Abstract
Advances in nanotechnology have impacted the field of therapeutics delivery significantly. This is evidenced by the increase in the number of nanoparticle- based therapeutic products in development over the last two decades. A 2006 global survey conducted by the European Science and Technology Observatory (ESTO) revealed that more than 150 companies are developing nanoscale therapeutics, and 24 nanoparticles therapeutics are currently in clinical use. These drugs are being developed to treat a wide range of diseases, such as fungal or bacterial infections, HIV infections, diabetes and cancers. There are several advantages of using nanoparticles for therapeutics delivery. The use of materials on the nanoscale level provides unprecedented freedom to modify some of the most fundamental properties of therapeutic carriers, such as solubility, diffusivity, bio distribution, release characteristics and immunogenicity. Precise nanoparticle engineering has yielded longer circulation half-lives, superior bioavailability and lower toxicity. For example, the liposomal encapsulation of doxorubicin significantly reduces its most serious and dose-limiting side effect, cardiac toxicity.
Key words
Nanoparticles, biomedical, biological application
Introduction
Advances in nanotechnology have impacted the field of therapeutics delivery significantly. This is evidenced by the increase in the number of nanoparticle- based therapeutic products in development over the last two decades. A 2006 global survey conducted by the European Science and Technology Observatory (ESTO) revealed that more than 150 companies are developing nanoscale therapeutics, and 24 nanoparticle therapeutics are currently in clinical use [1]. These drugs are being developed to treat a wide range of diseases, such as fungal or bacterial infections, HIV infections, diabetes and cancers. There are several advantages of using nanoparticles for therapeutics delivery. The use of materials on the nanoscale level provides unprecedented freedom to modify some of the most fundamental properties of therapeutic carriers, such as solubility, diffusivity, bio distribution, release characteristics and immunogenicity. Precise nanoparticle engineering has yielded longer circulation half-lives, superior bioavailability and lower toxicity. For example, the liposomal encapsulation of doxorubicin significantly reduces its most serious and dose-limiting side effect, cardiac toxicity [2]. One strategy to further improve the therapeutic index of nanoparticle therapeutics is to functionalize nanoparticles with targeting ligands. The addition of targeting ligands allows the delivery of drug-encapsulated nanoparticles to uniquely identified sites while having minimal undesired effects elsewhere. Since biologically targeted nanoparticles have the potential to be the optimal drug delivery vehicles, there has been tremendous amount of interest in developing novel targeted nanoparticles for therapeutic applications. This chapter reviews recent advances in the development of biofunctionalized targeted nanoparticles. The authors discuss the existing nanoparticle platforms for therapeutic applications, targeting ligands that can be used to functionalize the nanoparticles, and the various targeted nanoparticles in development. Over the last several decades, numerous nanoparticle platforms have been studied for their use in therapeutic applications. These nanoparticle platforms include liposomes, polymer therapeutic conjugates, polymeric micelles, dendrimers, nanoshells and nucleic acid-based nanoparticles. The two dominant classes of nanoparticles, liposomes and polymer-drug conjugates, account for > 80% of the available nanoparticle therapeutics in clinical use. Liposomes have been used widely as pharmaceutical carriers in the past decade, with 11 formulations approved for clinical use and many more in clinical development [3]. Some of the commonly used therapeutics include liposomal amphotericin, liposomal doxorubicin and liposomal daunorubicin. Liposomes are spherical vesicles that contain a bilayered membrane structure composed of natural or synthetic amphiphilic lipid molecules. Their biocompatible and biodegradable composition, as well as their unique ability to encapsulate both hydrophilic and hydrophobic therapeutic agents, make liposomes excellent therapeutic carriers. Liposomes can also be coated with biocompatible and antibiofouling polymers, such as polyethylene glycol (PEG), to prolong their circulation half-life. The polymer coating of the liposomes can also be engineered to carry a functional group, which can be used for targeting ligand conjugation. Another nanoparticle drug delivery platform, polymer-drug conjugates, has been studied extensively [4]. Small molecule therapeutic agents and proteins usually have two unfavorable properties: short circulation half-life, leading to the need for frequent administration, and non-site-specific targeting, resulting in undesired systemic side effects. The conjugation of drugs to polymeric nanocarriers can reduce these undesirable adverse effects. Polymer-drug conjugates not only prolong the in vivo circulation time from several minutes to several hours but also reduce cellular uptake along the endocytic route. This characteristic enhances the passive delivery of drugs to tissues with leaky blood vessels, such as tumors and atherosclerotic plaques.
Many polymers have been proposed as drug delivery carriers but only a few, with linear architecture, have been used in clinic. The major challenges of most polymer-drug conjugates include polymer toxicity, immunogenicity, nonspecific biodistribution, in vivo circulation instability, low drug carrying capacity, rapid drug release and manufacturing challenges. PEG, which was first introduced into clinical use in the early 1990s, enhances plasma stability and drug solubility while reducing drug immunogenicity. There are currently six examples of PEG-drug conjugates in clinical use. In addition to PEG, other linear polymers such as polyglutamic acid, N-(2-hydroxypropyl) methacrylamide (HPMA), polysaccharide and poly(allylamine hydrochloride), have been harnessed as polymeric drug delivery carriers [5-8]. The polymers in the polymer-drug conjugates can be used for conjugation to a targeting ligand, in turn creating biologically targeted therapeutics. Dendrimers are well defined, regularly branched macromolecules that are 2.5-10 nm in size. They are synthesized from either synthetic or natural building blocks such as amino acids, sugars and nucleotides. The core of a dendrimer is denoted generation zero and each additional level of branching adds another generation. Dendrimers’ characteristics as carriers of therapeutics include nanoscale spherical architecture, narrow polydispersity, multifunctional surface chemistry and large surface area. Many dendrimer families have been reported. Among the families, the polyamidoamine (PAMAM) and poly(propylenemine) (PPI) families have been most widely used for biomedical applications [9].
The specific molecular structure of dendrimers enables them to carry various drugs through their multivalent surfaces by covalent conjugation or electrostatic adsorption. Alternatively, dendrimers can be loaded with drugs, by using the cavities in their cores through hydrophobic interaction, hydrogen bonding or chemical linkage. Their surface can be engineer to provide precise spacing of surface molecules and to conjugate targeting molecules. Biodegradable polymer nanoparticles have been extensively investigated as therapeutic carriers. They are generally formed by the self-assembly of copolymers consisting of two or more polymer blocks with different hydrophobicity. These copolymers spontaneously assemble into a core-shell micellar structure in an aqueous environment. Specifically, the hydrophobic blocks form the core to minimize their exposure to aqueous surroundings while the hydrophilic blocks form the corona-like shell to stabilize the core. This core-shell structure provides an ideal drug delivery nanocarrier. Its hydrophobic core is capable of carrying therapeutics with varying loading capacity (5-25% by weight). The hydrophilic shell not only provides a steric protection for the micelle but also provides functional groups for further particle surface modifications. Polymeric nanoparticles have been formulated to encapsulate either hydrophilic or hydrophobic small drug molecules, as well as macromolecules such as proteins and nucleic acids. The release of encapsulated drugs occurs at a controlled rate in a time or environment dependent manner. Furthermore, the rate of drug release can be controlled by modification of the polymer side chain, development of novel polymers or synthesis of copolymers. In general, these biodegradable polymer systems can provide drug levels at an optimum range over a longer period of time than other drug delivery methods, thus increasing the efficacy of the drug and maximizing patient compliance, while enhancing the ability to use highly toxic, poorly soluble or relatively unstable drugs. Poly(d,l -lactic acid), poly(d,l-glycolic acid), poly(ε-caprolactone), and their copolymers at various molar ratios diblocked or multiblocked with PEG are the most commonly used biodegradable polymers, while PEG is the polymer used most commonly to engineer the polymeric micelle surface.
For example, poly lactide-co-glycolide (PLGA) encapsulated antibiotics have been investigated for the treatment of tuberculosis using murine models [10]. Metallic nanoshells are characterized by a dielectric core coated with a thin metallic shell to improve their biocompatibility and optical absorption. These particles possess a highly tunable plasmon resonance mediated by the size of the core and the thickness of the shell, which in turn determines their absorbing and scattering properties over a broad range of the spectrum from the near-ultraviolet to the mid- infrared.
