Abstract
Introduction: There is mounting evidence to suggest that hyper-homocysteinemia (eHct) correlates with cognitive decline severity and cortical atrophy in AD. Hydrogen Sulfide (H2S) exerts neuroprotection through regulating Homocysteine (eHcy) signaling. Elemental sulfur can mediate H2S neuroprotection through the trans-sulfuration pathway and correct the the dysregulation of H2S /Hct signaling in AD. Sublimed Sulfur, formulated as SULMEDOL and approved for the treatment of lactose intolerance, activates gut beta-galactosidase (GALAC). We repurpose Sulmedol from lactose intolerance to likely CNS effect, since gut GALAC cross- talks with senescence-related GALAC expression in the brain. In view of the known regulatory function of GALAC in CNS endosomal-lysosomal autophagy system , we propose that SULMEDOL may rescue aberrant autophagy in AD.
Methods: We hypothesize that sublimed Sulfur (SS), via targeting H2S/Hcy and GALAC, may be efficacious in AD. In study I, we examined the 30-day treatment effects of SS at daily dosage of 200 mg to be administered at in altering the plasma Hcy levels in a small cohort of cognitive normal subjects. We stratified the cohort of cognitively normal control subjects to be administered SS into three groups with pre-determined range of basal fHCy levels as follows: Group 1 (n=16): range of fHcy 10.0-22.1 µmol/L; Group 2(n=15) fHcy range:7.1-9.9 µmol/L;Group 3 (n=15); range of fHcy 2.3-7.0 µmol/L . Primary outcome was defined as change in Hcy from baseline to 24-hr and 30-day post-treatment period. In study II, we evaluated the safety and tolerability of SS in two AD patients were treated with 200 mg for 3 months, with Clinical Global Impression–Global Improvement (CGI –I) scale as the primary outcome measure.
Results: In Study I, Group I subjects, fHcy decreased significantly by 36.1%, 24 hours after treatment and by 29.3%, 30 days later.In Group 2 fHcy did not change significantly. Group 3 exhibited paradoxical non-significant increase in fHcy, without change in erythrocyte folic acid, serum lipids and vitamin B12. SS was well tolerated in all patients. In Study II, clinical vignette analysis of two AD patients revealed that 3-month SS treatment ameliorated cluster of AD symptoms: nocturnal wandering, paranoid delusions, disorientation and confusion. CGI-I scale score was “very much improved”. No serious adverse events were reported.
Conclusions: SS in dual targeting H2S-homocysteine inflammation signaling and GALAC autophagy pathway related to AD, holds promise in therapeutic development of AD. Our findings warrant controlled trial of SS in AD.
Key words
Autophagy, homocysteine, cardiovascular disease, dementia , galactosidase, Alzheimer disease, elemental sulfur
Introduction
Despite advances in neurobiology of Alzheimer Disease or Alzheimer Dementia (AD), current AD drugs fail to modify the course of AD or improve significantly the decline in activities of daily living (ADL) in AD. The extent to which early identification of risk and protective factors can directly translate to preventive measures in cognitive aging remains controversial [1,2]. For the past decade, there is a large body of evidence to support the role of elevated level of the sulfur-containing amino acid: homocysteine level: Hcy as a risk factor common to both cardiovascular diseases (CVD) and AD [3]. Cross-talks between cardiovascular system and the brain through neuro-hormones: neurosteroids and neuro-modulators have become fascinating areas of research. Imaging studies support the model of dysregulation of neuro-vascular coupling in AD [4].
Elevated Hcy levels confer increase in mortality from coronary heart disease (CHD), cardiovascular disease (CVD) and all-cause mortality [5-8]. A meta-analysis of twelve studies comprising 23623 subjects calculated the core elements of Hcy risk estimate: the pooled adjusted risk ratio (RR) and the corresponding 95% confidence levels [5]. In comparing the highest to the lowest total Hcy, CHD mortality increased by 66 % (RR: 1.66, 95% CL 1.12-2.47) and cardiovascular (CVD) mortality increased by 68 % (RR: 1.68; CL 1.04-2.70). The approach takes into account the change in Hcy level as contributing towards the CVD and CHD risk. In general, the cutoff for “normal” Hcy has been defined to be from 5 -15 μmol/L and hyper-homocysteinemia (h-Hcy) is set at fHcy greater than 15.00 μmol/L. Despite the lack of universal agreement regarding the cut-off level to classify the condition as h-Hcy, hHcy is accepted as the independent cardiovascular risk factor. The complex roles of hyper-homocysteine in cardiovascular functions may be summarized in the context of impaired endothelial function [7]. Nitric oxide orchestrates the oxidative stress load and the coupling of nitric oxide synthetase activity. Furthermore a hyper-homocysteinemia seal the eventual fate of apoptosis of endothelial cells is precipitated by endothelium stress and shifts the phenotype of endothelium towards pro-inflammatory and pro-thrombotic state. Homocysteine is metabolized by two pathways: re-methylation and trans-sulfuration , which are dependent upon the availability of stores of folate and vitamin B groups: Vitamin B-6 and vitamin B-12 (cyanocobalamin) [8] (Figure 1). Transmethylation reactions depend upon S- adenosylmethionine (SAM) as a methyl donor. The release of the potent inhibitor S- adenosylhomocysteine (SAH) is the common denominator underlying SAM-dependent transmethylation reactions. Studies to examine the likely beneficial effects of folates and vitamins B supplementation to reduce hyper-homocysteinemia and alter the cardiovascular risks have met with mixed results: both positive and negative findings have been reported [3,9-11]. In analyzing the data from 19 vitamin B supplementation studies recruiting 47921 participants, a positive protective trend was observed for stroke but not for CHD [9]. Co-existing kidney diseases do not interfere with the safety and the beneficial effects of vitamin B and folate supplementation. There is consensus that folic acid appears to exhibit similar favorable effects in reducing stroke risk [10,11]. Critical re-analysis of studies on Hcy suggests that the ratio of reduced S-adenosyl-methionine (rSAM) to S-adenosyl-homocysteine (SAH) related to hyper-homocysteinemia , may be the more reliable biomarker for predicting CVD risk.
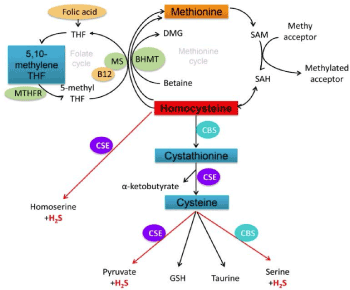
Figure 1. Molecular Footprints of Homocysteine/Hydrogen Sulfide Signaling relevance to Vitamin B-12, Folate and DNA methylation
The emerging role of homocysteine in predicting cognitive decline independent of cardiovascular risks has drawn equal attention in the context of cognitive aging and AD [12-17]. In the seminal Framingham study recruiting 1092 normal healthy subjects (677 female and 425 male; mean age: 76 years) were followed for 8 years for repeated plasma Hcy monitoring [13]. Dementia developed in 111 subjects of which 83 subjects were given the AD diagnosis. While the Hcy level exceeded 14 mol/L the AD risk doubled. The relative risk of AD was 1.8 per increased of standard deviation (SD) at baseline. A recent meta-analysis of 111 pertinent studies [18] reveals a positive trend between cognitive decline and increased plasma Hcy levels in general population and in patients with cognitive impairments and Folstein Mini-Mental State scores (MMS). The threshold cut-off value of Hcy level for AD risk is set at 10.0 μmol/L. Studies on effects of folate and Vitamin B group have shown mixed and somewhat inconsistent effects in rescuing cognitive deficits in hyper-homocysteinemia [19-21]. A systematic review found that 6/7 randomized controlled trials of subjects who were followed for 2-5 years, reported cognitive benefits with high Hcy or low folate levels at baseline [22]. The heterogeneity of the study population, the variations in methodology in outcome measures and the relative short duration of intervention may account for the discrepant findings. It is noteworthy that the clinical trials did not include subjects who were experiencing cognitive decline. Besides, folate and vitamin B group intervention studies have not standardized the basal level of omega-3-fatty acid levels which synergize with folate in reducing cognitive decline risk in AD. Furthermore, a meta-analysis of forty one case control association studies in AD concludes that the key enzyme in the folate/homocysteine pathway ,methylenetetrahydrofolate reductase (MTHFR) MTHFR C677T polymorphism, conferred significant susceptibility to AD in all genetic models [23], independent of the specific ethnic subgroups.
