion
A new complex chronic mental stress (CCS) mouse model was used in the present study. This mouse model consisted of physical and psychosocial stressors of different types and intensities, presented in a random order similar to the stressors that develop in humans. Our data demonstrate that the process of atherosclerosis can be controlled by CCS in mic fed standard chow diet and the atherogenic diet. The synergy between CCS and atherogenic diet may also accelerate the atherosclerosis development in mice. CCS could induce endothelial dysfunction and excessive leukocyte accumulation at the aortic wall, leading to the process of atherosclerosis in mice. These effects may be modulated in part by CCS-induced hyperactivity of the adrenal gland and the proinflammatory cytokine (Tnf-α) related co-up-regulation of aortic Nr3c1 and Nfkb1 gene expressions.
Animal stress models for atherosclerosis study
Insight into the link between stress and atherosclerosis disease can be gained from studies in animals. Animal stress models are typically divided into physical stress models and psychological stress models. It is important for animal models to mimic the natural courses of disease and stress responses found in humans; unfortunately, stress in humans is complex, and the animal models of stress used in previous studies are very limited in complexity [see review in reference 6]. Referring to methods in previous studies, we used a chronic complex stress mouse model in the present study [6,14]. This mouse model consisted of the physical and psychosocial stressors of different types and intensities, presented in a random order, similar to what human’s experience. Restraint stress is a physical method of stress induction. A previous study reported that a restraint stress (120 min/day, 14 days) induced endothelial dysfunction by oxidative stress [23]. Conspecific exposure is a psychosocial stress that can abate social interaction and increase anxiety-derived behaviors similar to stress-induced psychopathology in humans. The social defeat test is associated with sympathetic hyperactivity and tachyarrhythmia in rat studies [see review in reference 24]. In a mouse study, the volume fraction of fibrosis was six folds larger than in control animals due to the appearance of more microscopic scarring [25]. Unpredictable chronic mild stresses: In a study with ApoE -/- mice, unpredictable chronic mild stress promoted plaque features associated with vulnerable lesions that cause myocardial infarction and stroke in humans [26]. Three kinds of unpredictable chronic mild stresses (water avoidance, damp bedding, and cage tilting) were used in the present study. Our chronic complex stress mouse model applied various stress modules in a random order, with one stressor per day for a duration of 6-h each for 5 consecutive days (from Monday to Friday) followed by 2 days of rest (Saturday and Sunday) during the last month of the study. CCS were shown to accelerate the process of atherosclerosis, suggesting that this chronic complex stress mouse model may be useful in gaining insight into the link between stress and atherosclerosis in humans.
CCS accelerated the development of atherosclerosis
The link between stress and cardiovascular diseases has not been clarified completely. Overreaction of the autonomic nervous system to a chronic or unexpected stressor can produce cardiovascular dysfunction. Previous studies have shown that acute restraint stress caused lethal bradycardia associated with the shift of cardiac sympathovagal balance toward sympathetic predominance in rats, and patients with major depressive disorder were shown to have extraordinarily high sympathetic nervous activity, including the sympathetic outflow to the heart [14,27]. In the present study, 5 kinds of stressful situations were used in the CCS project, and the effects of CCS on the progress of atherosclerosis, a major underlying cause of cardiovascular disease, were assessed. Our data suggest that CCS may have accelerated the progress of atherosclerosis (Figure 2) along with the leukocyte overresponses (Figure 3A and 3E) and aortic endothelial dysfunction (Figure 4) in mice. These results were associated with overreaction of the autonomic nervous system based on the increased secretion of adrenal PNMT and plasma noradrenaline concentration (Figure 6C, 6E, and 6F), and activation of the HPA axis as evidenced by increases in secretion of adrenal NR3C1 (Figure 6C and 6D). Our results indicate that the HPA axis and the autonomic nervous system, the first two responders to stressful situations, play a role in CCS-accelerated development of atherosclerosis.