Gold nanoshells have been developed for in vivo photothermal therapy using near infrared light. Similarly, thermally sensitive polymeric hydrogels and optically active nanoshells have been developed for the purpose of photothermally modulated drug delivery. Nanoshell particles with a magnetic core (carbonyl iron) and a biodegradable poly(butylcyanoacrylate) (PBCA) shell have also been developed for controlled release of 5-fluorouracil [11]. In nucleic acid based nanoparticles, DNA and RNA macromolecules can be used as substrates for developing therapeutic and imaging nanocarriers. By rationally constructing nucleic acid chains that can yield shapes other than the traditional linear or circular shapes, researchers have been able to formulate novel nanoparticles using nucleic acids as building blocks. A multivalent DNA delivery vehicle, with an average size of 100 nm was recently reported for simultaneous targeted drug delivery, imaging and gene therapy [12]. Targeted multifunctional RNA nanoparticles (25-40 nm) have also been developed with a trivalent RNA core, RNA aptamers for targeting, and small interfering (si)RNAs for therapeutic effect [13]. Monoclonal antibodies (mAbs) have been the preferred class of targeting molecules for the last several decades. Artificially engineered mAbs have been used commonly for molecular targeting purposes. In order for the engineered antibodies to function in the human body, they have to evade the immune system. Current development of mAbs has thus been focused on chimeric, humanized and fully humanized derivatives to decrease their immunogenicity. The ability of engineered monoclonal antibodies to target disease processes has been demonstrated by the success of several monoclonal antibody therapeutics, including rituximab, trastuzumab, cetuximab and bevacizumab [14]. Despite the vast effort expended on their development, mAbs have their share of limitations. They are large, complex molecules that require significant engineering at the molecular level to be effective. They are expensive to manufacture and there is variation from batch to batch, which limits their efficiency as targeting molecules. Aptamers are small nucleic acid ligands that can bind to targets with high sensitivity and specificity. Aptamers fold by intra-molecular interaction into unique conformations with ligand binding characteristics. For a particular target, aptamers are selected through an in vitro process called systemic evolution of ligands by exponential enrichment (SELEX) [15].
This process uses the principles of evolution, where a library of 10-15 random oligonucleotides is enriched to identify those aptamers that can bind to the target with the highest affinity and specificity. Aptamers have potential advantages as targeting ligands. They are small in size (15 kDa), and generally have less immunogenicity which leads to better biodistribution. Most importantly, SELEX is a chemical process that can be scaled up with ease, without batch-to-batch variations and with lower costs. More than 200 aptamers have been isolated. For example, RNA aptamers to the VEGF 165 isoform with 2′-O -methylpurine and 2′-F pyrimidines have been reported [16]. It was found that VEGF aptamers not only can lead to regression of tumor vessels but the aptamers also exhibited a remarkable stability in plasma in monkeys. Pegaptanib, an aptamer targeted against VEGF 165, was approved by the FDA in December 2004 for the treatment of neovascular macular degeneration, underscoring the rapid progress of aptamers from their original conception to clinical application [17]. Aptamers’ major shortcomings are their low serum stability and their high production cost. Peptides are an attractive alternative targeting molecule due to their smaller size, lower immunogenicity, higher stability and ease of manufacture. The development of peptide phage libraries (10-11 different peptide sequences), bacterial peptide display libraries, plasmid peptide libraries, and new screening technologies have made their selection much easier, contributing to their popularity as targeting ligands. Peptides can also bind to their targets with high specificity and affinity. For example, cilengitide is a cyclic peptide that binds to integrins, which is currently in Phase II clinical trials for the treatment of non-small cell lung cancer and pancreatic cancer [18].
There are a handful of oligopeptides that are distinct in their characteristics. These include A-domain proteins, AdNectins, and affibodies. A-domain proteins are 40 amino acid oligopeptides that bind to cell surface through multiple points of attachment. The first A domain protein was found in the low-density lipoprotein receptor (LDLR) by Tschopp and Mollnes [19]. AdNectins represent another distinct type of peptides. They are thermostable and protease resistant oligopeptides that were initially derived from the 10FN3 domain of human fibronectin. Each AdNectin typically has three distinct loop structures. A large library of AdNectins has been created by introducing diversity into these loops. Recently, an AdNectin for human VEGF receptor 2 (VEGFR2) named Angiocept been isolated by AdNexux Pharmaceuticals, and entered Phase I clinical trials for treating advanced solid tumors and non-Hodgkin’s lymphoma in 2006. Affibodies are small polypeptides derived from an antibody binding domain of staphylococcal protein A. Affibodies targeted against a specific cell marker can be selected using phage-display technology. For example, a 6kDa affibody with selective binding to human EGF receptor 2 (HER2) was found to have subnanomolar affinity [20]. Due the limitations and challenges of using mAbs discussed earlier, there is increasing interest in using antibody fragments as targeting molecules while retaining the high antigen binding specificity of antibodies. These include the Fab fragments, single chain variable fragments (scFV), minibodies, diabodies and nanobodies [21]. The Fab fragment is composed of one constant and one variable domain of each of the heavy and the light chains; scFV is a fusion of the variable regions of the heavy and light chains. Minibodies are engineered antibody fragments that is a fusion between scFV and a C H -3 domain that self-assembles into a bivalent dimer [22]. Diabodies are covalently linked dimers or non-covalent dimers of scFvs. Nanobodies, which are the smallest of all fully functional antigen-binding fragments, evolved from the variable domain of heavy-chain antibodies. Nanobodies are typically evolved from single-domain antibodies, antibodies carrying only a functional heavy chain without the light chain. These antibody fragments are engineered to retain high affinity for target antigens but have less immunogenicity and a smaller size, and thus are better suited for molecular targeting.
Most recently, small antibody mimetics were formulated by Qiu et al. [23]. They fused two complementarity-determining regions that retained the antigen recognition of their parent molecules. These 3-kDa mimetics showed better biodistribution than their parent molecules, suggesting their potential as a new class of targeting ligands. Small molecules have shown great promise as a class of targeting molecules because of their small size and low cost of production. One of the most extensively studied small molecule targeting moieties in targeted drug delivery is folic acid (folate). The high-affinity vitamin folate is a commonly used ligand for cancer targeting because folate receptors (FRs) are frequently overexpressed on tumor cells. Folate specifically binds to FRs with a high affinity (K D = ∼ 10-9 M), enabling a variety of folate derivatives and conjugates to deliver molecular complexes to cancer cells without causing harm to normal cells [24]. It has been used as a targeting moiety combined with a wide array of drug delivery vehicles including liposomes, protein toxins, polymeric NPs, linear polymers, and dendrimers to selectively deliver drugs into cancer cells using FR-mediated endocytosis. Molecular targeting has been a key concept in recent years. Drugs such as trastuzumab, bevacizumab and rituximab have achieved great results that have eluded conventional therapeutics [25]. Most of the success can be attributed to targeting, as targeted therapeutics can selectively treat diseases without affecting normal tissue. There has been increasing interest in applying molecular targeting to nanoparticle therapeutics and formulating biofunctionalized targeted nanoparticles. Targeted nanoparticles, when compared with non-targeted nanoparticles, have several potential advantages: the ability to partition more of the nanoparticles within target tissue, increased uptake into target cells, higher therapeutic efficacy and lower toxicity. Although there are no clinically approved targeted nanoparticle therapeutics yet, many are in preclinical and clinical development. Almost all the combinations between the nanoparticle platforms and targeting ligands mentioned in the previous sections have been formulated. Data obtained from these targeted nanoparticles so far have supported their theoretical advantages. The most significant effect of functionalizing nanoparticles with targeting ligands is increased intracellular uptake by the target cells. Kim et al. showed that folate-targeted polymeric nanoparticles had more than 6.7 times more cell uptake than non-targeted nanoparticles [26]. Our own data showed that aptamer-targeted nanoparticles displayed a 77-fold increase in intracellular uptake by prostate cancer cells in vitro when compared with non-targeted nanoparticles [27]. In a separate study, we demonstrated that by varying the targeting ligand density on the nanoparticle surface (0-10%), targeted nanoparticles had more than seven times the intracellular uptake of the non- targeted nanoparticles after two hours of incubation [28]. Oyewumi et al. also studied uptake of folate targeted polymeric nanoparticles into KB cells. Folate coated nanoparticles showed higher uptake in comparison to PEG-coated nanoparticles. At a nanoparticle concentration of 180 mg/ml, KB cell uptake of folate coated nanoparticles was 20-fold higher than that of non-targeted nanoparticles [29].