These considerations strongly suggest that hyper-homocysteinemia per se cannot account for the complex pathway leading to rate of cognitive decline in AD. Within the 1-carbon metabolism involving methionine recycling and transulfuration pathway, S-adenosyl-homocysteine (SAH) as the culprit of homocysteine-related chronic inflammatory state underlying cardiovascular and cerebrovascular disorders. Hcy may be the putative functional link to yet another important neuro-modulator: hydrogen sulfide (H2S) [24-27]. Accumulating studies on H2S as the unique gaseous neuromodulator highlight the role of H2s in regulating oxidative stress, parallel inflammatory and apoptosis pathways exerting feedback control over Hcy metabolism [24,25]. Recent failures in therapeutic trials targeting Abeta amyloid cascade comprising of alpha-, beta- and gamma-secretase have led some investigators to shift their attention to exploring autophagy signaling in AD. Autophagy is a major intracellular highly-coordinated clearance and degradation pathway involved in recycling of cellular debris through the activities of lysosomes in various stages of maturation within the neurons and in and around synapses [28-30]. Aberrant accumulation of toxic Abeta amyloid and Tau aggregates, damaged organelles and inclusions results from faculty autophagy-lysosomal system. Intriguing findings have been reported on changes in lysosomal enzymes and autophagy-lysosomal (auto-Lys) proteins in relation to the severity of AD core symptoms [30-32]. In both AD and Down’s syndrome, leukocytes showed a statistically significant increase of beta-galactosidase activity when compared with age-matched controls [33].
Elemental sulfur: Sublimed Sulfur [SS](patent and biotechnology inventor: A Khan) was formulated as oral compound and submitted to Health Canada Ottawa Canada for approval as a Natural Health Product(NHP) for lactose intolerance: Lac-INT. Lactose intolerance can be primary or secondary lactase deficiency and is characterized by a host of gastrointestinal symptoms: abdominal pain and distension, borborygmi, flatus, and diarrhea induced by lactose in dairy products [34,35]. We have previously shown in a controlled clinical study [36] showing that Sulmedol was efficacious in reducing the symptoms of Lac-INT in a cohort of Lac-INT. Sulfur most likely activates beta-galactosidase responsible for hydrolyzing lactose to glucose and galactose through the disulfide bond of the GALAC in the gut for reducing the gasto- intestinal symptoms of lactose intolerance.
SulmedolR is currently marketed in Canada for the health claim benefits of improving Lac- INT. A very recent study found hydrogen sulfide-containing sulfurous water (SW) containing hydrogen sulfide to be neuroprotective in the in vitro clinical model of AD [37]. An in vitro therapeutic trial of SW was carried out in peripheral blood mononuclear cells from AD patients compared with matched controls using the comet assay (to measure oxidative DNA damage) and the MTT assay (as an indicator of cell viability). Hydrogen peroxide and homocysteine were used to induce oxidative DNA damage. SW protected them against enhanced oxidative stress in AD and control populations in response to pro-oxidant homocysteine exposure. The results confirm the role of hydrogen sulfide in protecting against Hcy-induced oxidative stress. H2S confers to SW a more potent antioxidant capacity than other known antioxidants. Hitherto no study has been conducted whether elemental sulfur in dual targeting hydrogen sulfide/homocysteine and galactosidase (GALAC) /autophagy signaling can reduce hyper- homocysteinemia in cognitively normal subjects. We repurpose Sulmedol with indication for lactose intolerance to potential CNS drug and test the hypothesis whether Sulmedol is safe and efficacious in rescuing the core symptoms of AD. In addition, we examine whether oral treatment with elemental sulfur is safe and beneficial in patients diagnosed as AD. We propose that Sulmedol ‘s possible beneficial effects in AD patients can be interpreted in light of the pharmacological effects of elemental sulfur in facilitating the clearance of neurotoxic Abeta- amyloid and tau aggregates and related cellular organelles via activating beta-galactosidase/autophagy system and hydrogen sulfide/homocysteine pathway in AD.
The objective of our study is threefold:
1. To determine whether 30-day oral treatment with SS, at the daily dosage of 200 mg, reduces fHCy to levels free from increased cardiovascular risks ;
2. To determine the metabolic effect of ingested SS on erythrocyte folic acid, serum vitamin B12 and serum lipids.
3. To determine the treatment responses of AD patients (n=2) towards oral dosing regimen of Sulmedol in an open-label exploratory design.
Methods
A Study protocol
By making use of a patented biotechnology for upscale manufacturing and quality control, the product has achieved a purity of 99.5% [18]. Pharmacokinetics data indicated that 10% to 40% of ingested sulfur is absorbed from the gastrointestinal tract as sulfides and excreted as sulfates in the urine. Given the physio-chemical properties of galactosidase are similar in the brain as in the gut, we consider it valid to adopt the heuristic repurposing paradigm to examine the CNS effects of Sulmedol in AD. In stratifying the cohort of cognitively normal subjects with diverse medical histories involving cardio-vascular diseases, we stratified three groups with range of fHcy levels based on previous studies of elevated Hcy levels as independent cardiovascular risk factor and cognitive impairment related to AD [3,5,6,12,16-18,20]. With advances in our understanding of the multiple role of hHcy in regulating cerebral vasculature and cardiovascular homeostasis, the original normal value of Hcy has to be refined to predict both cardiovascular risk and AD risk as for each 5 μmol/L of Hcy increment , the pooled RR was 1.52 for CHD mortality and 1.32 for CVD mortality. The risk of atherogenesis is doubled when plasma homocysteine levels are >10.2 µmol/L, even though Hcy level is at the normal level. The risk of carotid stenosis is increased significantly with parallel changes in fasting homocysteine level (fHcy) from 11·4 µmol/L at baseline to to 14·3 µmol/L. Cardiovascular events can adversely affect brain health in general, especially the cognitive domain with aging. it is more pragmatic and informative to consider fHcy as a continuous variable and has to be adjusted for impaired renal function [5,6]. In basis of what is known about the central role of homocysteine in modulating cardiovascular and cognitive outcome and the results of previous studies on fHcy levels to discriminate AD, and Mild Cognitive impairment (MCI) from normal control subjects we choose to define the cut-off Hcy to be >/=10.00 µmol/L, rather than the common accepted normal range of Hcy to fall within the range of 5 -15 μmol/L. We adopted the following pre-determined range of basal fHCy levels fHcy for the stratification of our cohort of participants in our study as follows: Group 1 (n=16): range of fHcy 10.0-22.1 µmol/L; Group 2 (n=15) range of fHcy 7.1-9.9 µmol/L; Group 3: ( n=15) range of fHcy 2.3-7.0 µmol/L
The 46 subjects in this study ranged from 22 to 76 years of age; 31 (67·4%) were male and 15 (32·6%) female, with various medical problems listed in Table 1. The exclusion criteria for the study were inflammatory bowel disease, peptic ulcer, severe constipation, cholelithiasis, hyperthyroidism, untreated hypothyroidism, acute or chronic renal or hepatic disease, anaemia and pregnancy. The subjects were required to avoid the use of folic acid, vitamin B12, vitamin B6 and betaine, or combinations of these vitamins, and multi-vitamins for at least 30 days prior to the study, during the 30 days of treatment with SS, and for 30 days afterwards. Signed, informed consent was obtained from subjects willing to participate in the study.
