A 51-years-old male with malignant pheochromocytoma underwent resection of the right adrenal tumor in May 2007. The pathological diagnosis was pheochromocytoma, suspected of being malignant tendency, which contained neuroblastoma cells. Recurrence and metastasis was observed 4 years after the operation. During the treatment, 131I- metaiodobenzylguanidine (131I-MIBG) treatment, palliative cytoreductive surgery, somatostatin treatment, TKI (sunitinib) targeted therapy, combined chemotherapy was administered with the overall survival time reaching 87 months. Like literature reports, surgery is the main treatment of malignant pheochromocytoma, 131I-MIBG treatment is an effective method, molecular targeted therapy is the most promising therapy. Even so, treatment of malignant pheochromocytoma is still lack of unified standard, successful treatment requires large sample of clinical observation and a multidisciplinary team.
malignant pheochromocytoma, systematic treatment,131I- metaiodobenzylguanidine, targeted therapy, palliative cytoreductive surgery
Malignant pheochromocytoma is a rare tumor, originating from adrenal medulla chromaffin cells with the highest incidence observed in the age group of 40-50 (1). Distant metastases can be found in lymph, bones, liver and lungs, there is a little systematic study of malignant pheochromocytoma due to its rareness and the best treatment strategy is still being debated. The natural course of malignant pheochromocytoma or paragangliomas remains unknown. A retrospective study established the progression-free survival time at 1 year in treatment-naïve patients. 26 of 30 (87%) patients believed to have progressed 1 year later actually initially presented progression at a median of 5.7 months, diagnosed in the imaging workup. No prognostic parameter was definite (2). Patients with metastases receive no curative treatment and their 10-year survival rate drops to just 20% (3). In light of poor prognosis for the disease, it is of great value to choose an appropriate method to improve survival rate. In this report, we present a malignant pheochromocytoma case and review relevant literature report. At the end of article, we summarize our clinical experience for treating malignant pheochromocytoma.
This is a case of malignant pheochromocytoma in the right adrenal gland. A 51- years-old man was admitted to the hospital with adrenal masses found during a regular abdominal examination in May 2007. He complained of headache, palpitations and sweating, but was free from syncope, amaurosis, chest tightness, chest pain or asthma. His tumor markers were within normal range. Electrocardiogram (ECG) presented a sinus tachycardia. His past medical history included hypertension for 5 years and diabetes for 4 years, without any family history. Once diagnosed, surgical treatment was scheduled in Jiangsu province hospital of Traditional Chinese Medicine. Postoperative pathology: right adrenal pheochromocytoma, suspected of being malignant in nature, with neuroblastoma cell. The maximum diameter is 7 cm. Immunohistochemistry: S100 (+), CD56 (+), CgA (+), SYN (+), Vimentin local (+) (Figure 1). After curative resection, blood glucose and blood pressure were within normal range and re-examinations were conducted on a regular basis. In December 2011, CT examination revealed enlarged post-peritoneum lymph nodes (Figure 2). Also revealed in the 131I-MIBG whole body scan was abnormal tracer concentration accumulating in multiple retroperitoneal lymph nodes. This prompted consideration of recurrence and metastasis. After 4 times 131I-MIBG treatment, the lymph nodes were shrinking significantly. RECIST therapeutic effective evaluation was partial responded (PR). In January 2013, enlarged post-peritoneum lymph nodes were observed again. The Patient received Laparoscopic Retroperitoneal Lymph Node Dissection. Postoperative pathological diagnosis confirmed lymphatic metastasis. In August 2013, not only enlarged post-peritoneum lymph nodes but also enlarged mediastinal lymph nodes were observed (Figure 3). To control the disease long acting somatostatin (20mg, every 4 weeks) treatment was given for 4 months. In December 2013, another site of metastasis was identified. This time sunitinib targeted therapy (50mg, once a day for a total of 4 weeks with 6 weeks being a cycle) was administered. CT scan arranged after one-month treatment and the result showed that the lymph nodes were shrank significantly once again (Figure 4). Because of third-degree nausea and vomit, the second cycle of sunitinib to be reduced to 37.5mg once a day. In March 2014 symptoms of adverse events such as third-degree rash, oral mucositis, nausea and vomit led to therapy cessation. The Disease progressed in May 2014. In May 9th, 2014 and May 30th, 2014, 2 cycles chemotherapy (CVD combined with thalidomide) were administered. After 2 cycles of chemotherapy, efficacy evaluation was established as Stable Disease(SD) (Figure 5). In late June 2014, the patient presented incomplete intestinal obstruction, with physical status scoring below 60 points, and received supportive therapies such as gastrointestinal decompression and parenteral nutrition support. However, the patient died on August 31, 2014. The overall survival time was 87 months.