Kirpotin et al. formulated anti-HER2 mAb-liposome conjugates to study tumor targeting, where targeted liposomes had a sixfold higher intracellular uptake when compared with non-targeted nanoparticles. Biofunctionalized targeted nanoparticles also preferentially accumulate in tumors when compared with non-targeted nanoparticles. Kukowska-Latallo et al. demonstrated that folate targeted PAMAM dendritic polymers concentrated in KB tumor xenograft in SCID mice over 4 days [30]. Our own experience also showed a higher concentration of targeted nanoparticles in tumors when compared with that of non-targeted nanoparticles. We studied the bio distribution of aptamer targeted polymeric nanoparticles and non-targeted nanoparticles. Tumor accumulation for targeted nanoparticles was 2.5 times higher than that of non- targeted nanoparticles. On the other hand, Kirpotin et al. showed that antibody- targeted lipidic nanoparticles did not increase tumor localization but did increase internationalization in tumor cells (sixfold) [31]. The tumor accumulation of targeted nanoparticles is highly dependent on the characteristics of the targeting ligands and the nanoparticles. As more in vivo studies on targeted nanoparticles are reported, we will obtain more information on the factors determining tumor localizations. Targeted delivery of therapeutics has also been shown to achieve greater efficacy. Park et al. studied anti-HER2 immunoliposomes encapsulating doxorubicin in tumor xenograft models. They demonstrated that in four different xenograft models, immunoliposome-doxdoxorubicin was significantly superior to free doxorubicin, liposomal doxorubicin, and anti-HER2 monoclonal antibody [32]. Bartlett et al. showed that transferrin-targeted siRNA nanoparticles are more effective than non-targeted siRNA nanoparticles despite the similar biodistribution and tumor accumulation of the two nanoparticles. Using mouse xenograft tumors expressing luciferase and siRNA against luciferase, they showed that transferring targeted nanoparticles reduced luciferase activity to 50% of the levels with non- targeted nanoparticles. Increased efficacy was also seen in the Kukowska-Latallo study. Folate targeted methotrexate (MTX) lead to statistically slower tumor growth compared with non-targeted MTX [33]. In our own experience, we have developed aptamer-targeted nanoparticles (NP-Apt) that target the prostate specific membrane antigen (PSMA) on prostate cancers. Using NP-Apts encapsulating docetaxel and a murine xenograft model of prostate cancer, it was demonstrated that the targeted nanoparticles effectively decreased tumor size following a single intra-tumor injection while non-targeted nanoparticles did not [34].
Biomedical applications of gold nanoparticles functionalized using hetero-bifunctional poly(ethylene glycol) spacer
Nanotechnology offers unique approaches to probe and control a variety of biological and medical processes that occur at nanometer length scales, and is expected to have a revolutionary impact on biology and medicine. Among the approaches for exploiting nanotechnology in medicine, nanoparticles offer some unique advantages as sensing, image enhancement, and delivery agents. Several varieties of nanoparticles with biomedical relevance are available including, polymeric nanoparticles, metal nanoparticles, liposomes, micelles, quantum dots, dendrimers, and nanoassemblies. To further the application of nanoparticles in disease diagnosis and therapy, it is important that the systems are biocompatible and capable of being functionalized for recognition of specific target sites in the body after systemic administration.
Nanoparticles based on gold chemistry have attracted significant research and practical attention recently. They are versatile agents with a variety of biomedical applications including use in highly sensitive diagnostic assays, thermal ablation and radiotherapy enhancement, as well as drug and gene delivery. For instance, antibody-modified gold nanoparticles when used for detection of prostate specific antigen, had an almost a million-fold higher sensitivity that conventional ELISA- based assay. Near-infrared radiation absorbing gold-silica nanoshells have been prepared and evaluated for thermal ablation of tumors after systemic administration [35].
For biomedical applications, surface functionalization of gold nanoparticles is essential in order to target them to specific disease areas and allow them to selectively interact with cells or biomolecules. Surface conjugation of antibodies and other targeting moieties is usually achieved by adsorption of the ligand to the gold surface. Surface adsorption, however, can denature the proteins or, in some cases, limit the interactions of the ligand with the target on the cell surface due to steric hinderance. Additionally, for systemic applications, long-circulating nanoparticles are desired for passive targeting to tumors and inflammatory sites. Poly(ethylene glycol) (PEG)-modification of nanoparticles affords long circulating property by evading macrophage-mediated uptake and removal from the systemic circulation[36]. Surface modification of gold nanoparticles through the PEG spacer would, therefore, allow the modified nanoparticles to remain in the systemic circulation for prolonged period and provide flexibility to the attached ligand for efficient interaction with its target.
The research describes the synthesis of hetero-bifunctional PEG with a thiol group on one terminus and a reactive functional group on the other for conjugation to a biologically-relevant targeting moiety. Using thiol-PEG-coumarin, a model fluorescent dye, describe the “proof of concept” for surface functionalization of gold nanoparticle, cellular cytotoxicity evaluations, and fluorescence confocal analysis of cell uptake and nanoparticle distribution.
Magnetic nanoparticles for biomedical NMR-based diagnostics
Rapid and sensitive measurement of clinically relevant biomarkers, pathogens and cells in biological samples would be invaluable for disease diagnosis, monitoring of malignancy, and for evaluating therapy efficacy in personalized medicine. To translate such molecular measurements into clinical settings, however, an assay would need to 1) provide high sensitivity and specificity, 2) minimize sample preparation and sample volume, and 3) ideally allow concurrent detection of diverse target moieties through multiplexed measurements. Biosensing strategies based on magnetic nanoparticles (MNPs) have recently received considerable attention, since they offer unique advantages over traditional detection methods. Specifically, because biological samples exhibit negligible magnetic background, MNPs can be used to obtain highly sensitive measurements in turbid samples with reduced sample preparation. In contrast, traditional detection strategies based on optical techniques, for example, are often affected by scattering, absorption, autofluorescence, and require extensive sample purification before measurements can be made. To detect biomarkers using MNPs, several technologies have been developed. These include techniques that use magnetometers, such as superconducting quantum interference device (SQUID), magnetoresistive sensors, and Hall sensors, which directly measure the magnetic fields from magnetically- labeled biological targets. Another technology that has achieved considerable success is diagnostic magnetic resonance (DMR). Based on nuclear magnetic resonance (NMR) as the detection mechanism, DMR exploits MNPs as proximity sensors, which modulate the spin–spin relaxation time of water molecules adjacent to the molecularly-targeted MNPs. The latter create a local magnetic field and induce a change in proton relaxation rate in billions of neighboring water molecules. Direct detection of magnetic moments with magnetometers requires MNP-labeled targets to be closely positioned to the sensing elements. DMR assays, however, are faster and simpler since the analytical signal is generated from the entire sample volume.
By developing optimized MNPs, DMR detection sensitivities for various target moieties have been considerably improved. To date, numerous magnetic biosensors have been designed to identify and quantify a wide range of targets including DNA/mRNA, proteins, small molecules/drugs, bacteria, and tumor cells [37]. More recently, the development of miniaturized, chip based NMR detector systems has served to further enhance DMR technology. Such detectors can perform highly sensitive measurements on microliter sample volumes and in a multiplexed format. With the integration of key components (i.e., microcoils, microfluidic networks, NMR electronics, and a portable magnet), the DMR systems have now demonstrated their potential for portable, sensitive and rapid operation in a point-of-care setting have tremendous potential in the field of biomedical applications, primarily on account of their similar size comparable to biological molecules, and because their properties can be fine-tuned during chemical synthesis. In particular, MNPs can be synthesized in such a way as to possess unique superparamagnetic properties, to be biocompatible, and to remain inert with respect to cells and molecules of interest. As the size of magnetic objects shrinks to the nanometer scale, it becomes energetically more favorable for them to have a single magnetic domain than to form domain walls and a consequent multidomain structure. The upper limit for a single domain [~(A/2K)½] is determined by the material properties: the exchange stiffness (A) and the anisotropy constant (K). For most magnetic materials (e.g., ferrite and iron), MNPs with a diameter <20 nm will have a single domain with magnetic moments aligned in a particular direction defined by magnetic anisotropy. At sufficiently high temperatures (above blocking temperature), thermal energy can induce free rotation of the magnetic moment. Thus, when MNPs are grouped together, they display a form of paramagnetic behavior, known as superparamagnetism: MNPs assume overall magnetic moments when placed in an external magnetic field but lose their moments when the field is removed. Distinct from paramagnetism, which arises from individual spins at the atomic or molecular level, superparamagnetism applies to magnetic elements that already assume a magnetically-ordered spin state (typically ferromagnetic or ferrimagnetic). This superparamagnetic property enables MNPs to avoid spontaneous aggregation in solution, a feature that makes them suitable for many biomedical applications. In its simplest form, an MNP is comprised of an inorganic magnetic core and a biocompatible surface coating that stabilizes the particle in physiological conditions. By applying suitable surface chemistry, functional ligands can be integrated and confer the MNP with molecular specificity.
Synthetic methods for MNPs have been recently reviewed [38]. A variety of chemical methods, ranging from traditional wet chemistry to high-temperature thermal decomposition, have been employed to synthesize MNPs. Colloidal iron oxide nanoparticles, which are used as clinical magnetic resonance imaging (MRI) contrast agents, are generally prepared via an aqueous co-precipitation method. During these hydrolytic processes, control of solution pH and the addition of suitable coating surfactants are critical for regulating the nanoparticle size as well as the magnetic properties [39]. Unfortunately, depending on the synthesis procedure used, magnetization can vary significantly among nanoparticles of similar sizes. More recently, high quality MNPs have been prepared through thermal decomposition of organometallic precursors, in nonhydrolytic organic solutions containing surfactants. Monomers are generated via high-temperature thermal decomposition of precursors. Above a supersaturation level, these monomers then aggregate to induce nucleation and nanoparticle growth. By tuning the growth conditions during this procedure (such as precursor choice, monomer concentration, growth temperature and time), it is possible to control the size, composition, and crystallinity of the nanoparticles. While high-temperature decomposition markedly improves size control, size distribution and crystallinity of MNPs, the resulting particles are encased in a hydrophobic coating. In order to achieve nanoparticle stability in aqueous media, this approach requires additional modifications. Techniques, such as the addition of an amphiphilic polymer or surfactant exchange strategies, have been examined for their ability to transfer the hydrophobic MNPs into the aqueous phase [40].