Table 1. Clinico-characteristics of participating subjects
Total number of participants |
n = 46 |
n = 46 |
Gender: Male/female |
31/15 |
31/15 |
Age ( mean +/- SD) years |
58/2 +/- 11.7 |
58/2 +/- 11.7 |
Age range |
22 - 76 |
22 - 76 |
Medical diagnosis |
|
|
Generalized Atherosclerosis |
27 |
Hypertension |
25 |
Angina Pectoris |
18 |
Myocardial Infarction |
4 |
Coronary Angioplasty |
1 |
Peripheral Vascular Disease |
2 |
Diabetes Mellitus |
6 |
Hyperlipidemia |
15 |
Hypothyroidism |
2 |
Postmenopausal |
4 |
Hiatus Hernia |
3 |
Osteoarthritis/Osteoporosis |
8 |
Depression |
1 |
Essential Tremor |
1 |
Obstructive Airways Disease |
3 |
Bronchial Asthma |
1 |
Past History of Carcinoma |
2 |
Multiple Diagnoses |
24 |
Lactose Intolerance |
1 |
The results of the baseline fHcy were blinded to investigators prior to treatment with Sublimed Sulfur. Each subject was treated with a course of SS 200 mg. postprandially, daily, for 30 days beginning immediately after a baseline blood sample was obtained for measurement of fHcy. Post-treatment fHcy levels were measured 24 hours and 30 days after the last dose of the 30 day course of SS. The blood samples for measurement of plasma fHcy were drawn by two qualified licensed laboratories, centrifuged for 15 minutes, the plasma immediately frozen to -20°C and transported at -20°C to two accredited laboratories, two laboratories specializing in the measurement of plasma homocysteine. The level of homocysteine was measured by two independent laboratories using different methods: the Simplified Simultaneous Assay of Total Plasma Homocysteine via High-Pressure Liquid Chromatography (HPLC) [38] and an Electrochemical Detector with Pulsed Integrated Amperometry and the IMX Homocysteine Assay using Fluorescence Polarisation Immunoassay Technology (FPIA) [39]. The two methods yielded very good correlation coefficient with no significant differences in intra- and inter-laboratory variation. Basal and post-treatment fHcy for each subject were measured at the same laboratory. Both laboratories were blinded to the treatment received by the patients.
Baseline and post-treatment serum vitamin B12 (n=37), erythrocyte folic acid (n=30), total serum cholesterol, serum triglycerides, calculated LDL cholesterol, HDL cholesterol and total cholesterol/HDL cholesterol ratio (n=41) were compared. In order to screen for side effects, baseline and post-treatment comparisons were made of complete blood counts, urinalysis, serum creatinine, electrolytes, uric acid, glucose, liver function tests and electrocardiograms (n=44).
When all the samples were collected, the final laboratory results reviewed and certified by the licensed clinical chemist and pathologist were sent to the research team. In conducting statistical analysis, we used the Wilcoxon t-test was used to evaluate the quantitative data. Numerical values indicate the mean and standard error of the mean.
Case study of SSul in AD
Two patients diagnosed as AD were referred to Dr. A Khan MD medical clinic. The patients were explained the rationale of likely benefits of SS in AD in terms of altering the hydrogen sulfide/homocysteine signaling and beta-galactosidase-mediated autophagy involved in AD. Clinical Global Impression (CGI): global improvement was used to evaluate the clinical efficacy and side effects of SS. The proprietary natural health product (NHP) was administered at the daily oral dosage of 200 mg for a period of 3-4 months.
Results
Part a: As shown in Table 1, the participants represented a wide range of age groups from young adult to elderly groups. We did not make any specific age-related adjustment in the level of fHcy. The heterogeneous unselected groups had a wide variety of medical diagnosis. None of the subjects were screened for cognitive impairment and found to be cognitively normal with no evidence of cognitive impairment.
The baseline fHcy of the subjects ranged from 2·3 to 22·1 µmol/L and averaged 8·7 ± 1·29 µmol/L (mean ± standard error of the mean). The effect of SS therapy on fHcy varied according to the initial baseline fHcy concentrations. In general, low basal fHcy increased and high basal fHcy decreased after the 30 day course of treatment with oral SS Overall, there was a significant inverse correlation (+0·40, n=46, p <0·01) between the baseline fHcy and the degree of change of fHcy produced by SSul therapy. Higher levels of baseline fHcy were associated with larger decreases . On the other hand, a lower baseline fHcy predicted higher increases in Hcy level.
The Group I subjects had fHcy decreased significantly by 36·1%, 24 hours after SSul treatment (t15,0·05=–3·9, p=0·0014), and by 29·3%, 30 days later (t15,0·05=-3·80, p=0·0018)
in Group 2 the fHcy did not change significantly. Group 3 demonstrated paradoxical increase in fHcy but the relative change did not reach clinically relevant level in the context of cardiovascular: CV and AD dementia risk threshold. The vitamin B12 levels fluctuated somewhat during the treatment period with SSul, as the basal serum B12 level decreased significantly by 8% at 24 hours and non-significantly increased to 95% by 30 days. Erythrocyte folic acid and serum lipids did not change significantly, regardless of whether the participants were maintained on lipid-lowering drugs: statins. We used the non-parametric test: Mann-Whitney tests to compare the differences in fHcy among the three groups at the baseline level and at 24 hr and 30-day post-treatment period. Our results revealed no significant differences of fHcy at 24 hours and 30 days after discontinuation of SS. Taken together, we found that 30 day treatment course of SS eliminated the initial fHcy differences among the three groups of the participants and essentially converted elevated and low fHcy to clinically normal levels (Table 2), hence restoring the fHcy to the normal level.
Table 2. Changes from Basal of Fasting Plasma Homocysteine Levels to 24 hr to 30 day post- Treatment with Sublimed Sulfur
Plasma Homocysteine
mmol/L |
n |
Basal |
24 Hours1 mmol/L |
Mean Change % |
P Value |
30 Days2 mmol/L |
Mean Change % |
P Value |
Group 1: 10·0 to 22·1 |
16 |
13·30 ± 0·85 |
8·50 ± 0·99 |
- 36·1 |
<0·0014 |
9·40 ± 0·92 |
- 29·3 |
<0·0018 |
Group 2: 7·1 to 9·9 |
15 |
8·50 ± 0·20 |
7·50 ± 0·61 |
NS |
NS |
8·20 ± 0·95 |
NS |
NS |
Group 3: 2·3 to 7·0 |
15 |
4·10 ± 0·33 |
6·50 ± 0·79 |
+ 58·5 |
<0·001 |
7·60 ± 0·62 |
+ 85·4 |
<0·001 |
1. 24 hours after discontinuation of a 30 day course of Sublimed Sulfur. |
2. 30 days after discontinuation of a 30 day course of Sublimed Sulfur. |
Numerical values indicate the mean and standard error of the mean. Probability values according to Wilcoxon are sure test comparing basal levels with levels after therapy. |
(NS, non-significant) |
The changes in fHcy levels induced by Ssul treatment occurred independent of concomitant alterations in the vitamin B12, folic acid (Table 3a and Table 3b), suggesting that the dynamics of 1-carbon metabolism involving recycling of methionine-homocysteine and transulfuration pathways were not adversely impaired. The basal serum vitamin B12 decreased significantly by 8% from baseline (320 ± 24) at 24 hours (t 36,0·05=-2.43, p=0.0200), and increased to 95% of basal to (304 ± 23), 30 days later (NS) (Table 3). In the absence of vitamin B12 supplementation, the serum vitamin B12 increased to 95% (304 ± 23) of baseline levels (NS), 30 days after SS was discontinued. There were no significant changes from baseline of the erythrocyte folic acid levels, total serum cholesterol, HDL cholesterol, calculated LDL cholesterol and serum triglycerides (Table 3). Lipid lowering drugs: statins did not alter the responses towards SS treatment compared with the control group. No changes were found in glycemic control, kidney and liver functions throughout the treatment period. There were no significant changes from baseline of the complete blood count, urinalysis, serum glucose, creatinine, electrolytes, uric acid, and aspartate transaminase. There were no adverse effects on the pulse rate and rhythm, blood pressure and electrocardiograms. There were no cases of cyanosis due to sulfhemoglobinemia.
