The Objectives were twofold, first to evaluate the efficacy of steroids in refractory epilepsy in children, and second, to assess the effectiveness of the steroid treatment protocols used. We included 30 patients with medically refractory epilepsy, according to the international definition for the disorder. The data analyzed included epilepsy type and etiology, seizure reduction, and type of the steroid treatment protocols associated with the best seizure outcome. Our data showed a statistically significant seizure reduction in children with structural etiology and immune-mediated/infectious etiologies at 15 days after the steroid treatment initiation which was still sustained after at 30 days after starting steroids. In comparison, refractory epilepsy secondary to genetic causes did not show similar response to steroid treatment. While there isn’t significant steroid response on seizure control to follow-up at 6 months, regardless of the etiological cause. The second aim of this study was to evaluate the efficiency of the 4 steroid treatment protocols used for seizure control in children with medically refractory epilepsy. Out of the four therapeutic used in our study, the protocols with best efficacy were the IV methylprednisolone 30 mg/Kg/day for 3 days and IV methylprednisolone 15 mg/Kg/day for 5 days. In the DRE, inflammatory mediators are likely to significantly contribute to the onset and recurrence of seizures. What our study adds, is providing supportive evidence on efficacy of steroids in drug resistant epilepsy. Use of steroids for refractory seizures appears to be safe, and it is associated with clinical reduction of seizures as well as EEG improvement.
drug resistant epilepsies, steroid-therapy, neuroinflammation
Drug resistant epilepsies (DRE) are defined as epilepsies resistant to two or more antiepileptic drugs (AEDs), appropriately chosen and used whether as monotherapies or in combination [1]. DRE is challenging condition affecting 10-15% of all children with epilepsy; it is associated with a significant morbidity due to the high seizure burden and multiple medications used. Recent studies have shown that neuroinflammation may both precipitate and sustain seizures, implying that inflammation may be involved not only in epileptogenesis, but also in the development of the drug-resistant profile. Due to their role in the immunomodulation and immunosuppression, steroids have been considered potential candidates in the ongoing search for novel anti-epileptic therapies [2]. The mechanism of action of steroids is thought to be the interference with the inflammatory cascade believed to trigger ongoing seizures [3-6]. With this hypothesis in mind, several different treatment protocols have been used in studies with the goal to break the seizure refractoriness. Currently, there is no clear consensus which treatment protocols is the most efficacious. The objectives of this study were twofold, first to evaluate the efficacy of steroids in refractory epilepsy in children, and second, to assess the effectiveness of the steroid treatment protocols used.
This was a retrospective study that included all DRE patients treated with steroids in Pediatric Neurology Centers of Catania, Padova, Chieti, Troina, Palermo and Rome between 2015-2017. The following data information were collected for all study participants: gender, epilepsy etiology, age at epilepsy onset, seizure type, seizure frequency, video-EEG in wakefulness and sleep, presence or absence of developmental delay, age at treatment initiation, antiepileptic drugs tried, steroid type, steroid treatment dose, duration of steroid therapy, effect of steroid therapy on seizure frequency and EEG (Table 1).
Table 1. Analyzed data in all 30 patients
Patient |
Gender |
Etiology |
Seizure type (s) |
Age. at onset (m) |
Frequency (daily) |
Psychomotor delay/mental delay |
EEG background |
EEG: Epileptic anomalies |
Steroid Therapy |
1 |
M |
C |
1 |
90 |
20 |
no |
a |
2 |
A |
2 |
M |
A |
1 |
4 |
40 |
yes |
b |
2 |