When placed in an external field, each MNP creates a local magnetic field, which increases the field inhomogeneity. When water molecules diffuse within the periphery of the MNPs, the coherent precessions of water proton spins are perturbed. The net effect is a change in the magnetic resonance signal, which is measured as a shortening of the longitudinal (T1, spin–lattice) and transverse (T2, spin–spin) relaxation times. The capacities of MNPs to decrease T2 and T1 are respectively defined as the transverse (r2) and the longitudinal (r1) relaxivities. Typically, because the transverse relaxivities (r2) of MNPs are greater than their longitudinal relaxivities (r1), T2 is used for NMR-based biosensing applications. With a higher r2 relaxivity, fewer numbers of nanoparticles are required to produce detectable T2 changes.
Within an ensemble of MNPs, magnetic relaxation properties depend on more than simply the particles’ relaxivities; the organizational state of the ensemble is also important. Unlike evenly dispersed MNPs, aggregates of nanoparticles (self- assembled magnetic clusters) have been shown to enhance the net rate of transverse relaxation. This unique phenomenon, known as magnetic relaxation switching (MRSw), is a cooperative process in which the interacting nanoparticles become more efficient at dephasing the spins of neighboring water protons, leading to a decrease in T2 relaxation time. The phenomenon can be explained by the outer- sphere theory. For a given volume fraction of MNPs in solution, T2 of the sample is inversely proportional to the cross-sectional area of the particles. Thus, the same amount of magnetized material is much more effective when dispersed as fewer large nanoparticles than as a greater number of smaller ones. In MRSw, nanoparticles aggregate to form self-assembled clusters, and the consequent increase in cross-sectional area of the particles shortens T2 relaxation times.
Analogous to MRI, DMR exploits targeted MNPs to modulate the spin–spin T2 relaxation time of biological samples. Depending on the size of the target biomarker, DMR assays can take two forms. For small molecular targets with sizes less than or comparable to that of the MNPs, MRSw assays can be used effectively for their detection and quantification. Small molecular analytes, such as drugs, metabolites, oligonucleotides, and proteins, can cross-link MNPs to promote relaxation switching. As indicated in Figure 1a, MRSw assays can be designed to cause forward switching: a process whereby molecular targets are used as cross- linking agents to assemble MNPs into clusters, thus effecting a corresponding decrease in T2. Alternatively, the assays can cause reverse switching, where enzymatic cleavage or competitive binding of molecular targets disassembles preformed clusters to cause an increase in T2. Note that MRSw assays are carried out without removing excess unbound MNPs. For larger biological structures, such as bacteria, entire mammalian cells or cellular components, targeted MNPs can be used to tag cell surface markers to impart a magnetic moment (Figure 1b). The change of 1/T2 is proportional to the number of MNPs bound, and also indicative of the abundance of relevant surface biomarkers. Unlike MRSw assays, this magnetic tagging strategy requires washing steps to remove excess unbound MNPs from the tagged biological targets. To enhance DMR detection sensitivity, MNPs should possess the following characteristics: 1) exhibit superparamagnetic properties; 2) have high stability in aqueous media to avoid spontaneous aggregation, which could mimic target-induced clustering; 3) have high magnetization and transverse relaxivity (r2) to induce pronounced T2 changes; and 4) have good surface chemistry to simplify conjugational procedures for attaching affinity molecules, such as antibodies and peptides. The MNPs and their representative strategies described below have been shown to be uniquely suited for DMR applications.
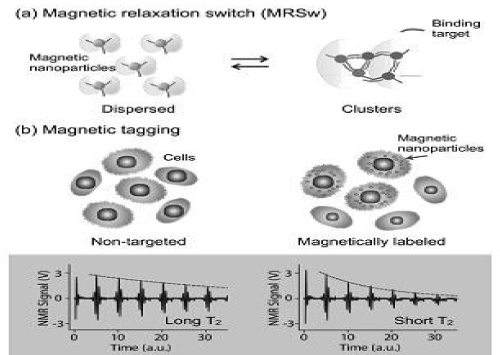
Figure 1. DMR assay configurations with magnetic nanoparticles(MNPs). (a) Magnetic relaxation switching assays detect the clustering of MNPs. (b) Magnetic tagging assays detect the presence of bound MNPs on larger biological entitles.
Cross-linked iron oxide (CLIO) nanoparticles have been widely used for DMR applications on account of their excellent stability and biocompatibility. CLIO nanoparticles contain a superparamagnetic iron oxide core (3–5 nm monocrystalline iron oxide) composed of ferromagnetic magnetite (Fe3O4) and/or maghemite (γ-Fe2O3). The metallic core is subsequently coated with biocompatible dextran, before being cross-linked with epichlorohydrin and activated by ammonia to provide primary amine group functionality. The amine groups can then be easily reacted with various agents containing anhydride, hydroxyl, carboxyl, thiol, or epoxide groups, to confer molecular specificity to the nanoparticle through bioconjugation. Amine-terminated CLIO nanoparticles have an average hydrodynamic diameter of 25–40 nm, approximately 40–80 amines per nanoparticle for bioconjugation, and a r2 of ~50 s−1·mM−1 [Fe]. Despite their relatively low r2, their unique coating makes CLIO nanoparticles exceedingly robust for use in biological applications [41].
The magnetization of ferrite nanoparticles can be further enhanced by doping the ferrite with ferromagnetic elements such as manganese (Mn), cobalt (Co) or nickel (Ni). Among the singly-doped ferrite MNPs, Mn Fe2O4 nanoparticles were found to exhibit the highest magnetization and r2 value, on account of their electron spin configurations, followed by FeFe2O4, CoFe2O4, and NiFe2O4. More recently, it has been demonstrated that magnetization can be further enhanced via additional Zn2+ dopant control in MnFe2O4 nanoparticles.
In addition, nanoparticle magnetization is known to increase with particle size. Ideally, each magnetic spin within a bulk magnetic material would be aligned parallel to the external magnetic field. However, in the nanoscale regime, surface spins tend to be tilted, a feature that reduces the overall magnetic moment. By increasing the MNP size, this surface effect is decreased, which in turn increases the magnetization. It has also been noted that transverse relaxivity r2 is proportional to the cross-sectional area of the magnetic core. Thus, increasing MNP size is an efficient method for enhancing r2, since this strategy increases both the magnetization as well as the particle cross-sectional area. Both magnetic doping and sizing strategies were recently employed to produce MnFe2O4 nanoparticles with superior r2 relaxivity, for DMR biosensing applications. These particles were synthesized by reacting iron(III) acetylacetonate [Fe(acac)3], manganese(II) acetylacetonate [Mn(acac)2] and 1,2-hexadecanediol at high temperature (300°C). A seed-mediated growth approach was used to increase the size of the magnetic core from 10 nm to 12, 16, or 22 nm. MnFe2O4 nanoparticles with a diameter ≤16 nm were found to be highly monodisperse and superparamagnetic at 300 K (Figure 2a). The MNPs were subsequently rendered water-soluble using the small molecule, meso-2,3-dimercaptosuccinic acid (DMSA). DMSA has a terminal carboxylic acid group at one end which interacts directly with the magnetic core, and a sulfhydryl group at the other end which cross-links with other DMSA molecules to increase stability [42].
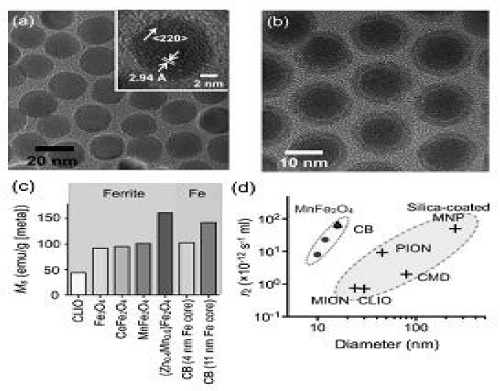
Figure 2. Higher r2-relaxivity MNPs developed to improve detection sensivity of in vitro diagnostics.