Table 3a. Changes from Basal of Fasting Erythrocyte Folic acid, Serum vitamin B-12 and Lipids to post-Treatment with Sullimed Sulfur in subjects. Group 3a: maintained on lipid-lowering drugs (statin); Group 3 b; without lipid-lowering drug
Vitamins and Lipids |
Normal Range |
N |
Basal |
24 Hours1 |
30 Days2 |
|
|
|
|
|
|
Vitamin B123 |
> 150 pmol/L |
37 |
320 ± 24 |
295 ± 22 |
304 ± 23 |
|
|
|
|
|
|
Erythrocyte Folic Acid |
> 215 nmol/L |
30 |
862 ± 50 |
859 ± 43 |
877 ± 46 |
|
|
|
|
|
|
Participants on Lipid Lowering Drugs |
Group 3a |
|
|
|
|
|
|
Total Cholesterol |
< 5·2 mmol/L |
12 |
4·80 ± 0·23 |
5·08 ± 0·23 |
5·19 ± 0·34 |
|
|
|
|
|
|
High-Density Lipoprotein Cholesterol |
0·77-1·68 mmol/L |
12 |
1·27 ± 0·03 |
1·27 ± 0·08 |
1·33 ± 0·07 |
|
|
|
|
|
|
Low-Density Lipoprotein Cholesterol |
< 3·4 mmol/L |
12 |
2·65 ± 0·22 |
2·82 ± 0·21 |
2·87 ± 0·28 |
|
|
|
|
|
|
Total/High-Density Lipoprotein Cholesterol |
up to 5.0 mmol/L |
12 |
3·93 ± 0·30 |
4·24 ± 0·36 |
4·06 ± 0·35 |
|
|
|
|
|
|
Triglycerides |
0·70 - 3·2 mmol/L |
12 |
1·94 ± 0·24 |
2·19 ± 0·36 |
2·20 ± 0·40 |
In sum, we found SS restored fHcy in cognitively healthy control subjects and exhibited a highly favourable safety and tolerability of SS. The patented product of elemental sulfur:SULMEDOL, was well tolerated and with no serious adverse events were noted.
Part b: In the two clinical vignettes of two AD patients, we reported on the effects of treatment with SS on moderately severe core neurobehavioral symptoms of AD. In case report 1, the patient presented with nocturnal confusion and wandering behavior in the community. In case report, the patient exhibited eating refusal. We used the Clinical Global Impression scale: Improvement scale to evaluate the responses towards SS and in both cases, we rated the extent of global improvement as “very much improved”.
Case Vignette 1: A 72 -year-old man of Caucasian descent developed 10-yr history of progressive memory deficit with features of Alzheimer’s disease with the age of onset around 62 years old. Diagnostic workup showed cerebral cortical atrophy on CT scan, a normal EEG and extensive normal blood test results. With regard to risk factors of AD, he presented with an extensive list of cardio-vascular and metabolic risk factors. He admitted to prolonged heavy cigarette smoking, Type II diabetes mellitus, hypercholesterolemia, generalized and coronary atherosclerosis, angina pectoris, and ventricular arrhythmia. Past history included excessive alcohol intake suggesting of alcohol dependence. He quit drinking after a recent episode of recent loss of consciousness which had caused him to discontinue alcohol. He was not certain whether he had a history of stroke or syncope contributing to his altered sensorium with no motor or sensory deficits.
When he was assessed at the geriatric medical clinic, he developed significant memory deficit and abnormal cognitive function. He was started on dilantin for an unspecified type of seizure, metformin for his diabetes mellitus and diclofenac for his unspecified pain syndrome.
His wife volunteered that his overall level of functioning declined substantially. He scored16/30 on the Folstein Mini-Mental State Examination and showed poor orientation and inability to recall the 3 objects after 3 minutes. He was able to complete the simple 3-stage command but definitely had difficulty with attention and calculation. He was unable to properly order the numbers in the clock face test, was unsuccessful in responding to that abstract request, and was unable to fill in the hands of the clock, writing the time by marks of x’s and lines. His wife described his mood was not depressed. He did not present any neuro-vegetative features of depressio . Subjectively he felt quite satisfied with his life and failed to describe any negative or depressed mood state which would be consistent with his history of major
depressive disorder or any other mood swings. His history was inconsistent with the category of mood disorder under the recent DSM V (Diagnostic and Statistical Manual). He denied perceptual disturbances of hallucinations, aberrant thought disturbances of delusions. During the daytime did not display any disorganized bizarre behavior. He did not report any delusions or suicidal or homicidal ideation.
Although retired, he began to wake up at 3:00-4:00 AM, get dressed for work and leave his apartment. Sometimes, when he was redirected, he undressed, went back to bed and fell asleep. More frequently, he would wake up in the middle of the night, get dressed, leave the apartment and wander the streets or get on the bus without knowing where he was going.
He was brought home by police cruisers on several occasions, as he had informed the police, when asked, that he did not know where he was and did not know how to get home. His wife indicated that he had circular conversation and spent more time in the past than in the present, quickly forgot the present, and appeared confused and disoriented for place.
At follow-up examination about one week following his initial visit, the examining geriatrician suggested to his wife to purchase and install a hotel emergency alarm in her apartment door so that she would be awakened if her husband tried to leave the apartment at night. Drug therapy was not prescribed. A recommendation was made for the patient was to be registered with the Alzheimer Wandering Patient Registry.
Three days following the initial visit, he was seen in general medicine clinic patient, accompanied by his wife. His wife was concerned that the installation of an emergency alarm on her apartment door was not a viable option, since her husband became confrontational whenever she tried to prevent him from leaving the apartment at night.
At the general medicine clinic, the alternative treatment option with SS was discussed with him and his wife. They were advised of the results of previous investigations, showing that elevated plasma homocysteine levels were associated with Alzheimer’s disease. In addition, the couple was introduced to the data from a recently completed study indicating that Sublimed Sulfur markedly reduced elevated plasma homocysteine to normal levels of <10 µmol/litre. In view of the lack of sustained efficacy of AD medication targeting wandering behavior for AD, the patient agreed to a course of treatment with SS.
Within 12 hours after SSul was started at oral dosage of 200 mg , the patient’s wife telephoned to report that after taking the first dose of SS at noon the previous day, the patient had remained asleep throughout the night and all the next morning and that she had not been able to arouse him for breakfast or lunch. Hypersomnolence was identified as the likely side effect of SS. She was advised to allow him to remain asleep for as long as he wished, and to get him up for his meals and medications. By day 3 of SS treatment , the patient woke up fully alert He stopped wandering and slept normally at night during the treatment with SS, subsequently, did not wander at any time after the 4 week course of treatment with Sublimed Sulfur was completed.