B |
3 |
F |
A |
5 |
3 |
20 |
yes |
b |
1 |
B |
4 |
M |
C |
3 |
neonatal seizures |
50 |
yes |
b |
2 |
B |
5 |
F |
B |
2 |
20 |
25 |
yes |
c |
2 |
B |
6 |
M |
C |
3 |
3 |
10 |
yes |
a |
1 |
B |
7 |
F |
B |
3 |
36 |
15 |
yes |
a |
1 |
B |
8 |
F |
A |
2 |
8 |
30 |
yes |
c |
3 |
B |
9 |
F |
C |
3 |
48 |
10 |
no |
a |
1 |
B |
10 |
M |
C |
5 |
neonatal seizures |
120 |
yes |
c |
1 |
C |
11 |
M |
C |
5 |
3 |
30 |
yes |
a |
1 |
C |
12 |
F |
B |
4 |
neonatal seizures |
50 |
yes |
a |
1 |
A |
13 |
M |
C |
2 |
10 |
10 |
yes |
b |
3 |
A |
14 |
M |
C |
5 |
12 |
2 |
yes |
b |
3 |
A |
15 |
M |
C |
5 |
36 |
10 |
no |
a |
1 |
A |
16 |
F |
B |
5 |
72 |
20 |
no |
c |
1 |
A |
17 |
M |
A |
3 |
1 |
10 |
yes |
c |
2 |
C |
18 |
M |
C |
3 |
52 |
20 |
yes |
a |
2 |
C |
19 |
F |
C |
5 |
48 |
30 |
yes |
a |
2 |
D |
20 |
M |
C |
3 |
7 |
5 |
yes |
c |
1 |
D |
21 |
F |
C |
3 |
96 |
4 |
yes |
b |
1 |
C |
22 |
F |
C |
3 |
12 |
2 |
yes |
a |
1 |
A |
23 |
M |
B |
4 |
118 |
100 |
yes |
b |
1 |
B |
24 |
F |
B |
1 |
24 |
20 |
yes |
b |
1 |
D |
25 |
M |
A |
2 |
96 |
4 |
yes |
b |
3 |
D |
26 |
F |
A |
2 |
3 |
20 |
yes |
c |
1 |
D |
27 |
M |
A |
4 |
72 |
30 |
yes |
b |
1 |
D |
28 |
F |
A |
4 |
18 |
30 |
yes |
a |
1 |
C |
29 |
M |
A |
5 |
2 |
20 |
yes |
c |
3 |
D |
30 |
F |
C |
2 |
8 |
20 |
yes |
c |
1 |
C |
Legends:
Etiology |
A |
Genetic/unknown |
| |
B |
Immune/infectious |
| |
C |
structural |
| |
|
|
seizure type |
1 |
generalized tonic seizures |
| |
2 |
generalized tonic-clonic seizures |
| |
3 |
focal tonic seizures |
| |
4 |
focal clonic seizures |
| |
5 |
focal onset to generalized tonic clonic seizures |
| |
|
|
EEG Background |
a |
Normal |
| |
b |
disorganized |
| |
c |
Slow |
| |
|
|
EEG: Epileptic anomalies |
1 |
Focal |
| |
2 |
Multifocal |
| |
3 |
Generalized |
Objectives
The primary objective was to define whether the administration of steroids, regardless of the treatment protocol used, is associated with a significant reduction in the seizure frequency, and which steroid treatment protocols was the most efficacious.
The secondary objective was to determine whether the effect of steroids differs based on the etiology of the refractory epilepsy.
Inclusion criteria
Children between 6 months and 14 years, with drug resistant epilepsy and meeting the following criteria were included:
- Diagnosis of DRE according to the classification of the International League Against Epilepsy (Commission on Classification and Terminology of the International League Against Epilepsy) [1].
- Treatment with steroids as add-on agent to the other antiepileptic drugs (AEDs).
- Treatment with at least two AEDs for minimum of 4 months.
- No change in AEDs for at least 8 weeks immediately prior to enrollment to our study.
Exclusion criteria
- Patients over 14 years of age.
- Patients diagnosed with any of the following: West syndrome (infantile spasm), syndrome of electrical status epilepticus during slow wave sleep (ESES) or Landau-Kleffner syndrome (LKS).
- Patients with history of malignancy within past 3 years prior to randomization.
- Patients with a moderate to severe hepatic and/or renal impairment.
- Patients with any severe and/or uncontrolled medical conditions at randomization such as: symptomatic congestive heart failure, cardiac arrhythmias, congenital heart diseases or any other clinically significant cardiac disease; significant symptomatic deterioration of lung function.
- Patient with uncontrolled diabetes or with uncontrolled hyperlipidemia.
- Patients with a history of organ transplant.
- Patients receiving any anti-cancer therapies or who have received anti-cancer therapy within 4 weeks of study entry; including chemotherapy, radiation therapy, antibody-based therapy.
- Patients receiving chronic, systemic treatment with corticosteroids or another immunosuppressive agent at study entry.
Methods
Steroid treatment was used as an add-on therapy to the conventional anticonvulsants.
Steroid treatment was initiated if the conventional anticonvulsants were ineffective after 4 months of treatment.