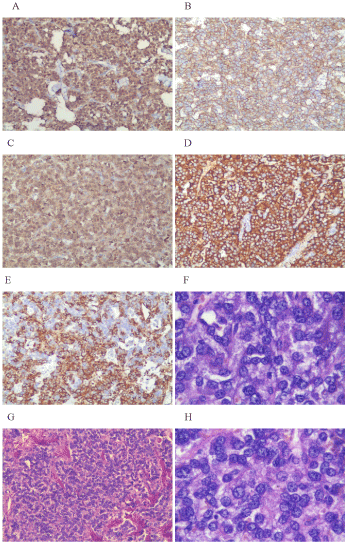
Figure 1a,b. A: nucleus and cytoplasm S100 (+) ; B: membrane CD56 (+) ; C: cytoplasm CgA (+); D: cytoplasm SYN (+); E: cytoplasm Vimentin (+); F: Abundant cytoplasm, moderate amphophilic, HE stain; G: cells with nest-like distribution HE stain; H: spherical, ovoid nuclei and prominent nucleoli. HE stains.
a: Figure 1 represent tumor cell b: A, B, C, D and E are 100-fold; F and H are 400-fold
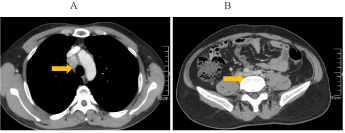
Figure 2. CT appearances: multiple enlarged lymph nodes around retroperitoneal artery, maximum nodule 1.7cm*1.0cm.
Figure 3. A: enlargement of mediastinal lymph nodes; B: multiple enlarged retroperitoneal lymph nodes were larger than before.
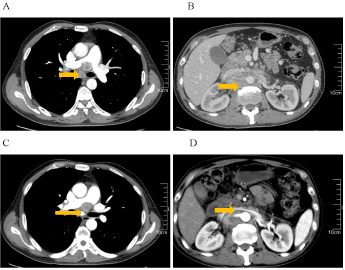
Figure 4. A and B: After somatostatin treatment for four months, lymph nodes of mediastinal and retroperitoneal significantly increased, fusion of lymph nodes can be recognized at abdominal and retroperitoneal, maximum nodule 4.3cm*3.0cm. Tumor invades and surrounds with abdominal aorta, portal vein, right renal artery, narrows portal vein. C: The size and number of mediastinal lymph nodes reduced after one-month targeted therapy. D: After targeted therapy, lymph nodes of abdominal and retroperitoneal reduced, particularly obvious at retroperitoneal.
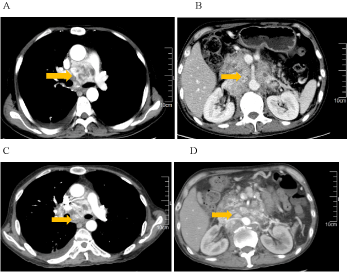
Figure 5. After 2 cycles of CVD chemotherapy, metastatic lymph node at mediastinal, abdominal and retroperitoneal is stable. A and B: Before CVD chemotherapy. C and D: After 2 cycles of CVD chemotherapy.