Case Vignette 2: An 81-year-old female patient of Caucasian descent presented with eating refusal in the context of mood and behavioral changes after she was diagnosed as AD. We noted her past medical history included ischemic stroke related to her severe generalized and cerebral arteriosclerosis. Previous MRI scan of her head revealed 80% left-sided internal carotid artery stenosis, cortical atrophy with old posterior parietal lobe infarcts and bilateral lentiform and cerebellar infarcts. It is evident that her long term and short-term memory deficits followed the onset of her cerebrovascular (CVA) event. Her clinical picture was consistent with AD with mixed subtype of vascular dementia. Her progressive long-term and short-term memory deficit for several years was recently accompanied with moderately severe episodic confusion and affective lability. She became worse after she was started on citalopram and continued for 4 months. She failed to respond to citalopram. On the contrary, her confusion, affective lability and wandering behavior worsened markedly while she was on citalopram. While she was prescribed trazodone, citalopram and memantine, her confusion and emotional lability worsened markedly resulting in her wandering behavior. Consequently, she was discontinued from the approved AD medication: memantine, while she was continued on citalopram, and trazodone. Galantamine was added to treatment within the recommended dosage.
She appeared to respond better to the medication regimen of galantamine, citalopram and trazodone with improvement in her mood. She reduced her crying spells and increased her nutritional intake. Despite the apparent behavioral and mood improvement, she continued to deteriorate. Three years later, she had severe cognitive impairment and urinary and fecal incontinence. At follow-up assessment, she had discontinued the galantamine without much deterioration. She was continued on trazodone, citalopram and coenzyme Q with satisfactory control of agitation, maintenance of improved nutritional intake and behaviour. Her hypertension was controlled with Ramipril. In reviewing her medical profile, she was continued on trazodone, ramipril, supplemental folic acid, vitamin B1 and vitamin B6 for the next 2 years. At the end of the two years, her anti-hypertensive medication was changed to metoprolol. Her atrial fibrillation was switched to digoxin. Meanwhile, she was continued on trazodone and supplemental vitamins.
Her overall condition continued to fluctuate and later on, she became increasingly withdrawn from her surroundings. She refused any nutritional intake, and persistent diarrhea, Sublimed Sulfur: SS 200 mg daily was added to treatment, and diarrhoea subsided. She became more alert and resumed nutritional intake within 24 hours. She tolerated the addition of donepezil to her medication regimen. She remained in stable condition and was continued on daily oral dosage of 200 mg of SSul. She was stable and continued on donezepil, trazodone, metoprolol, digoxin and supplemental vitamins for 3.5 months. Her caregiver decided to discontinue the trial of SSul afterwards.
Table 3b. Changes from basal of Fasting Erythrocyte Folic Acid, Serum Vitamin B12 and Serum Lipids after Treatment with Sublimed Sulfur in Subjects Group 3b:without prescribed Lipid-lowering drugs: statins
Participants not on Lipid Lowering Drugs |
Group 3b |
|
|
|
|
|
|
Total Cholesterol |
< 5·2 mmol/L |
29 |
5·46 ± 0·15 |
5·54 ± 0·17 |
5·54 ± 0·17 |
|
|
|
|
|
|
High-Density Lipoprotein Cholesterol |
0·77 - 1·68 mmol/L |
29 |
1·37 ± 0·08 |
1·37 ± 0·07 |
1·36 ± 0·07 |
|
|
|
|
|
|
Low-Density Lipoprotein Cholesterol |
< 3·4 mmol/L |
29 |
3·33 ± 0·15 |
3·42 ± 0·16 |
3·51 ± 0·18 |
|
|
|
|
|
|
Total/High Density Lipoprotein Cholesterol |
up to 5 mmol/L |
29 |
4·37 ± 0·25 |
4·36 ± 0·26 |
4·42 ± 0·27 |
|
|
|
|
|
|
Triglycerides |
0·70 - 3·2 mmol/L |
29 |
1·61 ± 0·12 |
1·63 ± 0·14 |
1·47 ± 0·10 |
|
|
|
|
|
|
1. 24 hours after discontinuation of a 30 day course of Sublimed Sulfur. |
2. 30 days after discontinuation of a 30 day course of Sublimed Sulfur. |
3. Significant reduction from baseline (p=0·002) 24 hours after discontinuation of Sublimed Sulfur. |
|
Numerical values indicate the mean and standard error of the mean. Probability values according to Wilcoxon test comparing basal levels with levels after therapy show no significant difference. |
Discussion
In our study, we demonstrate for the first time elemental sulfur, formulated as the proprietary product, Sulmedol with 99% purity, attenuates fHcy level from baseline level of fHCy > 10.0 μMol/L in cognitive normal subjects. The positive effect of SS on fHcy occurred without markedly altering the dynamics of Vitamin B and folate metabolism. Our results provide the basis of fHcy as the pharmacological engagement biomarker of SS to be further validated in clinical trial of AD. Furthermore, we reported in two AD patients at the moderate-advanced stage of AD, SS at 200 mg daily dosage of 200 mg brought about marked improvement in the neurobehavioral symptoms: eating refusal and wandering behavior. We have demonstrated for the first time beneficial effects of 3-month treatment with oral SS at 200 mg dosage, on the neurobehavioral symptoms of AD, SS was highly tolerated with no adverse events.
Our preliminary study of SS in AD is consistent with pivotal roles of Hcy and vitamin B12 and folate as AD risk factors [19,40-42]. A recent meta-analysis of 67 studies [42] using the stata 12.0 statistical software conclude that AD patients distinguish from age-adjusted matched control in having higher levels of Hcy, and lower levels of age-dependent vitamin B12 and folate. High Hcy and low folate levels correlate with the magnitude of AD risk occurrence. Earlier studies on the association of vitamin B12 focus on cyanocobalamin as the functional status of metabolic vitamin B12 storage and utilization; however, functional biomarkers of vitamin B12 consisting of holotranscobalamin (TC) , along with methymalonic acid and total plasma level of Hcy, may be produce a more accurate picture of metabolic vitamin B12/Hcy utilization [43]. From the therapeutic perspective,methycobalamin rather than cyanocobalamin, may be preferable in elderly patients with impaired renal function. A very recent study of the AD risk showed that both total Hcy and holoTranscobalamin (TC) may be involved in normal aging and AD : both plasma biomarkers are correlated with MRI imaging indices of brain volume loss and hyper-density white matter [44,43]. The odds ratios (ORs)
(95% confidence interval [CI]) for AD were 1.16 (1.04-1.31) per increase of 1 μmol/L of tHcy at baseline and 0.980 (0.965-0.995) for each increase of 1 pmol/L baseline holoTC.The putative Hcy-AD may be mediated in part by the serum holoTC. Taken together, converging evidence suggests that plasma level of 10.0 μMol/L may be threshold level of Hcy at risk of AD. Our findings suggest that ingested SS likely induces increase in methionine synthase activity.
Recent advances in our understanding of the multiple complex CNS actions of hydrogen sulfide have shed new lights on the role of Hcy/H2S coupling in aging and AD [24,25,45,46]. There is growing body of evidence to support the likely role of H2S as molecular “switch” that activates downstream pathways that persist long after H2S decay [45]. H2S is usually stored as bound sulfane sulfur in neurons and astrocytes. In this respect, homocysteine diversifies at the Intersection of transulfuration and methionine recycling , as well as participates at H2S/Hcy cross-talks underlying neuro-vascular coupling. As a prototypal gaseous neurotransmitter, hydrogen sulfide is synthesized from the amino acids: cysteine (Cys) and homocysteine (Hcy) by three enzymes: cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and mercaptopyruvate sulfurtransferase (3-MST). H2S modulates NMDA glutamate receptors and regulates intracellular cAMP levels and intracellular Ca(2+) . In this respect H2S functions as a key anti-inflammatory, antioxidant, anti-apoptotic and neuroprotection signaling molecule. Studies show cross-talks between Homocysteine (Hcy) and Hydrogen sulfide are involved in memory paradigm. H2S plays the role of neuromodulator to facilitate the long term potentiation (LTP) correlates of learning. In vitro neuronal culture, H2S regulates intracellular calcium concentration and pH level. In vivo AD model, H2S prevents cognitive impairment in AD model and rescues Abeta toxicity. H2S restores memory function and impaired cerebral blood flow induced by the intracerebral administration of Hcy AD model.