CCS may via Tnf-α–related co-up-regulation of Nr3c1 and Nfkb1 mRNA expressions induced endothelial dysfunction and excessive leukocyte accumulation in aortic wall especially when combined with atherogenic diet
The vascular endothelium plays an important role in the initiation and development of atherosclerotic diseases. The primary role of the endothelium is to adjust vascular function by producing NO, which decreases vascular resistance, inhibits platelet adhesion, and decreases vascular smooth muscle cell proliferation [28]. Stress activates the HPA axis, leading to increased secretion of adrenal GCs [see review in reference 12], and the actions of GCs are mediated through the glucocorticoid receptor (GR), a transcription factor that belongs to the nuclear receptor superfamily (NR3C1). Incubation of human endothelial cells with cortisol reduced NOS3 mRNA and protein to 60–70% of control expression levels, and these responses were prevented by the glucocorticoid receptor antagonist mifepristone [29]. The sugar-protein glycocalyx coats the vascular endothelium. Damage to the glycocalyx in the vascular endothelium causes impaired endothelial barrier function and an increase in platelet and leucocyte adhesion and shear stress in the vessel [30]. Higher levels of syndecan-1 (Sdc-1, a biomarker of endothelial glycocalyx damage) were observed during acute coronary syndrome, suggesting that damage to endothelial glycocalyx may participate in the atherosclerotic plaque vulnerability process in patients [31]. Thrombomodulin, an endothelial membrane glycoprotein, is an essential part of the protein C anti-coagulant pathway and has a role in the regulation of fibrinolysis [32]. Our results are consistent with previous studies showing that CCS-induced up-regulations of Nr3c1 mRNA both in the adrenal gland and aorta were accompanied by the down-regulation of aortic Nos3 mRNA expression. Furthermore, CCS-induced up-regulation of Nr3c1 mRNA was also accompanied with the up-regulation of Sdc1 and Thbd mRNA expressions, suggesting impaired endothelial barrier function and Thbd-mediated fibrinolysis regulation. In addition, binding of activated NR3C1 on glucocorticoid-responsive elements and interaction with NF-КB are the main mechanisms of regulation associated with glucocorticoid-mediated transactivation and trans repression [33].
NF-КB is a family of constitutively expressed transcription factors that impact cell growth, proliferation, and development, and inflammatory and immune responses [see review in reference 34]. The NF-КB pathway has been considered as a prototypical proinflammatory signaling pathway due to the role of NF-КB in the expression of proinflammatory genes including cytokines, chemokines, and adhesion molecules. In response to diverse internal and external stressors, NF-КB is response to inflammatory stimuli, such as the proinflammatory cytokine TNF-α signaling, and the activated NF-КB also drives proinflammatory cytokines, chemokines, receptors, and adhesion molecules productions including TNF-α [34,35]. Previous studies reported that chronic mild stress increased the circulating level of TNF-α in rats, and a higher level of TNF-α occurs in school teachers who are under a greater degree of psychological stress [36,37]. In addition to this study, several findings have demonstrated that anti-TNF-α therapies may improve endothelial dysfunction in various pathophysiological conditions [38,39]. These findings are consistent with our results. We found that CCS-induced Nos3 mRNA down-regulation was accompanied by up-regulation of aortic Tnf-α, Nr3c1, and Nfkb1 mRNA expressions. Thus, CCS activates the HPA axis leading to increased production of NR3C1 both in the adrenal gland and aorta, increases aortic proinflammatory mediators (TNF-α in the present study), and results in the endothelial dysfunction observed in CCS.
Excessive leukocyte accumulation in the aortic wall contributes to the progress of atherosclerosis. Our results showed that CCS-induced endothelial dysfunction (Figure 4) was associated with excessive leukocyte accumulation (Figure 3A and 3E) in the aortic wall. Endothelial dysfunction and the subsequent inflammatory response induce production of adhesion molecules such as PECAM-1 and recruitment of immune cells such as leukocytes, which in turn perpetuate the vicious cycle of leukocyte infiltration, atherosclerotic plaque formation, and adverse thrombotic events. The process of leukocyte emigration begins with the generation of inflammatory mediators (such as TNF-α) and upregulation of endothelial cell adhesion molecules. These support the initial attachment of leukocytes to endothelial cells and their migration through junctions between adjacent endothelial cells in vessel walls, and the changes in levels of numerous endothelial cell junctional molecules including PECAM-1 [see review in reference 20]. The NF-КB signal transduction pathway in artery endothelial cells is primed for atherosclerotic lesion formation, and PECAM-1-deficient mice showed no detectable activation of NF-КB or evidence of downstream inflammatory genes in regions of disturbed flow [40,41]. Oxidized LDL upregulated PECAM-1 and downregulated VE-cadherin at the endothelial junctions, and then promoted monocyte entry by enhanced homophilic binding to monocyte PECAM-1, and by a disrupted junctional barrier, respectively [42]. Furthermore, previous studies showed that hyperinsulinemia accelerated atherosclerosis by directly enhancing neutrophil trans-endothelial migration via increased endothelial PECAM-1 expression [43,44]. An in vitro study showed that rapamycin, which can inhibit oxidized LDL accumulation in human umbilical vein endothelial cells, reduced mechanistic target of its phosphorylation and subsequently inhibit NF-κB activation and suppresses LOX-1 resulted in a reduction in oxidized LDL uptake in human umbilical vein endothelial cells [45]. The present study showed that the CCS-induced Pecam1 mRNA up-regulation was accompanied with Olr1 (the receptor of oxidized LDL) mRNA up-regulation in the mice aorta. Together with the previous studies, up-regulated aortic Pecam1 and Olr1 mRNA expressions during the progress of atherosclerosis may at least in part be responsible for the leukocyte accumulation in the aortic wall observed in CCS.