Pheochromocytomas are rare neuroendocrine tumors that arise from chromaffin cells of the adrenal medulla, with ten percent of all cases being extra adrenal paraganglioma (4). It’s estimated annual incidence is about 8 per 100,000 people a year (5). The highest incidence is in the age of 40-50 with no significant different between males and females (6). 10% of all pheochromocytomas cases are malignant (7). Extra-adrenal pheochromocytoma is more likely to be malignant in nature, reaching 29%-40% of the total. It is difficult to distinguish benign and malignant pheochromocytoma, depending only on histopathological examination (8). Because there are no molecular or histologic markers specifically for malignancy, patients often don’t get diagnosed with malignant pheochromocytoma until the unresectable disease has developed. Therefore, other methods for early diagnosis are still being considered, such as the percentage of adrenal chromaffin cells, Ki-67 label index, the level of Activator of Transcription 3 or HSP 90 (heat shock protein 90). Unfortunately, among all the methods, the most effective and practical is still in dispute (9-11). At present, according to the WHO classification standards in 2004 (12), metastatic lesions located in the non-chromaffin tissue is the golden standard of malignant pheochromocytoma diagnosis. There are two main clinical features of malignant pheochromocytoma. Firstly, symptoms and complications due to increasing catecholamine release, such as Typical clinical triad (headaches, palpitations, excessive sweating, Hypertension and Diabetes, and so on. Secondly, symptoms related to the tumors metastasizing and spreading, for example, aches, caused by compression of the mass, obstruction and multiple organ failure.
Currently, malignant pheochromocytoma study is mainly based on case reports, or clinical observations of small samples of a single treatment method. Thus, optimal treatment approach is still unsettled (13-14). Treatment options for unrespectable metastatic pheochromocytoma, including chemotherapy, 131I-MIBG and radiation, can produce limited effect, although none of them prove special efficacious (15). Other methods such as transcatheter arterial chemoembolization (TACE) has been reported to treat liver metastasis lesion (16). Like other tumor therapies, surgery plays a significant role in the management of malignant pheochromocytoma and may be the only way to achieve radical cure (17). While surgical removal of metastatic lesions is still advocated for alleviation of symptoms (18), if patients are asymptomatic with stable disease then close surveillance with no active treatment may be appropriated for a period of time. For other patients with symptomatic or progressive disease, who are not amenable to or appropriate for undergoing surgery, then additional systemic therapy is required.
131I-MIBG imaging remains helpful in localizing pheochromocytoma and, since 1983, has been used in the treatment of malignant pheochromocytoma in patients with 131I-MIBG-avid disease (19). 131I-MIBG can be selectively absorbed by pheochromocytoma, before accumulating in tumor cells and producing beta rays, finally achieving antitumor effect. Rutherford MA has collected the clinical data of 22 patients with metastatic pheochromocytoma or paraganglioma and made a retrospective analysis (20). According to the results, more than 50% of patients can achieve SD or PR, with 5-years survival rate reaching 68%. The efficacy and safety of MIBG treatment has been confirmed by related multicenter cohort study.131I-MIBG is an effective and safe method in treating malignant pheochromocytoma, without any severe complications (21). Thus, 131I-MIBG is considered as the most valuable treatment except surgery. However, as mentioned above, a universally applicable standard is still in dispute. The clinical validity of 131I-MIBG still lacks evidence of a large sample of randomized controlled trials.
Somatostatin is present in many tissues, such as pancreas, adrenal glands and intestines. In vivo and in vitro experiments showed that somatostatin could inhibit the growth of tumor. Pheochromocytoma has been confirmed with the character of somatostatin subtype-specific. Therefore, if patients test positive in octreotide scan, good effect can be achieved by using somatostatin (22). While, the efficacy of Long-acting somatostatin is limited in case of recurring and metastasizing malignant pheochromocytoma, this may be related to the different expression levels of somatostatin subtype-receptor in Pheochromocytoma. Unger found that when given a somatostatin scan, adrenal tumor with the membrane associated somatostatin receptor 3 would have a higher uptake rate (23). Therefore, somatostatin treatment may be only effective for this receptor 3 positive tumors.
Only a few reports feature Chemotherapy for malignant pheochromocytoma. There has been no conclusive evidence to support that combination chemotherapy involving cyclophosphamide, vincristine, and dacarbazine (CVD) improves survival in patients with malignant pheochromocytoma. A retrospective analysis breaks down data of 23 patients with metastatic and unrespectable malignant pheochromocytoma who received at least 2 cycles of this regimen, and finds that CVD therapy achieved a complete tumor response(CR) in 1 patient (4%), a partial response(PR) in 5(22%), stable disease(SD) in 5(22%), and progressive disease(PD) in 13(52%) (24).