The in vitro and in vivo translational studies of H2S are highly consistent with clinical findings. In postmortem AD brains, H2S levels were found to be markedly reduced compared with non- AD matched control [26]. A recent clinical AD study [27] showed that plasma level of hydrogen sulfide was significantly lower than in control: the severity of AD was inversely correlated with H2S and directly correlated with the plasma total homocysteine levels. These considerations expand the pharmacology of Sulmedol beyond regulating homocysteine level. We propose that elemental sulfur to function in vivo as a potential hydrogen sulfide releasing action to exert concerted anti-oxidant, anti-inflammatory and anti-apoptotic actions in synaptic remodeling in aging and in AD.
In our clinical vignette, we did not have any data on changes in H2S and Hcy to correlate with the beneficial effects in AD. Nevertheless, our preliminary findings on Sulmedol on lowering Hcy levels in normal subjects, and in improving the neurobehavioral symptoms in moderate- advanced AD stage, raise the relevance of Sulmedol in targeting Hcy/H2S link in development of novel AD therapeutics. Intervention trials using B-vitamin supplementation, however, have produced somewhat inconsistent findings on cognitive function [18-22]. In reviewing the plethora of many cross-sectional and prospective studies, increased levels of plasma total homocysteine are related to the severity of cognitive impairment and AD and vascular dementia. Methodological differences in the heterogeneous, patient population, the duration and dosage of B-vitamin may partially explain the discrepant findings.
Differences in baseline levels of Hcy in the various clinical studies may be the important determinant in the direction of therapeutic responses. Elevated baseline fHcy subjects may be more likely to show positive cognitive responses to B-vitamin supplementation [47]. In the randomized placebo-control trial: VTIACOG trial (47, 48, 49, 50), 24-month treatment with combined folic acid (0.8 mg/d) with vitamin B(12) (0.5 mg/d) and vitamin B(6) (20 mg/d) significantly slowed the rate of cognitive atrophy as measured by MRI in subjects diagnosed as MCI compared with placebo control group. The treatment response was related to baseline homocysteine levels: the rate of atrophy in participants with homocysteine >13µmol/L was 53% lower in the active treatment group (P= 0.001). The accelerated rate of cortical atrophy was related to the lower final cognitive test scores. A greater rate of atrophy was associated with a lower final cognitive test scores. In addition, baseline level of omega 3-fatty acids(eicosapentaenoic acid and docosahexaenoic acid) may influence cognitive response towards Vitamine B supplementation [49,50]. A randomized control study in MCI elderly subjects showed that the B vitamin treatment had no significant effect on the rate of atrophy among subjects with low baseline ω-3 fatty acids (<390 μmol/L). High baseline ω-3 fatty acids were linked with a slower rate of brain atrophy only in the vitamin B group but not in the placebo group [49,50]. While low folate level may be the risk factor for cognition decline, supplementation studies with folate remain inconclusive [22,51]. Taken together, diet enriched in omega-3-fatty acids and folate can synergize with Vitamin B supplementation supplementation in preventing cognitive decline.
Our current study expands our earlier study of SS in lactose tolerance to AD subjects. Since GALAC is involved in both AD and lactose intolerance, the effects of SS in AD are not dependent on any prior history of lactose intolerance. GALAC is the signaling molecule linking gut-brain nexis mediating the pivotal role of lysosomes in regulating cellular homeostasis in the intestine and in the brain. Dysregulation of lysosomal function has been shown to give rise in lysosomal storage diseases: ganglionsidosis disorders with devastating consequences in the pediatric population. In aging , the phenomenon of lysosomal dysregulation reemerge in neurodegenerative disorders , reflecting epigenetic regulation of lysosome enzyme complexes. in both disorders [52]. In lysosomes, the degradation of complex, macromolecular substrates depends upon the synergistic action of multiple hydrolases working in a well-defined stepwise manner. We propose that AD may be mimicry of lysosomal dysregulation disorder driving the autophagy system in a chaotic manner among the elderly. We are not aware of specific drug leads or pipeline drugs targeting GALAC-driven lysosomal autophagy system in AD. Elemental sulfur sets the central stage in resetting the defects in GALAC in AD through the thiol redox signaling and the synthesis of cystine and disulfide bridges. Tau hyper- phosphorylation can be reduced through restoring the activities of kinases and phosphatases.
Previous is growing evidence suggesting that clearance of toxic Abeta or phospho-Tau can further attenuate the inflammatory process and maybe modify the course of AD.
Our approach in repurposing Sulmedol’s approved indication for lactose intolerance for potential AD therapeutics is strongly based on translational studies of the emerging role of lysosomal autophagy in AD. In the localization of β-hexosaminidase and β-galactosidase glycohydrolases, both involved in step by step degradation of the GM1 to GM3 gangliosides, in lipid microdomains from the cortex of both an early and advanced TgCRND8 mouse model of Alzheimer's disease. For the first time in AD patients, β-hexosaminidase and β-galactosidase were found to be associated with post-synaptic vesicles, and at early and advanced stage of AD [31]. The activities of both lysosomal enzymes are found to be increased and that their activities are increased at both the early and the advanced stage of Alzheimer's disease. A recent study [30] using the innovative non-invasive technique of processing and harvesting neutrally derived blood exosomes found that the levels of exosome-derived auto-Lysosome sharply discriminated AD patients from control subjects, at the prodromal phase of AD. Levels of cathepsin D, and ubiquitinylated protein also were significantly higher for AD patients than for frontotemporal dementia patients. The early increase of lipid microdomain-associated β- hexosaminidase and β-galactosidase activities may have relevant implications for the course of AD. Type II diabetes mellitus (T2DM) is known to be a risk factor for AD. A recent study found evidence for upregulation of lysosomal enzymes [32]. Plasma β-Galactosidase and β-Hexosaminidase levels were higher in patients with AD-T2DM compared to those with T2DM, suggesting that both β-Galactosidase and β-Hexosaminidase activities, either intracellular or plasma-secreted, may be used to discriminate AD patients from controls and AD-T2DM from T2DM patients.
A recent study found homocysteine levels correlate with the severity of neurobehavioral symptoms : behavioral agitation , delusions, and hallucinations in AD [53], calling into question whether fHcy and H2S levels can be used to monitor treatment responses of neurobehavioral symptoms, beyond the cognitive symptoms , in the early, mid- and advanced stages of AD. Furthermore a critical level of homocysteine reflecting the neurotoxic burden on homocysteine on synaptic plasticity and tau phosphorylation and beta-amyloid accumulation, has been demonstrated to correlate with brain atrophy in AD [48-50]. We have yet to measure the level of H2S in plasma of AD to determine whether the elevated levels of Hcy occur in parallel with reduced plasma level of H2S or derived measures of H2S.