The study participants were randomized into four groups according to four steroid therapy protocols:
- IV methylprednisolone 30 mg/Kg/day for 3 days (P1)
- IV methylprednisolone 15 mg/Kg/day for 5 days, followed by oral prednisolone 1.2 mg/Kg/day for 4 weeks (P2)
- Hydrocortisone 10mg/kg for 3 months (P3)
- Dexamethasone 6 mg/day for 5 days (P4)
Follow-up was performed by a pediatric neurologist who documented the therapeutic response. The response to steroid treatment was based on seizure diaries filled by the patients’ parents. Seizure diaries included seizure characteristics, frequency, intensity and duration, similarly to the study performed by Fisher, et al. [7]. The reduction of seizure frequency was assessed prior to the initiation of the steroid therapy (T0),15 (T15) and 30 (T30) days after the steroid treatment initiation and 6 months (T180) after the steroid treatment initiation. For our study purposes, therapeutic outcomes were considered as complete (no seizures), good (>50% seizure reduction), fair (<50% seizure reduction), or none (no response).
Statistical analysis
Statistical analysis was performed using JMP® 14.3.0 program for Mac by SAS Institute inc.
For all variables the approximation to normal of the distribution of the population was tested by Kolmogorov-Smirnov One-Sample Test and statistics for kurtosis and symmetry. As results were asymmetrically distributed, non-parametric tests were used. Data are expressed as mean ± standard deviation.
We used the Kruskal-Wallis nonparametric one-way analysis of variance to examine the changes in seizure frequency at T0, T15, T30 and T180. The null hypothesis was that the groups of the study all came from the same distribution. When the Kruskal-Wallis test was significant we used the Wilcoxon test to compare the intragroup differences at the four observation times (T0, T15, T30, T180). A p value <0.05 was considered significant (Table 2).
Table 2. Means and StdDeviations
Level |
Number |
Mean |
StdDev |
StdErrMean |
Lower 95% |
Upper 95% |
T0 |
30 |
17,7 |
6,4120038 |
1,1706664 |
15,305718 |
20,094282 |
T15 |
30 |
12,3 |
5,5531911 |
1,0138693 |
10,226404 |
14,373596 |
T30 |
30 |
8,8 |
4,5288348 |
0,8268483 |
7,1089053 |
10,491095 |
T180 |
30 |
6,4666667 |
4,7469288 |
0,8666667 |
4,6941343 |
8,239199 |
Frequency: number of convulsive episodes per day
Kruskal-wallis test p < 0.0001
T0 vs T15 p < 0.0072
T0 vs T30 p < 0.0001
T0 vs T180 p < 0.0001
T15vsT180 p < 0.0003
T15 vs T30 p NS
T30 vsT180 p NS
30 participants were included in the study; 16 were males, 14 were females, mean age of 34.48 ± 36.77 months. All patients were diagnosed with DRE, according to the standard international criteria. The etiologies were as follows: 1. Group A -genetics/unknown (30 %), 2. Group B - immune-mediated/infectious (20 %), and3. Group C - structural abnormalities (50 %) of cases.
The patients presented with five main types of seizures semiology: 1. generalized tonic seizures (GTS) in 10 % cases(S1),2. generalized tonic-clonic seizures (GTCS) in 20 % patients(S2), 3. focal tonic seizures in 30% cases(S3), 4. focal clonic seizures in 13,33% cases(S4) and5. focal onset evolving into generalized tonic-clonic seizures (FOGTCS) in 26,66 % cases(S5).
In all the 30 patients undergoing steroid therapy, regardless of the treatment protocol, there was a statistically significant reduction in seizure frequency at 15 days after the treatment initiation which remained statistically significant at 30-day follow up. While there isn’t significant steroid response on seizure control, regardless of the etiological cause (Figure 1).
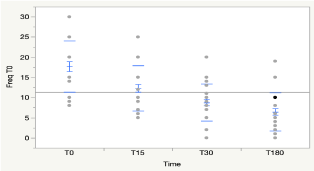
Figure 1. Frequency of daily episodes of convulsions in all patients subjected to steroid therapy
Table 3 shows the frequency trend of daily seizures at T0, T15 and at T30 points in all three etiologic groups. While patients in groupA (9 patients) did not show a statistically significant reduction in seizure frequency following the treatment with steroids at any point, the children in group B (6 patients) showed a statistically significant reduction in seizure frequency after 30 days of treatment with steroids, and the patients in group C (15 patients) showed a statistically significant reduction of the seizure frequency at 15 day follow up that remained statistically significant at 30 day follow up.