Anti-angiogenic drugs can inhibit the growth of tumor, to achieve the anti-tumor purpose. There have been reports of increased expression of VEGF in malignant pheochromocytoma (25). By contrast, lower VEGF expression can be observed in benign tumor. Anti-angiogenic drugs are considered to be one of the most promising treatments for malignant pheochromocytoma. Sunitinib, a molecular targeted drug, is a potential new treatment strategy for malignant pheochromocytoma (26). Aya1a-Ramirez reported on 17 malignant pheochromocytoma cases treated with sunitinib, 8 of which got a stable condition (27). Attempts are also being made to conduct treatment with the use of other medicines of similar functioning, such as thalidomide, imatinib and evrolimus (28).
In recent years, the immunotherapy of tumor is one of the research hotspots, but there are few reports about immunotherapy of malignant pheochromocytoma. Data clearly demonstrate that CgA peptide-based immunotherapy induces a cytotoxic immune response in experimental pheochromocytoma, indicating potential for therapeutic applications in patients with malignant pheochromocytoma (29).
After this patient given surgical treatment, 131I -MIBG treatment, palliative lymph node dissection, somatostatin therapy, targeted therapy and chemotherapy, the overall survival time was 87 months. As reported in the literature, surgery was the primary course of treatment for this patient, when recurrence and metastasis presented, 131I-metaiodobenzylguanidine (MIBG) scan was arranged which turned out to be high uptake. According to the result, 131I-MIBG treatment became a preferred method. Combined with palliative lymph node dissection, the patient had nearly 2 years of progression-free survival. Palliative surgery can thus be considered as a treatment option, even in the case of multiple metastases. According to literature, sunitinib, a targeted drug for inhibiting tumor angiogenesis, can be used after somatostatin treatment. We prescribed sunitinib and achieved PR (RECEST evaluation criteria: sum of maximum length reduced by 37%), but the remission period was short. Because of the severe adverse reactions, we had no choice but to reduce the dosage. Other methods, such as chemotherapy and somatostatin treatment had limited efficacy in this patient.
According to the diagnosis and treatment of this patient, we summarize our experience, as follows: Firstly, it is difficult to distinguish the malignant and benign pheochromocytoma only by histopathological examination. Early diagnosis and potential malignant assessment are necessary. Secondly, malignant pheochromocytoma has poor cure rate. Most of these can only receive palliative care to ease the signs of their sickness. Thirdly,131I-MIBG treatment is effective, but large scale clinical verification is still needed. Chemotherapy is only suitable for those patients in good physical condition, and the treatment efficacy is limited. Molecular targeted therapy is one of the most promising therapy for malignant pheochromocytoma. An effective molecular targeted therapy aiming at permanent control of these highly complex neoplasms should be the aim of future efforts. In clinical setting, investigatory trials with multiple drug therapies targeting a variety of different parallel pathways should be pursued. Successful management requires a multidisciplinary teamwork (30). Fourthly, tumor immunotherapy has recently sprung up. More clinical research has been expected on immunotherapy of malignant pheochromocytoma.
- Jimene C, Rohren E, Habra MA (2013) Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep 15: 356-371 [Crossref]
- Hescot S, Leboulleux S, Amar L (2013) One-year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 98: 4006-4012 [Crossref]
- Mohammed AA, E-Shentenawy AM, Sherisher MA (2014) Target Therapy in Metastatic Pheochromocytoma: Current Perspectives and Controversies. Oncol Rev. 8: 249 [Crossref]
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K (2005) Phaeochromocytoma. Lancet 366: 665-675 [Crossref]
- Arnas-Leon C, Sánchez V, Santana Suárez AD (2016) Complete Remission in Metastatic Pheochromocytoma Treated with Extensive Surgery. Cureus. 8: e447 [Crossref]