Atherogenic diet is also responsible for the development of atherosclerosis
In a study with male ApoE-/- mice, mice fed the atherogenic diet of 12% carbohydrate, 43% fat, 45% protein, and 0.15% cholesterol showed the more extensive progress in atherosclerosis than mice fed Western’ diet (43% carbohydrate, 42% fat, 15% protein, and 0.15% cholesterol), despite of the similar dietary fat and cholesterol content in the diet, and reduced weight gain [13]. This conclusion was evidenced by the significantly more atheroma and the larger cross-sectional plaque area at the level of the aortic valve. In the present study, wild type mice (C57BL/6j) fed AD of 12% carbohydrate, 52% protein, 21% fat also showed the more extensive progress in atherosclerosis, as evidenced by the increased max IMT and hypoechoic plaque formation, aortic mean area, aortic max Oil-Red-O content, and aortic triglyceride level (Figure 2 and 3), furthermore, CCS in mice fed the AD showed the highest max IMT, hypoechoic plaque formation, Oil-Red-O content, and triglyceride level in the four groups. Our results indicated that the synergy between CCS and AD may responsible for the atherosclerosis development in mice. However, the changes of the aortic Pecam1, Olr1, Nos3, Thbd, Sdc1, and Tnf-α at the message level are insufficient to address the synergy (between CCS and AD)-induced the atherosclerosis development completely, further study for the underlying mechanisms is necessary.
Limitations
The chronic complex stress (CCS) mice model used in the present study only induced mild atherosclerotic lesions in the wild type mice. In a recent study, a chronic immobilization stress (4 hours in an immobilization stress tube daily for 12 weeks) enhanced vascular endothelial senescence and atherosclerotic plaque growth in ApoE-/- mice [46]. To further investigate the impact of CCS and AD on atherosclerosis, an established atherosclerosis mice model regarding CCS and AD in an ApoE-/- and Ldlr-/- mice is necessary.
In the present study, descending and abdominal aorta were used; however, arteriosclerosis is relatively hard to develop in descending and abdominal aorta compared to aortic root. This limitation may be responsible at least in partly for the not evident changes of mean aorta wall areas evaluated by HE is staining images in mice with CCS, in contrast with other results, such as the max IMT (Figure 2A and 2C) and % of CD45+ cells in the whole aorta (Figure 3A and 3E). However, to investigate the full particulars of CCS and AD on atherosclerosis, all of the aortic fractions including the aortic root, outflow tract are essential, even the generic noninvasive ultrasound bio microscopy and flow cytometry methods were used.
In the present study, we evaluated the relationship between gene phenotypes and CCS-accelerated atherosclerosis development. However, CCS accelerated the process of atherosclerosis in mice are not only account for the gene phenotypes. The further molecular and biology study for the atherosclerosis-related cells in the aorta are necessary.
Conclusions
Psychological stress may lead to adverse atherosclerosis; however, the mechanisms have not been well understood. Nervous and humoral systems are activated with variations in the type and intensity of stressors, suggesting the importance of creating chronic complex stress animal models. The CCS used in the present study may constitute a better model of the kind of unexpected stressful stimuli that develop in human lives. CCS, especially when combined with an atherogenic diet, accelerated the process of atherosclerosis via endothelial dysfunction and excessive leukocyte accumulation despite a decrease in blood TCHO in mice. The hyperactivity of adrenal gland function, the co-up-regulation of aortic proinflammatory genes (Tnf-α, Pecam1, and Olr1) and Nfkb1 mRNA expressions may be involved in endothelial dysfunction and the activation of the aortic inflammatory response.
Acknowledgement
The authors would like to thank Kenichi Sasamoto (Miltenyibiotec Tokyo), Ihoko Fujimoto (Yamaguchi University), Yuki Tanaka (Yamaguchi University), and Fuka Haruse (Yamaguchi University) for technical assistance.
Funding and grants
This study was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (no. 25460869 to Jinyao Liu and no. 17K09265 to Jinyao Liu) for data collection and analysis.