Due to DMSA’s small size, the hydrodynamic diameter of Mn Fe2O4 nanoparticles was found to be smaller than that of CLIO nanoparticles, despite their larger magnetic core. More importantly, these MnFe2O4 nanoparticles possessed superior relaxivities with r2 values as high as 420 s-1·mM-1 [metal] (equal to 6 × 10-11 s-1·[particle/mL]-1), more than 8 times greater than CLIO nanoparticles in metal basis (50 s-1·mM-1[metal] or 0.7 × 10-12 s-1·[particle/mL]-1). The synthesis of elemental iron-based nanoparticles (i.e., with elemental iron rather than iron oxides) and their stable dispersion in aqueous media, has remained one of the most attractive goals in magnetic nanomaterial engineering. Elemental iron (Fe) has a higher magnetization than that of metal oxides, which consequently motivates the creation of Fe-core MNPs to achieve high r2 relaxivities. Because the Fe cores are extremely reactive and subject to rapid oxidation, they need to be encased by a protective shell in order to maintain their magnetic properties. Recently, a 16 nm Fe-core/ferrite shell MNP, known as “cannonball”, was developed for DMR applications (Figure 2b). The cannonballs were synthesized by thermal decomposition of iron(0) pentacarbonyl [Fe(CO)5] to form the Fe core. A protective ferrite shell was formed by controlled oxidation with oxygen gas; this method resulted in a thinner shell than that previously produced by chemical oxidizers, and thus the nanoparticles retained a larger Fe core. The cannonballs were then coated with DMSA as described above. Because of their large Fe core, superparamagnetic cannonballs showed high magnetization (139 emu·g-1 [Fe]) when compared to other published Fe core–shell structures (Figure 2c). The relaxivity of cannonballs is similar to that of the MnFe2O4 nanoparticles (6 × 10-11 s-1·[particle/mL]-1), which is considerably higher than other commercially available or previously reported ferrite nanoparticles (Figure 2d). Bioorthogonal “click” chemistry has emerged as a novel method to label small molecules in complex biological media. Most reported applications, however, rely on either the azide–alkyne cycloaddition, which requires a copper catalyst, or the strain promoted azide–alkyne cycloaddition, which has relatively slow kinetics. A new bioorthogonal chemistry have developed based on the Diels–Alder cycloaddition between a 1,2,4,5-tetrazine (Tz) and a transcyclooctene (TCO). The reaction is fast, irreversible (covalent) and can be performed at room temperature without using a copper catalyst. Recently, this chemistry has been successfully adapted to magnetic targeting, so as to improve nanoparticle binding efficiency and detection sensitivity [43].
Termed ‘bioorthogonal nanoparticle detection’ (BOND), this technique provides a novel targeting platform in which Tz and TCO act as the coupling agents. In a two- step labeling strategy (BOND-2; Figure 3), antibodies against biomarkers of interest are first modified with TCO, which is then used as a target to facilitate the coupling of Tz-modified nanoparticles onto mammalian cells. Because of the small size of the coupling reagents, their high multiplicity on antibodies/nanoparticles resulted in higher nanoparticle binding to cells. In comparison to alternative standard techniques, such as the avidin/biotin method, BOND-2 not only amplifies the biomarker signals but also significantly improves the detection sensitivity. Moreover, this platform is broadly-applicable and scalable for biomedical use. BOND-2 has already been successfully adapted for molecular profiling of cell samples by DMR, and has now established itself as a major targeting method. Table 1 lists a library of cellular makers tested with BOND-2 and DMR.
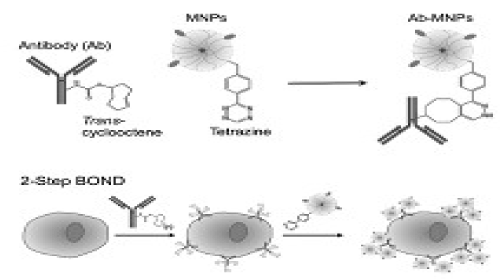
Figure 3. Biorthogonal nanoparticle detection strategy for DMR detection.
Nuclear magnetic resonance (NMR) can be detected with instruments such as clinical MRI scanners (routinely used for deep tissue whole body imaging), and NMR spectroscopy (used to study proteins and small molecules). Both of these techniques have been used to measure T2 relaxation time for DMR biosensing. However, because these conventional instruments are bulky and expensive, they remain as specialized equipment in hospitals and laboratories. Benchtop relaxometers, which operate at lower NMR frequencies (100 kHz–50 MHz) with a permanent low-field magnet (<1 T), provide a lower-cost alternative for DMR biosensing [44]. However, these systems lack the capability for performing multiplexed measurements, and require large sample volumes (>100 μL) to achieve accurate measurements.
To overcome the limitations of conventional detectors and to address the need for fast, simple and high-throughput biosensing, recently have developed a chip-based microNMR (μNMR) device. This miniaturized DMR device consists of an NMR probe containing microcoils for both radio-frequency (RF) excitation and NMR signal detection, on-board NMR electronics, a microfluidic network for sample handling, and a small permanent magnet for generating an external magnetic field. The first μNMR prototype was designed with a 2 × 4 planar microcoil array that was lithographically patterned onto a glass substrate (Figure 4a). This array format enabled the performance of parallel measurements, and each microcoil held 5–10 μl of sample. In the second-generation μNMR, changed the design to solenoidal coils, as such geometry provides higher signal-to-noise ratio (SNR) by producing more homogeneous radio-frequency magnetic fields for sample excitation. The SNR could be further increased by integrating the coil with a microfluidic channel (Figure 4b). The solenoidal coils were first wound around polyethylene tubes and subsequently immersed into a polymer (polydimethylsiloxan). Following polymer curing, the tubes were retracted to open up fluidic channels. The entire bore of the solenoid thus can be filled with sample to achieve maximal filling factor (≈1), the fraction of the coil volume occupied by the sample. Due to the larger cross- sectional area of the winding wires, the solenoidal coils also have smaller less electrical resistance than lithographically patterned coils. With these advantages, the sample volume for DMR detection could be reduced by a factor of ~10 (to 1 μL) compared to the previous devices (~10 μL).
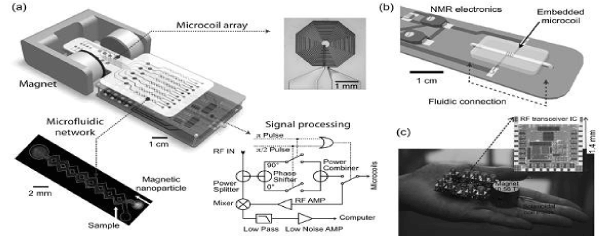
Figure 4. The devices developed for DMR biosensing.
The microfluidic networks in the DMR system facilitate the handling of biological fluids, the effective mixing of MNPs with small sample volumes, and the distribution of small volumes to different coils for parallel sensing. The networks also serve to confine the samples to the most sensitive region of a given microcoil. Furthermore, a membrane filter can be inserted at the outlet of the solenoidal microcoil to retain large biological targets, whilst removing smaller contaminants such as unbound MNPs. This configuration enables both the concentration of scant samples from large volumes, as well as the performance of on-chip washing steps. The NMR electronics generate versatile RF pulse sequences to measure the longitudinal (T1) and transverse (T2) relaxation times, process raw NMR signals (amplification, frequency conversion, filtering) for acquisition by a computer, and handle the multiplexed operation of an array of coils. In the first prototype, the NMR electronics was constructed as a tabletop system using discrete RF chips (e.g., AD9830 for RF generation and AD604 for NMR signal amplification; Analog Devices) and off-the-shelf RF components (e.g., ZAD-1 mixer, ZMSC-2 power splitter, and ZYSWA-2 RF switch; Mini-Circuits) [45]. In newer versions, these functionalities have been integrated onto a single CMOS IC chip. This chip was designed to overcome the adverse effects associated with system miniaturization during NMR measurements, including 1) low NMR signals resulting from small sample volumes, and 2) fast signal decay due to field inhomogeneity produced by the portable magnet. These challenges were addressed by implementing low noise RF amplifiers with high voltage gain, and by developing an on-chip digital pulse generator for various pulse sequences. The latter enabled the accurate measurement of transverse (T2) relaxation times by generating Carr–Purcell Meiboom–Gill (CPMG) sequenes to compensate for the inhomogeneity of the polarizing magnetic field.
In the latest implementation, the entire DMR system was packaged as a handheld unit for portable operation (“palm” NMR system; Figure 4c). When benchmarked against conventional NMR systems, these miniaturized devices provided both superior detection sensitivities and capabilities for multiplexed measurements on small sample volumes. In view of such advantages, the miniaturized DMR technology is well suited for fast, simple and high-throughput analysis of scant biological samples within a point-of-care setting. DMR applications DMR has been successfully applied to sensitively identify and quantify a wide range of biological targets including DNA/ mRNA, proteins, enzyme activities, small molecules/drugs, bacteria, viruses and mammalian tumor cells. As described previously, the detection mode of DMR depends on the size of its target.