- Adjallé R1, Plouin PF, Pacak K, Lehnert H (2009) Treatment of malignant pheochromocytoma. Horm Metab Res 41: 687-696 [Crossref]
- Strajina V, Dy BM, Farley DR (2017) Surgical Treatment of Malignant Pheochromocytoma and Paraganglioma: Retrospective Case Series. Ann Surg Oncol 24:1546-1550 [Crossref]
- Hodin R, Lubitz C, Phitayakorn R (2014) Diagnosis and management of pheochromocytoma. Curr Probl Surg. 51: 151-187 [Crossref]
- Thompson LD (20022021 Copyright OAT. All rights reservnd Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 26: 551-566 [Crossref]
- de Wailly P, Oragano L, Radé F, Beaulieu A, Arnault V (2012) Malignant pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg 397: 239-246 [Crossref]
- Tavangar SM, Shojaee A, Moradi Tabriz H (2010) Immunohistochemical expression of Ki67, c-erbB-2, and c-kit antigens in benign and malignant pheochromocytoma. Pathol Res Pract. 206: 305-309 [Crossref]
- DeLellis RA, Lloyd RV, Heitz PU (2004) Pathology and genetics of tumours of endocrine organs. World Health Organization classifieation of tumors. Lyon: IARC Press
- Chen H, Sipperl RS, O’Dorisio MS (2010) The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: phaeochromocytoma, paraganglioma and medullary thyroid cancer. Pancreas 39: 775-783 [Crossref]
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK (2014) Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99: 1915-1942 [Crossref]
- Fishbein L (2016) Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol Oncol Clin North Am 30: 135-150 [Crossref]
- Kim JK, Kim BH, Baek SM (2014) Incidentally detected inoperable malignant pheochromocytoma with hepatic metastasis treated by transcatheter arterial chemoembolization. Endocrinol Metab (Seoul). 29: 584-589 [Crossref]
- Gimm O, DeMicco C, Perren A, Giammarile F, Walz MK (2012) Malignant pheochromocytomas and paragangliomas: a diagnostic challenge. Langenbecks Arch Surg 397: 155-177 [Crossref]
- Lehnert H, Mundschenk J, Hahn K (2004) Malignant pheochromocytoma. Front Horm Res 31: 155-162 [Crossref]
- Harari A1, Inabnet WB 3rd (2011) Malignant pheochromocytoma: a review. Am J Surg 201: 700-708 [Crossref]
- Rutherford MA, Rankin AJ, Yates TM (2015) Management of metastatic phaeochromocytoma and paraganglioma: use of iodine-131-meta- iodobenzylguanidine therapy in a tertiary referral centre. QJM. 108: 361-368 [Crossref]
- Yoshinaga K, Oriuchi N, Wakabayashi H (2014) Effects and safety of ¹³¹I-metaiodobenzylguanidine (MIBG) radiotherapy in malignant neuroendocrine tumors: results from a multicenter observational registry. Endocr J 61: 1171-1180 [Crossref]
- Forssell-Aronsson E, Bernhardt P, Wängberg B, Kölby L, Nilsson O (2006) Aspects on radionuclide therapy in malignant pheochromocytomas Ann N Y Acad Sci 1073: 498-504 [Crossref]
- Unger N, Serdiuk I, Sheu SY (2004) Immunohistochemical determination of somatostatin receptor subtypes 1, 2A, 3, 4 and 5 in various adrenal tumors. Endocr Res 30:931-934 [Crossref]
- Asai S, Katabami T, Tsuiki M, Tanaka Y, Naruse M (2017) Controlling Tumor Progression with Cyclophosphamide, Vincristine, and Dacarbazine Treatment Improves Survival in Patients with Metastatic and Unresectable Malignant Pheochromocytomas/Paragangliomas. Horm Cancer 8: 108-118 [Crossref]
- Salmenkivi K, Heikkila P, Liu J (2003) VEGF in 105 pheochromocytomas: enhanced expression correlates with malignant outcome. APMIS. 111: 458-464 [Crossref]
- Hata J, Haga N, Ishibashi K (2014) Sunitinib for refractory malignant pheochromocytoma: two case reports. Int Urol Nephrol. 46: 1309-1312 [Crossref]
- Ayala-Ramirez M, Chougnet CN, Habra MA (2012) Treatment with sunifinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J Clin Endocrinol Metab 97: 4040-4050 [Crossref]
- Kukla U, Labuzek K, Chronowska J (2015) Antiangiogenic therapy of malignant pheochromocytoma and paraganglioma with the view to the recent scientific developments. Pol Merkur Lekarski. 38: 191-195 [Crossref]
- Papewalis C, Kouatchoua C, Ehlers M (2011) Chromogranin A as potential target for immunotherapy of malignant pheochromocytoma. Mol Cell Endocrinol. 335: 69-77 [Crossref]
- Buzzoni R, Pusceddu S, Damato A, Meroni E, Aktolun C, et al. (2013) Malignant pheochromocytoma and paraganglioma: future considerations for therapy. Q J Nucl Med Mol Imaging 57: 153-160 [Crossref]