Conflict of interest
Not declared
References
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, et al. (2012) Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation 125: e2-2e220. [Crossref]
- Moss JW, Ramji DP (2016) Nutraceutical therapies for atherosclerosis. Nat Rev Cardiol 13: 513-532. [Crossref]
- Strike PC, Steptoe A (2004) Psychosocial factors in the development of coronary artery disease. Prog Cardiovasc Dis 46: 337-347. [Crossref]
- Gullette EC, Blumenthal JA, Babyak M, Jiang W, Waugh RA, et al. (1997) Effects of mental stress on myocardial ischemia during daily life. JAMA 277: 1521-1526. [Crossref]
- Rosengren A, Hawken S, Ounpuu S, Sliwa K, Zubaid M, et al. (2004) Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case-control study. Lancet 364: 953-962. [Crossref]
- Golbidi S, Frisbee JC, Laher I (2015) Chronic stress impacts the cardiovascular system: animal models and clinical outcomes. Am J Physiol Heart Circ Physiol 308: H1476-H1498. [Crossref]
- Schaaf MJ, Cidlowski JA (2002) Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol 83: 37-48. [Crossref]
- Farrell C, O'Keane V (2016) Epigenetics and the glucocorticoid receptor: A review of the implications in depression. Psychiatry Res 242: 349-356. [Crossref]
- Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E (2010) The human glucocorticoid receptor: molecular basis of biologic function. Steroids 75: 1-12. [Crossref]
- Rao NA, McCalman MT, Moulos P, Francoijs KJ, Chatziioannou A, et al. (2011) Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res 21: 1404-1416. [Crossref]
- Pan W, Yu H, Huang S (2016) Resveratrol protects against TNF-α-induced injury in human umbilical endothelial cells through promoting Sirtuin-1-induced repression of NF-KB and p38 MAPK. PLoS One 11: e0147034. [Crossref]
- Gu HF, Tang CK, Yang YZ (2012) Psychological stress, immune response, and atherosclerosis. Atherosclerosis 223: 69-77. [Crossref]
- Foo SY, Heller ER, Wykrzykowska J, Sullivan CJ, Manning-Tobin JJ, et al. (2009) Vascular effects of a low-carbohydrate high-protein diet. Proc Natl Acad Sci U S A 106: 15418-15423. [Crossref]
- Liu J, Hakucho A, Liu X, Fujimiya T (2016) Acute restraint stress provokes sudden cardiac death in normotensive rats and enhances susceptibility to arrhythmogenic effects of adrenaline in spontaneously hypertensive rats. Leg Med (Tokyo) 21: 19-28. [Crossref]
- Michel C, Duclos M, Cabanac M, Richard D (2005) Chronic stress reduces body fat content in both obesity-prone and obesity-resistant strains of mice. Horm Behav 48: 172-179. [Crossref]
- Gan LM, Grönros J, Hägg U, Wikström J, Theodoropoulos C, et al. (2007) Non-invasive real-time imaging of atherosclerosis in mice using ultrasound bio microscopy. Atherosclerosis 190: 313-320. [Crossref]
- Schiano V, Sirico G, Giugliano G, Laurenzano E, Brevetti L, et al. (2012) Femoral plaque echogenicity and cardiovascular risk in claudicants. JACC Cardiovasc Imaging 5: 348-357. [Crossref]
- Larsen AM, Leinøe EB, Johansson PI, Birgens H, Ostrowski SR (2013) High syndecan-1 levels in acute myeloid leukemia are associated with bleeding, thrombocytopathy, endothelial cell damage, and leukocytosis. Leuk Res 37: 777-783. [Crossref]
- Larsen AM, Leinøe EB, Johansson PI, Larsen R, Wantzin P, et al. (2015) Haemostatic function and biomarkers of endothelial damage before and after platelet transfusion in patients with acute myeloid leukaemia. Transfus Med 25: 174-183. [Crossref]
- Woodfin A, Voisin MB, Nourshargh S (2007) PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol 27: 2514-2523. [Crossref]
- Tai TC, Claycomb R, Siddall BJ, Bell RA, Kvetnansky R, et al. (2007) Stress-induced changes in epinephrine expression in the adrenal medulla in vivo. J Neurochem 101: 1108-1118. [Crossref]
- Tai TC, Wong DL (2003) Protein kinase A and protein kinase C signaling pathway interaction in phenylethanolamine N-methyltransferase gene regulation. J Neurochem 85: 816-829. [Crossref]
- Chung IM, Kim YM, Yoo MH, Shin MK, Kim CK, et al. (2010) Immobilization stress induces endothelial dysfunction by oxidative stress via the activation of the angiotensin II/its type I receptor pathway. Atherosclerosis 213: 109-114. [Crossref]