The concept of forward MRSw sensing can be demonstrated with proteins, for example avidin, in model applications. In one early series of experiments, biotinylated MNPs were incubated with varying amounts of avidin. As shown in Figure 5a, the binding of biotin to avidin resulted in clustering of MNPs and a concomitant avidin concentration-dependent change in T2. By varying the concentration of MNPs, four orders of dynamic ranges were achieved, indicating that the system has a robust working range. Likewise, specific antibodies can also be used to perform MRSw on target protein molecules [46]. As the second proof- of-principle analysis, green fluorescent protein (GFP)- sensitive nanoparticles were prepared by conjugating CLIO nanoparticles with anti-GFP polyclonal antibodies.
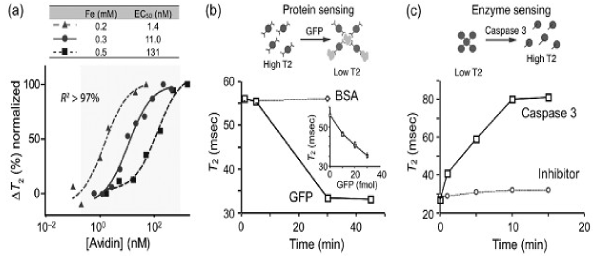
Figure 5. DMR detection of proteins and enzyme activities with MRSw sensors.
Using this system, GFP was rapidly and sensitivity detected in a dose dependent manner while the addition of bovine serum albumin (BSA) protein as a control did not elicit any change in T2 (Figure 5b). More recently, MRSw biosensors, capable of detecting soluble tumor biomarker proteins (such as CA-125, VEGF, and α- fetoprotein) were described, and used for parallel detection of multiple markers in blood samples with the μNMR device. Finally, using the BOND-2 method, many other cancer proteins have been detected. Reverse sensors have been designed to detect and quantify proteases, endonucleases and methylases. In these assays, the enzyme activity disassembles pre-formed clusters of MNPs; this disintegration translates the enzymatic activity into a detectable T2 signal. In the first demonstration of this strategy, MNP aggregates were formed with the peptide sequence biotin- GDEVDGC. This sequence served as a linker, binding both an avidin-conjugated CLIO population (via the biotin/avidin interaction) as well as a second CLIO population (via the thiol provided by the terminal cysteine on the peptide). The subsequent addition of caspase-3 disassembled the aggregates by cleaving within the DEVD site, which led to a corresponding increase in T2 relaxation time (Figure 5c). This dissociation was not observed when a specific caspase-3 inhibitor was added. A similar reverse switching strategy has been used to detect trypsin, renin, and matrix metalloproteinase 2 activities.
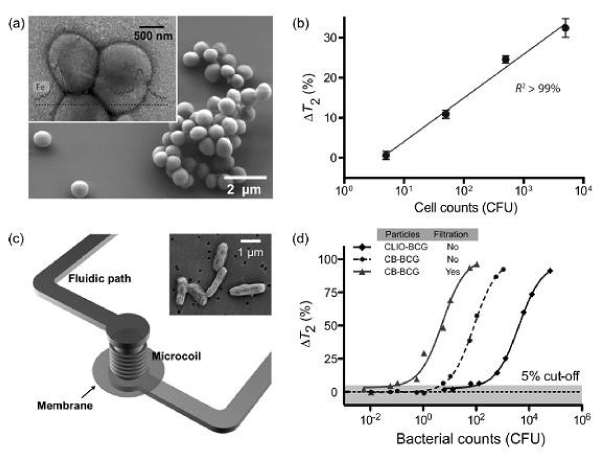
Figure 6. DMR detection of bacteria by tagging the bacterial samples with MNPs.
Forward MRSw assays on enzymatic activities have also been demonstrated via the assembly of nanoparticle biosensors (as a result of enzymatic reactions). For example, specific MNPs have been designed to assess human telomerase (hTERT) activity by hybridizing with the 30-base pair telomeric repeat sequences produced by hTERT activity. More recently, myeloperoxidase (MPO) sensors were generated by attaching phenol-containing molecules, such as dopamine or serotonin, to CLIO nanoparticles. In the presence of peroxidase activity, tyroxyl radicals were formed to cross-link the nanoparticles. Using the same assay configuration, leukocyte-derived MPO has been shown to play a critical role in the pathogenesis of atherosclerotic plaques.
Bacteria Detection and quantification of large pathogens have been successfully demonstrated using the DMR platform, primarily through magnetic tagging of targets. For example, detection of the bacterium Staphylococcus aureus was recently reported with the μNMR device. S. aureus were initially incubated with MNPs derivatized with vancomycin, a drug which binds to D-alanyl–D-alanine moieties in the bacterial cell wall to form dense clusters (Figure 6a). On account of the low sample volume required by the μNMR device, this first proof-of concept analysis demonstrated a detection sensitivity of only a few colony-forming units (CFUs) per microliter sample (Figure 6b). More recently, tuberculosis (TB) bacteria have been detected using DMR. In one study, the highly magnetic Fe- core/ferrite shell nanoparticles (CB; cannonballs) were used in combination with the second generation DMR device. To evaluate clinical utility of the DMR platform for TB detection, Bacillus Calmette-Guérin (BCG), used as a surrogate for Mycobacterium tuberculosis, was spiked into sputum samples. Following liquefaction, the biological samples were incubated with cannonballs conjugated to an anti-BCG monoclonal antibody. Unbound MNPs were then removed via a built- in membrane filter, embedded within the device (Figure 6c). This membrane (~100 nm size cut-off) not only removed excess unbound MNPs but also retained the BCG bacteria; thus was effective for both concentrating scant bacteria and removing background signal. In comparison to standard TB diagnostics, which involve time-consuming culture and acid-fast bacilli (AFB) smear microscopy, the DMR diagnostic technology showed unprecedented detection sensitivity and speed: as few as 20 CFUs could be detected in 1 ml of sputum sample, in less than 30 minutes (Figure 6d). Currently, this detection technology is being adapted to detect infectious pathogens in clinical sputum samples [47].
Sensitive detection and rapid profiling of tumor cell surface markers in unprocessed biological samples will undoubtedly have a significant impact on both the life sciences and clinical practice. DMR molecular profiling of Her2/neu, EGFR, and D326 (EpCAM) cancer markers on mammalian cells was first demonstrated using the first-generation DMR device. In these early experiments, CLIO nanoparticles were directly conjugated to monoclonal antibodies. More recently, the use of BOND-2 strategy has further advanced DMR profiling capabilities and Western blot analysis (Figure 7c). Importantly, the DMR detection platform not only required far fewer cells than either of the alternative approaches, but also produced results in a fraction of the time (<15 minutes). The DMR platform has since been shown to be adaptable to rapid multi-target detection, where putative cancer cells can be profiled for multiple biomarkers; DMR is ideally suited to this use since it can perform measurements on a few cells in small sample volumes and in a multiplexed manner. Fine-needle aspirate biopsies from a panel of mouse xenograft tumors have already been successfully analyzed for Her2/neu, EGFR, and EpCAM expression [48]. Furthermore, the multiple-marker targeting strategy has been shown to significantly improve the accuracy for correctly diagnosing cancer cells as malignant (Figure 7d). These, in addition to other advanced refinements to DMR sensing, are currently being applied to clinical trials of cancer cell profiling.
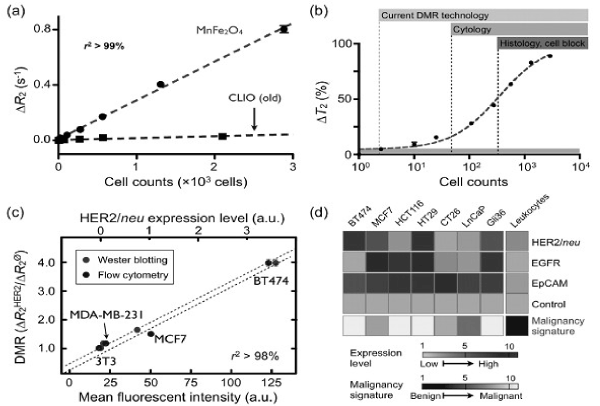
Figure 7. Tumor cell detection and profiling with the µNMR device.
One-pot synthesis of monodisperse iron oxide nanoparticles for potential biomedical applications
Magnetic nanoparticles with size up to 20 nm in diameter represent an important class of artificial nanostructured materials. Their magnetic properties change drastically with the sizes as magnetic anisotropic energy, KV, where K is the magnetic anisotropic constant and V is the particle volume, becomes comparable to the thermal energy, kT, resulting in moment randomization and superparamagnetism. Such superparamagnetic nanoparticles have great potential for biomedical applications. Their magnetic signal far exceeds that from any of the known bio-entities, making them readily identified in the ocean of biomolecules. Without external magnetic field, they show no net magnetic moment, facilitating their long-term stability in various dispersion media. They are smaller than or comparable to a cell (10–100 nm), a virus (20–450 nm), a protein (5–50 nm), or a gene (2 nm wide and 10–100 nm long). These, plus their capability of being manipulated under an external magnetic field, provide controllable means of magnetically tagging of all biomolecules, leading to highly efficient bioseparation/biodelivery and highly sensitive biolabeling and magnetic resonance imaging (MRI) contrast enhancement.