From the perspective of GALAC-mediated autophagy, we have to demonstrate whether in AD, the changes in lysosomal enzyme, GALAC are reversed with SS treatment and correlate with the clinical responses in AD. The efficacy of Sulmedol is most likely related to its action in restoring redox balance and buffering the oxidative stress load, as well as in shifting the host defense towards anti- inflammatory phenotype. Modulation of autophagy has entered to the main stage of AD drug development. Recently, the promising findings of the old compound, methylthioninium chloride (methylene blue) inhibits Tau aggregation through inducing autophagy [54,55]. At nanomolar concentrations, MB can reduce Tau levels in both organotype brain slices from the mouse model of frontal-temporal dementia (FTD). MB altered levels of cathepsin D and BECN1 the mammalian ortholog of the yeast autophagy-related gene 6 , consistent with MB’s action as the potent inducer of autophagy. Besides, MB has also been shown to inhibit mitochondrial caspase through oxidation (sulfenation) of the active site : 163 cysteine of Caspase 6. MB shares with elemental sulfur in targeting GALAC. The molecular mechanism of action of MB is reminiscent of the specific role of conformational changes related to the disulfide formation between cysteine residues: C500 and C536, in close proximity to the active site of GALAC [56]. GALAC is a model enzyme to illustrate the known evolutionary progression from the microbial world: E. coli to the mammalian system including humans. It is noteworthy that the complex functions and catalytic mechanism of lacZ β-galactosidase inspire Jacob and Monod's development of the operon model for the regulation of gene expression. GALAC regulating brain-gut nexus across life cycle represents novel landmark in creative drug development. These mechanistic considerations of the overlapping molecular sites may suggest that elemental sulfur behaves similarly to MB in inhibiting Tau and activating autophagy and hence may be efficacious in AD.
Conclusion
Our preliminary study has certain methodological limitations. We have not fully explored the differential pharmacological actions of SS in a wider range of fHCy levels. The findings from two case vignettes do not establish the efficacy of SS in AD. In designing randomized placebo-controlled trial of SS to evaluate the efficacy of SS in AD, we have to include relevant biomarkers to validate the mechanism of action of SS. However, our study of SS in cognitively normal subjects demonstrates that 30-day treatment with oral dosage of SS results in significant reduction in fHcy. The results of our pilot study of SS in AD raise the issue of biomarker to monitor pharmacological engagement paradigm of SS in the clinical arena. Our review of the accumulating evidence suggesting that elemental sulfur hits the dual targets: H2S/Hcy and GALAC, hit by SS, related to the onset and progression of AD. The preliminary study has galvanized our team with urgency and momentum to design RCT trial of Sulmedol in AD. The exploratory study calls into question the potential clinical utility of evaluating the key signal pathways: H2S/Hcy and GALAC as biomarkers in AD for diagnosis and therapeutic monitoring in AD trial. Both signal pathways will enhance the sensitivity, specificity and predictive validity of brain imaging: MRI and PET imaging to detect early AD and even at the prodromal phase of AD. We plan to design biomarker-based randomized controlled trial of Sulmedol in Phase II clinical trial to evaluate the efficacy of SS in AD. Our approach of drug discovery follows the recent innovative trend of repurposing SS approved for lactose intolerance to novel potential AD therapeutics. It remains to be seen whether SS belongs to the class of AD disease modifying agents capable of modifying the symptoms and course of AD and even to present the onset of AD in the stage of Mild Cognitive Impairment (MCI) among the elderly.
Acknowledgement
The patented compound, Sulmedol is owned by. Airudin Khan MD as the inventor of oral formulation of elemental sulfur. Dr. A. Khan designed and carried out the study of Sulmedol in hyper-homocysteinmia and in AD subjects. S Chiu MD PhD, A Khan MD and Vladimir Badmeav MD PhD contribute equally towards the interpretation of the results of the study. A Khan designed and carried out the study of Sulmedol in hyper-homocysteinemia and in AD subjects. Zack Cernvosky PhD provides statistical consultation. John Copen MD, Mariwan Husni MD, Mike Farina-Woodbury MD have participated in Epigenetics-based drug discovery program and have contributed towards methodology and targets in cognitive aging and AD. We appreciate the insightful comments from Ann S Johnson, B.Sc. M.Sc. (Engineering) on bio-inorganic chemical perspectives on elemental sulfur: sulfane and hydrogen sulfide as applied to our study. As research trainees and graduate students at Lawson Health Research Institute, University of Western Ontario London ON: Josh Vaughese B.Sc.(Honors) Hana Raheb B.Sc. (Honors) Kristen Terpstra B.Sc. (Honors), M.Sc. B.Sc. (nursing); Autumn Carriere (B.Sc. Honors) Zahra. Khazaeipool MD and Christina Chada B.Sc (Honors) have participated in research on cognitive aging and epigenetics regulation of AD. None of the contributing authors have potential conflict of interest to declare.
References
- Hickman RA, Faustin A, Wisniewski T (2016) Alzheimer Disease and Its Growing Epidemic: Risk Factors, Biomarkers, and the Urgent Need for Therapeutics. Neurol Clin 34: 941-953. [Crossref]
- Cummings J, Aisen PS, DuBois B, Frölich L, Jack CR Jr, et al. (2016) Drug development in Alzheimer's disease: the path to 2025. Alzheimers Res Ther 8: 39. [Crossref]
- Cacciapuoti F (2011) Hyper-homocysteinemia: a novel risk factor or a powerful marker for cardiovascular diseases? Pathogenetic and therapeutical uncertainties. J Thromb Thrombolysis 32: 82-88. [Crossref]
- Lourenço CF, Ledo A, Dias C, Barbosa RM, Laranjinha J (2015) Neurovascular and neurometabolic derailment in aging and Alzheimer's disease. Front Aging Neurosci 7: 103. [Crossref]
- Peng HY, Man CF, Xu J, Fan Y (2015) Elevated homocysteine levels and risk of cardiovascular and all-cause mortality: a meta-analysis of prospective studies. J Zhejiang Univ Sci B 16: 78-86. [Crossref]
- Pang H, Han B, Fu Q, Zong Z (2016) Association of High Homocysteine Levels With the Risk Stratification in Hypertensive Patients at Risk of Stroke. Clin Ther 38: 1184-1192. [Crossref]
- Lai WK, Kan MY (2015) Homocysteine-Induced Endothelial Dysfunction. Ann Nutr Metab 67: 1-12. [Crossref]
- Xiao Y, Su X, Huang W, Zhang J, Peng C, et al. (2015) Role of S-adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int J Biochem Cell Biol 67: 158-166. [Crossref]
- Huang T, Chen Y, Yang B, Yang J, Wahlqvist ML, et al. (2012) Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr 31: 448-454. [Crossref]
- Qin X, Huo Y, Xie D, Hou F, Xu X, et al. (2013) Homocysteine-lowering therapy with folic acid is effective in cardiovascular disease prevention in patients with kidney disease: a meta-analysis of randomized controlled trials. Clin Nutr 32: 722-727. [Crossref]
- Yang HT, Lee M, Hong KS, Ovbiagele B, Saver JL (2012) Efficacy of folic acid supplementation in cardiovascular disease prevention: an updated meta-analysis of randomized controlled trials. Eur J Intern Med 23: 745-754. [Crossref]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, et al. (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med 346: 476-483. [Crossref]
- Seshadri S (2006) Elevated plasma homocysteine levels: risk factor or risk marker for the development of dementia and Alzheimer's disease? J Alzheimers Dis 9: 393-398. [Crossref]
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, et al. (2006) Elevated plasma homocysteine levels: risk factor or risk marker for the development of dementia and Alzheimer's disease ? J Alzheimers Dis 2006 9: 393-398. [Crossref]
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, et al. (2005) Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr 82: 636-643. [Crossref]
- Wald DS, Kasturiratne A, Simmonds M (2011) Serum homocysteine and dementia: meta-analysis of eight cohort studies including 8669 participants. Alzheimers Dement 7: 412-417. [Crossref]
- Beydoun MA, Beydoun HA, Gamaldo AA, Teel A, Zonderman AB, et al. (2014) Epidemiologic studies of modifiable factors associated with cognition and dementia: systematic review and meta-analysis. BMC Public Health 14: 643. [Crossref]
- Setién-Suero E, Suárez-Pinilla M, Suárez-Pinilla P, Crespo-Facorro B, Ayesa-Arriola R (2016) Homocysteine and cognition: A systematic review of 111 studies. Neurosci Biobehav Rev 69: 280-298. [Crossref]
- Hankey GJ, Ford AH, Yi Q, Eikelboom JW, Lees KR, et al. (2013) Effect of B vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: a prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke 44: 2232-2239. [Crossref]
- Ford AH, Almeida OP (2012) Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. J Alzheimers Dis 29: 133-149. [Crossref]
- McCaddon A, Miller JW (2015) Assessing the association between homocysteine and cognition: reflections on Bradford Hill, meta-analyses, and causality. Nutr Rev 73: 723-735. [Crossref]
- Hinterberger M, Fischer P (2013) Folate and Alzheimer: when time matters. J Neural Transm (Vienna) 120: 211-224. [Crossref]
- Rai V (2016) Methylenetetrahydrofolate Reductase (MTHFR) C677T Polymorphism and Alzheimer Disease Risk: a Meta-Analysis. Mol Neurobiol.