- Sgoifo A, Koolhaas J, De Boer S, Musso E, Stilli D, et al. (1999) Social stress, autonomic neural activation, and cardiac activity in rats. Neurosci Biobehav Rev 23: 915-923. [Crossref]
- Costoli T, Bartolomucci A, Graiani G, Stilli D, Laviola G, et al. (2004) Effects of chronic psychosocial stress on cardiac autonomic responsiveness and myocardial structure in mice. Am J Physiol Heart Circ Physiol 286: H2133-H2140. [Crossref]
- Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, et al. (2014) Chronic variable stress activates hematopoietic stem cells. Nat Med 20: 754-758. [Crossref]
- Barton DA, Dawood T, Lambert EA, Esler MD, Haikerwal D, et al. (2007) Sympathetic activity in major depressive disorder: identifying those at increased cardiac risk? J Hypertens 25: 2117-2124. [Crossref]
- McMackin CJ, Vita JA (2005) Update on nitric oxide-dependent vasodilation in human subjects. Methods Enzymol 396: 541-553. [Crossref]
- Wallerath T, Witte K, Schäfer SC, Schwarz PM, Prellwitz W, et al. (1999) Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Natl Acad Sci U S A 96: 13357-13362. [Crossref]
- Alphonsus CS, Rodseth RN (2014) The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia 69: 777-784. [Crossref]
- Miranda CH, de Carvalho Borges M, Schmidt A, Marin-Neto JA, Pazin-Filho A (2016) Evaluation of the endothelial glycocalyx damage in patients with acute coronary syndrome. Atherosclerosis 247: 184-188. [Crossref]
- Salomaa V, Matei C, Aleksic N, Sansores-Garcia L, Folsom AR, et al. (2001) Cross-sectional association of soluble thrombomodulin with mild peripheral artery disease; the ARIC study. Atherosclerosis Risk in Communities. Atherosclerosis 157: 309-314. [Crossref]
- De Bosscher K, Vanden Berghe W, Haegeman G (2003) The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24: 488-522. [Crossref]
- Mitchell S, Vargas J, Hoffmann A (2016) Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med 8: 227-241. [Crossref]
- Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1: a001651. [Crossref]
- Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK (2005) Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav 84: 697-706. [Crossref]
- von Känel R, Bellingrath S, Kudielka BM (2008) Association between burnout and circulating levels of pro- and anti-inflammatory cytokines in schoolteachers. J Psychosom Res 65: 51-59. [Crossref]
- Hürlimann D, Forster A, Noll G, Enseleit F, Chenevard R, et al. (2002) Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation 106: 2184-2187. [Crossref]
- Fichtlscherer S, Rössig L, Breuer S, Vasa M, Dimmeler S, et al. (2001) Tumor necrosis factor antagonism with etanercept improves systemic endothelial vasoreactivity in patients with advanced heart failure. Circulation 104: 3023-3025. [Crossref]
- Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, et al. (2000) The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A 9: 9052-9057. [Crossref]
- Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, et al. (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426-431. [Crossref]
- Hashimoto K, Kataoka N, Nakamura E, Tsujioka K, Kajiya F (2007) Oxidized LDL specifically promotes the initiation of monocyte invasion during transendothelial migration with upregulated PECAM-1 and downregulated VE-cadherin on endothelial junctions. Atherosclerosis 194: e9-e17. [Crossref]
- Rattan V, Shen Y, Sultana C, Kumar D, Kalra VK (1996) Glucose-induced transmigration of monocytes is linked to phosphorylation of PECAM-1 in cultured endothelial cells. Am J Physiol 271: E711-E717. [Crossref]
- Okouchi M, Okayama N, Imai S, Omi H, Shimizu M, et al. (2002) High insulin enhances neutrophil transendothelial migration through increasing surface expression of platelet endothelial cell adhesion molecule-1 via activation of mitogen activated protein kinase. Diabetologia 45: 1449-1456. [Crossref]
- Zhou YD, Cao XQ, Liu ZH, Cao YJ, Liu CF, et al. (2016) Rapamycin Inhibits Oxidized Low Density Lipoprotein Uptake in Human Umbilical Vein Endothelial Cells via mTOR/NF-?B/LOX-1 Pathway. PLoS One 11: e0146777. [Crossref]
- Yang G, Lei Y, Inoue A, Piao L, Hu L, et al. (2017) Exenatide mitigated diet-induced vascular aging and atherosclerotic plaque growth in ApoE-deficient mice under chronic stress. Atherosclerosis 264: 1-10. [Crossref]