To apply superparamagnetic nanoparticles for biomedical applications, the nanoparticles should be monodisperse to have uniform physical and chemical properties for controlled biodistribution, bioelimination, and contrast effects. The magnetic nanoparticles should also have high magnetic moment, and can be so modified that they are capable of binding specifically to a biological entity and able to withstand various physiological conditions. Iron oxide nanoparticles, due to their chemical and magnetic stability and low level of toxicity in biological systems, have been widely tested for their use in biomedicine. However, some well-known material problems need to be solved before these nanoparticles can be utilized for any practical applications. The iron oxide nanoparticles used for the tests are often polydisperse with large variation not only in size, but also in shape. Consequently, the physical and chemical properties of these particles are not well controlled, and important data on biodistribution/ bioelimination in biological systems, which are essential for in vivo applications, are currently difficult to obtain.
Monodisperse ferrite MFe2O4 nanoparticles were recently made by a high- temperature reduction/ decomposition reaction of metal acetylacetonate. [49]. The size of the particles was controlled up to 8 nm from the one-step reduction/decomposition reaction. Larger size, up to 20 nm, was made possible by seed-mediated growth in which small MFe2O4 nanoparticles were used as seeds and more MFe2O4 was coated over the seeds. By controlling the heating parameters, the reaction further led to the ferrite nanoparticles with cube- or polyhedron-like shapes. This reduction/decomposition synthesis is complimentary to other reports in iron oxide nanoparticle syntheses from high-temperature decomposition reactions. The iron precursors used in this new synthesis are commercially readily available and less toxic than the iron pentacarbonyl, Fe(CO)5, a common precursor used in thermal decomposition reaction [50]. However, the synthesis of larger nanoparticles (>10 nm) via seed-mediated growth method involves multiple-step syntheses and as a result, the process leading to 15-20 nm nanoparticles is time-consuming. It is desired that, for practical applications, the iron oxide nanoparticles with a range of sizes can be prepared via a one-step synthesis. the reports show that a one-pot reaction of metal acetylacetonate with polyol in the presence of oleic and oleylamine to prepare Fe3O4, MnFe2O4, and Co Fe2O4 nanoparticles with the size tunable from 5 to 15 nm. The studies demonstrate that these nanoparticles are magnetically stable and can be readily functionalized for potential biomedical applications.
Advanced polymeric materials for biomedical applications
The use of polymers as biomaterials has grown tremendously in the past two decades. Currently, various types of polymeric materials are used for a variety of biomedical applications such as sutures, contact and intraocular lenses, drug carriers for sustained delivery, tissue and blood substitutes and artificial organs.
Development of intelligent nanoparticles for cancer detection and treatment
Cancer remains one of the leading causes of death in most parts of the world. It remains one of the world’s most devastating diseases; with more than 10 million new cases every year. Early diagnosis of cancer is essential to determine the treatment regime. Computerized tomography, positron emission tomography, magnetic resonance imaging and ultrasound imaging are some of the techniques used for cancer detection. However, these techniques are expensive, required lab facilities and highly trained technician, time consuming and can’t be taken to the field condition. Early diagnosis of the disease and better understanding of the pathophysiology of tumor progression has opened many new vistas as therapy options. In most solid tumors, after its surgical removal, the remaining cancer cells are managed with a variety of treatment options including, radiotherapy, chemotherapy, immunotherapy, etc. However, once the cancer is metastasized, the treatment options are limited, and chemotherapy remains the choice of treatment. The main reason for failure of chemotherapy is the poor accessibility of antineoplastic agents to the tumor, requiring higher doses, and the nonselective nature of these agents causes severe toxicity. Nanotechnology is in a unique position to produce a new generation of early cancer detection techniques with higher sensitivity and delivery of the drug to the target site.
Development of dendrimers/nanoparticles based systems for bacterial detection and mitigation
Rapid and sensitive identification of pathogenic bacteria is extremely important for drinking water supply, in path labs veterinary, food & beverage industry as well as in applications to microbiological threat detection and reduction. The several research groups are working on DBT sponsored research project entitled “Development of low cost biosensor for rapid detection of pathogenic bacteria” from Aug. 2007. Pathogenic bacterial strain of Salmonella typhi (S. typhi) was obtained from Rockland Hospital, Delhi and maintained by repeated subculturing and glycerol stock preparation. Polyclonal antiserum against the S. typhi was generated using whole bacterial cell as immunogen in NewZealand White Rabbit and purified using Protein A affinity column. Surface modification of nylon membrane & polyacrylonitrile fibre (PAN) was under taken with an aim to use them as solid matrices for ELISA/ FIA after immobilization with bacterial antibody. Modified membranes and fibres were characterized using various techniques. Both the matrices were standardized using RAG-IgG/GAR-IgG/GAR- HRP as a model antibody system. S. typhi bacteria detection in water samples were carried out with the S. typhi antibody immobilized nylon membrane (Figure 8). A detection limit of 104 cfu/ml of S.typhi could be primarily established as the lower detection limit for the developed Sandwich ELISA method using HRP labeled S. typhi antibodies within 2 h. Research activities are in progress to further decrease the detection limit in the range of 103-102 cfu/ml. Intraday and inter-day variations were also found to be below 10%. Specificity studies for the developed method showed the negligible cross reactivity with other Enterobacteriaceae members (S. paratyphi A, Klebsiella pneumonia, Shigella sonnei, E.coli andPseudomonas aeruginosa).

Figure 8. Scanning electron micrographs of (a) nylon membrane (b) GMA grafted nylon membrane.

Figure 9. (a) Confocal Laser Scanning Microscope image(CLSM) of S. typhi antibody conjugated QDs (b) CLSM image (right) and DIC image(left) of conjugated QDs.
Fluorescent core-shell QDs were also synthesized using hot injection method. Synthesized inorganic QDs were made hydrophilic by ligand exchange method for the conjugation with biomolecules. Antibody conjugated QDs were used as detector molecule for the detection of S. typhi. Preliminary experimental studies were carried out for detection of S. typhi in water using bacteria specific antibody (Sty IgG) bioconjugated to carboxy terminated CdSe/CdS/ ZnS QDs. Bacteria- bioconjugated QDs complex were seen as fluorescent aggregates under confocal laser scanning fluorescent microscope (Figure 9). Further studies towards the optimization of bacteria detection using bioconjugated QD’s and
Development of polymeric micro and nanoparticle based insulin delivery systems for diabetics
The prevalence of diabetes mellitus has been growing by leaps and bounds throughout the world. Major factors behind the increasing diabetic population are hereditary and environment. India is host to the largest diabetic population in the world with an estimated 35 million people, amounting to 8% of the adult population. Insulin dependent diabetic mellitus is an autoimmune disorder characterized by the destruction of the insulin secreting beta cells of langerhans of the pancreas and consequent inability to maintain blood glucose homeostasis. Presently the only feasible treatment is administration of insulin through subcutaneous injection. However, the problems encountered with subcutaneous insulin injections are pain, allergic reactions, hyperinsulinemia and chances of infection. The delivery of insulin by nonparenteral routes has gained significant attention over last two decades. The alternate routes explored are ocular, nasal, buccal, rectal, pulmonary and oral. The insulin delivery systems are developing as an alternative to the multiple insulin injections. Following two strategies have been adopted.
Oral insulin delivery system
Oral route is generally not feasible for peptide such as insulin due to rapid enzymatic degradation in the stomach, inactivation and digestion by proteolytic enzymes in the intestinal lumen and poor permeability across intestinal epithelial because of its high molecular weight. To overcome these problems, the pH sensitive mucoadhesive polymeric micro and nanoparticles of poly (ethylene glycol) dimethacrylate - methacrylic acid have synthesized and evaluated them as a carrier for oral delivery of insulin. Due the pH sensitive nature, these particles shrink at low pH in the stomach and protect the insulin from proteolytic enzymes and harsh acidic environment, while at basic pH in intestine the particles will swell and release the insulin. Polymeric micro and nanoparticles were synthesized by the free radical suspension and emulsion polymerization techniques. During in vitro insulin release studies, at pH 2.5, the release of insulin from polymeric particles was observed in the range of 5-8% while a significant higher release (20-35%) was observed at pH 7.4 during first 15 minutes of in vitro release studies. It was observed that polymeric microparticles were more effective than nanoparticles in reducing the blood glucose level in diabetic rabbits.
Biodegradable block copolymers of PLA-PEG with various chain length of PEG (575-4000) were synthesized and characterized by various techniques. Nanoparticles of PEG-PLA were prepared by double emulsion technique and insulin was encapsulated at the time of particle formation. Loading of insulin increased with increase in PEG chain length used during polymerization. Loading efficiency was calculated by HPLC and maximum loading efficiency of 52% was observed in the PLA-PEG nanoparticles with the PEG chain length of 4000. In-vitro release studies revealed a significant controlled insulin release up to 8 days. Injected insulin loaded nanoparticles controlled the blood glucose level of diabetic rabbits in the normal range up to 7 days.