- Kimura H (2011) Hydrogen sulfide: its production, release and functions. Amino Acids 41: 113-121. [Crossref]
- Wei HJ, Li X, Tang XQ (2014) Therapeutic benefits of Hâ‚‚S in Alzheimer's disease. J Clin Neurosci 21: 1665-1669. [Crossref]
- Eto K, Asada T, Arima K, Makifuchi T, Kimura H (2002) Brain hydrogen sulfide is severely decreased in Alzheimer's disease. Biochem Biophys Res Commun 293: 1485-1488. [Crossref]
- Liu XQ, Liu XQ, Jiang P, Huang H, Yan Y (2008) [Plasma levels of endogenous hydrogen sulfide and homocysteine in patients with Alzheimer's disease and vascular dementia and the significance thereof] 88: 2246-2249. [Crossref]
- Friedman LG, Qureshi YH, Yu WH (2015) Promoting autophagic clearance: viable therapeutic targets in Alzheimer's disease. Neurotherapeutics 12: 94-108. [Crossref]
- Wong YC, Holzbaur EL (2015) Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci 128: 1259-1267. [Crossref]
- Goetzl EJ, Boxer A, Schwartz JB, Abner EL, Petersen RC, et al. (2015) Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85: 40-47. [Crossref]
- Magini A, Polchi A, Tozzi A, Tancini B, Tantucci M, et al. (2015) Abnormal cortical lysosomal ?-hexosaminidase and ?-galactosidase activity at post- synaptic sites during Alzheimer's disease progression. Int J Biochem Cell Biol 58: 62-70. [Crossref]
- Tiribuzi R, Orlacchio A, Crispoltoni L, Maiotti M, Zampolini M, et al. (2011) Lysosomal β-galactosidase and β- hexosaminidase activities correlate with clinical stages of dementia associated with Alzheimer's disease and type 2 diabetes mellitus. J Alzheimers Dis 24: 785-797. [Crossref]
- Kalanj-Bognar S, Rundek T, Furac I, Demarin V, Cosović C (2002) Leukocyte lysosomal enzymes in Alzheimer's disease and Down's syndrome. J Gerontol A Biol Sci Med Sci 57: B16-21. [Crossref]
- Di Rienzo T, D'Angelo G, D'Aversa F, Campanale MC, Cesario V, et al. (2013) Lactose intolerance: from diagnosis to correct management. Eur Rev Med Pharmacol Sci 17 Suppl 2: 18-25. [Crossref]
- Deng Y, Misselwitz B, Dai N, Fox M (2015) Lactose Intolerance in Adults: Biological Mechanism and Dietary Management. Nutrients 7: 8020-8035. [Crossref]
- Khan AS (2003) The Effect of Ingested Sublimed Sulfur on Lactose Intolerance. Clin Invest Med
- Guzmán R, Campos C, Yuguero R, Masegù C, Gil P, et al. (2015) Protective effect of sulfurous water in peripheral blood mononuclear cells of Alzheimer's disease patients. Life Sci 132: 61-67. [Crossref]
- Cole DE, Lehotay DC, Evrovski J (1998) Simplified simultaneous assay of total plasma homocysteine and methionine by HPLC and pulsed integrated amperometry. Clin Chem 44: 188-190. [Crossref]
- Shipchandler MT, Moore EG (1995) Rapid, fully automated measurement of plasma homocyst(e)ine with the Abbott IMx analyzer. Clin Chem 41: 991-994. [Crossref]
- Smith AD, Smith SM, de Jager CA, Whitbread P, Johnston C, et al. (2010) Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One 5: e12244. [Crossref]
- de Jager CA (2014) Critical levels of brain atrophy associated with homocysteine and cognitive decline. Neurobiol Aging 35 Suppl 2: S35-39. [Crossref]
- Shen L, Ji HF (2015) Associations between Homocysteine, Folic Acid, Vitamin B12 and Alzheimer's Disease: Insights from Meta-Analyses. J Alzheimers Dis 46: 777-790. [Crossref]
- Hooshmand B, Mangialasche F, Kalpouzos G, Solomon A, Kåreholt I, et al. (2016) Association of Vitamin B12, Folate, and Sulfur Amino Acids With Brain Magnetic Resonance Imaging Measures in Older Adults: A Longitudinal Population-Based Study. JAMA Psychiatry 73: 606-613. [Crossref]
- Hooshmand B, Solomon A, Kåreholt I, Leiviskä J, Rusanen M, et al. (2010) Homocysteine and holotranscobalamin and the risk of Alzheimer disease: a longitudinal study. Neurology 75: 1408-1414. [Crossref]
- Kamat PK, Kalani A, Tyagi N (2015) Role of hydrogen sulfide in brain synaptic remodeling. Methods Enzymol 555: 207-229. [Crossref]
- Nagpure BV, Bian JS (2015) Brain, Learning, and Memory: Role of H2S in Neurodegenerative Diseases. Handb Exp Pharmacol 230: 193-215. [Crossref]
- Smith AD, Smith SM, de Jager CA, Whitbread P, Johnston C, et al. (2010) Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One 5: e12244. [Crossref]
- de Jager CA, Oulhaj A, Jacoby R, Refsum H, Smith AD (2012) Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: a randomized controlled trial. Int J Geriatr Psychiatry 27: 592-600. [Crossref]
- Oulhaj A, Jernerén F, Refsum H, Smith AD, et al. (2016) Omega-3 Fatty Acid Status Enhances the Prevention of Cognitive Decline by B Vitamins in Mild Cognitive Impairment. J Alzheimers Dis 50: 547-557. [Crossref]
- Jernerén F, Elshorbagy AK, Oulhaj A, Smith SM, Refsum H, et al. (2015) Brain atrophy in cognitively impaired elderly: the importance of long-chain ω-3 fatty acids and B vitamin status in a randomized controlled trial. Am J Clin Nutr 102: 215-221. [Crossref]
- Araújo JR, Martel F, Borges N, Araújo JM, Keating E (2015) Folates and aging: Role in mild cognitive impairment, dementia and depression. Ageing Res Rev 22: 9-19. [Crossref]
- Bonten EJ, Annunziata I, d'Azzo A (2014) Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci 71: 2017-2032. [Crossref]
- Zheng Z, Wang J, Yi L, Yu H, Kong L, et al. (2014) Correlation between behavioural and psychological symptoms of Alzheimer type dementia and plasma homocysteine concentration. Biomed Res Int 2014: 383494. [Crossref]
- Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, et al. (2015) Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis 44: 705-720. [Crossref]
- Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, et al. (2015) Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer's disease. J Pharmacol Exp Ther 352: 110-118. [Crossref]
- Juers DH, Matthews BW, Huber RE (2012) LacZ β-galactosidase: structure and function of an enzyme of historical and molecular biological importance. Protein Sci 21: 1792-1807. [Crossref]