References
- Wagner V, Dullaart A, Bock AK, Zweck A (2006) The emerging nanomedicine landscape. Nat Biotechnol 24: 1211-1217. [Crossref]
- Emerich DF, Thanos CG (2007) Targeted nanoparticle-based drug delivery and diagnosis. J Drug Target 15: 163-183. [Crossref]
- Groneberg DA1, Giersig M, Welte T, Pison U (2006) Nanoparticle-based diagnosis and therapy. Curr Drug Targets 7: 643-648. [Crossref]
- Harris L, Batist G, Belt R, Rovira D, Navari R, et al. (2002) Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as fi rst-line therapy of metastatic breast carcinoma. Cancer 94: 25-36
- Uziely B, Jeffers S, Isacson R, Kutsch K, Wei-Tsao D, et al. (1995) Liposomal doxorubicin: antitumor activity and unique toxicities during two complementary phase I studies. J Clin Oncol 13: 1777-1785
- Zhang L, Granick S (2006) How to stabilize phospholipid liposomes (using nanoparticles). Nano Lett 6: 694-698. [Crossref]
- Torchilin VP (2005) Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov 4: 145-160. [Crossref]
- Duncan R (2006) Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer 6: 688-701. [Crossref]
- Tanaka T, Shiramoto S, Miyashita M, Fujishima Y, Kaneo Y (2004) Tumor targeting based on the effect of enhanced permeability and retention (EPR) and the mechanism of receptor- mediated endocytosis (RME). Int J Pharm 277: 39-61. [Crossref]
- Deguchi JO, Aikawa M, Tung CH, Aikawa E, Kim DE, et al. (2006) Infl ammation in atherosclerosis – visualizing matrix metalloproteinase action in macrophages in vivo. Circulation 114: 55-62. [Crossref]
- Davis FF (2002) The origin of pegnology. Adv Drug Deliv Rev 54: 457-458. [Crossref]
- Svenson S, Tomalia DA (2005) Dendrimers in biomedical applications--reflections on the field. Adv Drug Deliv Rev 57: 2106-2129. [Crossref]
- Bosman AW, Janssen HM, Meijer EW (1999) About Dendrimers: Structure, Physical Properties, and Applications. Chem Rev 99: 1665-1688. [Crossref]
- Moghimi SM, Hunter AC, Murray JC (2001) Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 53: 283-318. [Crossref]
- Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, et al. (1994) Biodegradable long-circulating polymeric nanospheres. Science 263: 1600-1603. [Crossref]
- Torchilin VP (2007) Micellar nanocarriers: pharmaceutical perspectives. Pharm Res 24: 1-16.
- Tobío M, Gref R, Sánchez A, Langer R, Alonso MJ (1998) Stealth PLA-PEG nanoparticles as protein carriers for nasal administration. Pharm Res 15: 270-275.
- Perez C, Sanchez A, Putnam D, Ting D, Langer R, et al. (2001) Poly(lactic acid)-poly(ethylene glycol) nanoparticles as new carriers for the delivery of plasmid DNA. J Control Release 75: 211-224. [Crossref]
- Langer R (1990) New methods of drug delivery. Science 249: 1527-1533. [Crossref]
- Langer R, Tirrell DA (2004) Designing materials for biology and medicine. Nature 428: 487-492. [Crossref]
- Edelman ER, Mathiowitz E, Langer R, Klagsbrun M (1991) Controlled and modulated release of basic fi broblast growth factor. Biomaterials 12: 619-626.
- Langer R, Folkman J (1976) Polymers for the sustained release of proteins and other macromolecules. Nature 263: 797-800. [Crossref]
- Langer R (1998) Drug delivery and targeting. Nature 392: 5-10. [Crossref]
- Farokhzad OC, Cheng J, Teply BA, et al. (2006) Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA 103: 6315-6320.
- Kabanov AV, Batrakova EV, Alakhov VY (2002) Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release 82: 189-212. [Crossref]
- Pandey R, Khuller GK (2006) Nanotechnology based drug delivery system(s) for the management of tuberculosis. Indian J Exp Biol 44: 357-366. [Crossref]
- Hirsch LR, Gobin AM, Lowery AR, Tam F, Drezek RA, et al. (2006) Metal nanoshells. Ann Biomed Eng 34: 15-22. [Crossref]
- Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, et al. (2003) Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci USA 100: 13549-13554.
- (2001) Tissue Engineering: Technologies, Markets, and Opportunities (3rd Edition) Drug and Market Development Publishing, MA, USA.
- Mastrogiacomo M, Scaglione S, Martinetti R, Dolcini L, Beltrame F, et al. (2006) Role of scaffold internal structure on in vivo bone formation in macroporous calcium phosphate bioceramics. Biomaterials 27: 3230-3237.
- Rush SM (2005) Bone graft substitutes: osteobiologics. Clin Podiatr Med Surg North Am 22: 619-630.
- Giannoudis PV, Dinopoulos H, Tsiridis E (2005) Bone substitutes: an update. Injury 36 Suppl 3: S20-27. [Crossref]
- Ohgushi H, Caplan AI (1999) Stem cell technology and bioceramics: from cell to gene engineering. J Biomed Mater Res 48: 913-927. [Crossref]
- Ohlsson C, Bengtsson BA, Isaksson OG, Andreassen TT, Slootweg MC (1998) Growth hormone and bone. Endocr Rev 19: 55-79. [Crossref]
- Ramay HR, Zhang M (2004) Biphasic Calcium Phosphate Nanocomposite Porous Scaffolds for Load-bearing Bone Tissue Engineering. Biomaterials 25: 5171-5180. [Crossref]
- Ramay HR, Zhang M (2003) Preparation of porous hydroxyapatite scaffolds by combination of the gel-casting and polymer sponge methods. Biomaterials 24: 3293-3302. [Crossref]
- Zhang Y, Zhang M (2002) Three-dimensional macroporous calcium phosphate bioceramics with nested chitosan sponges for load-bearing bone implants. J Biomed Mater Res 61: 1-8. [Crossref]
- El-Ghannam A (2005) Bone reconstruction: from bioceramics to tissue engineering. Expert Rev Med Devices 2: 87-101. [Crossref]
- Anselme K (2000) Osteoblast adhesion on biomaterials. Biomaterials 21: 667-681. [Crossref]
- Puleo DA, Bizios R (1992) Formation of focal contacts by osteoblasts cultured on orthopedic biomaterials. J Biomed Mater Res 26: 291-301. [Crossref]
- Puleo DA, Bizios R (1992) Formation of focal contacts by osteoblasts cultured on orthopedic biomaterials. J Biomed Mater Res 26: 291-301. [Crossref]
- Scaglione S, Braccini A, Wendt D, Jaquiery C, Beltrame F, et al. (2006) Engineering of osteoinductive grafts by isolation and expansion of ovine bone marrow stromal cells directly on 3D ceramic scaffolds. Biotechnol Bioeng 93: 181-187. [Crossref]
- Yoshikawa H, Myoui A (2005) Bone tissue engineering with porous hydroxyapatite ceramics. J Artif Organs 8: 131-136. [Crossref]
- Mastrogiacomo M, Muraglia A, Komlev V, Peyrin F, Rustichelli F, et al. (2005) Tissue engineering of bone: search for a better scaffold. Orthod Craniofac Res 8: 277-284. [Crossref]
- Kim SS, Sun Park M, Jeon O, Yong Choi C, Kim BS (2006) Poly(lactide-co-glycolide)/hydroxyapatite composite scaffolds for bone tissue engineering. Biomaterials 27: 1399-1409. [Crossref]
- Cancedda R, Mastrogiacomo M, Bianchi G, Derubeis A, Muraglia A, et al. (2003) Bone marrow stromal cells and their use in regenerating bone. Novartis Found Symp 249: 133-143. [Crossref]
- Huang X, Miao X (2006) Novel Porous Hydroxyapatite Prepared by Combining H2O2 Foaming with PU Sponge and Modified with PLGA and Bioactive Glass. J Biomater Appl 21: 351-374. [Crossref].
- Morishita T, Honoki K, Ohgushi H, Kotobuki N, Matsushima A, et al. (2006) Tissue engineering approach to the treatment of bone tumors: three cases of cultured bone grafts derived from patients' mesenchymal stem cells. Artif Organs 30: 115-118.
- Tan KK, GH Tan, BS Shamsul, KH Chua, MH Ng, et al. (2005) Bone graft substitute using hydroxyapatite scaffold seeded with tissue engineered autologous osteoprogenitor cells in spinal fusion: early result in a sheep model. Med J Malaysia 60C: 53-58.
- Hunter A, Archer CW, Walker PS, Blunn GW (1995) Attachment and proliferation of osteoblasts and fibroblasts on biomaterials for orthopaedic use. Biomaterials 16: 287-295. [Crossref]