Table 3. frequency of daily convulsive episodes at T0, T15 and T30, in the three etiologic groups undergoing steroid therapy
Frequency |
T0 |
T15 |
T30 |
Group A |
13,56 ± 3.81 |
10,11 ± 2.42 |
8,89 ± 3.95 |
Group B |
22,5 ± 5.24 |
20,83 ± 3.76 |
11,00 ± 2.19 |
Group C |
18,27 ± 6.77 |
10,2 ± 4.06 |
7,87 ± 5.37 |
Frequency: number of convulsive episodes per day
Group A
Kruskal-Wallis Tests p NS
Wilcoxon Test T0 vs T15 p NS; T0 vs T30 p NS; T15 vs T30 p NS
Group B
Kruskal-Wallis Tests p < 0.0026
Wilcoxon Test T0 vs T15 p NS; T0 vs T30 p<0.0134; T15 vs T30p <0.0128
Group C
Kruskal-Wallis Tests p < 0.0001
Wilcoxon Test T0 vs T15 p< 0.0027; T0 vs T30 p<0.0006; T15 vs T30p NS
Table 4 illustrates the effect of the 4 steroid treatment protocols by evaluating the number of daily seizures at T15 and T30 points. The therapeutic scheme P1 (7 patients: 5 of group A and 2 of group C) and P2 (9 patients: 3 of group A, 3 of group B and 3 of group C) showed a significant reduction in the seizure frequency at 30 days following the initiation of therapy. On the contrary, the therapeutic scheme P3 (7 patients: 5 of group A and 2 of group B) and P4 (7 patients: 2 of group A, 4 of group B and 1 of group C) showed no statistically significant improvement in the daily seizure frequency at T15 and T30 points.
Table 4. effect of the 4 therapeutic protocols, evaluating the variation of the number of daily convulsive episodes at T15 and T30
Frequency |
T0 |
T15 |
T30 |
Protocol P1 |
17,86 ± 5.67 |
12,43 ± 7.28 |
8,43 ± 2.88 |
Protocol P2 |
18,33 ± 7.91 |
13,89 ± 6.90 |
10,22 ± 3.90 |
Protocol P3 |
19,14 ± 6.20 |
12,29 ± 3.95 |
10,57 ± 5.38 |
Protocol P4 |
15,29 ± 5.91 |
10,14 ± 2.79 |
5,57 ± 4.76 |
Frequency: number of convulsive episodes per day
Protocol P1
Kruskal-Wallis Tests p < 0.0301
Wilcoxon Test T0 vs T15p NS; T0 vs T30p NS; T15 vs T30 p <0.0239
Protocol P2
Kruskal-Wallis Tests p < 0.0486
Wilcoxon Test T0 vs T15 p NS; T0 vs T30 p NS; T15 vs T30 p <0.0394
Protocol P3
Kruskal-Wallis Tests p NS
Wilcoxon Test T0 vs T15 p NS; T0 vs T30 p NS; T15 vs T30 p NS
Protocol P4
Kruskal-Wallis Tests p NS
Wilcoxon T0 vs T15 p NS; T0 vs T30 p NS ; T15 vs T30 p NS
Analysis of the relationship between the main seizure semiology (S1-S2-S3-S4-S5) and steroid administration showed a positive response to treatment only for the group of focal tonic seizures (S3) at 30 days of steroid therapy (T0 = 16 ± 6.53; T15 = 10.77 ± 4,59; T30 = 7.11 ± 4.40 Kruskal-Wallis Tests p< 0.0093; Wilcoxon TestT0 vs T15 p NS; T0 vs T30 p<0.0175; T15 vs T30 p NS).
In this retrospective study we analyzed the efficacy of steroid treatment for children with DRE. We included 30 patients with medically refractory epilepsy, according to the international definition for the disorder [1]. The data analyzed included epilepsy type and etiology, seizure reduction, and type of the steroid treatment protocols associated with the best seizure outcome. Our data showed a statistically significant seizure reduction in children with structural etiology and immune-mediated/infectious etiologies at 15 days after the steroid treatment initiation which was still sustained after at 30 days after starting steroids. In comparison, refractory epilepsy secondary to genetic causes did not show similar response to steroid treatment. While there isn’t significant steroid response on seizure control, regardless of the etiological cause, to follow-up at 6 months.
The mechanism of action of corticosteroids in refractory epilepsy remains unclear. Recent studies have suggested that neuroinflammation may both, precipitate and sustain ongoing seizures, leading to theory that neuro inflammatory process may be involved not only in epileptogenesis, but also in the development of the drug-resistant profile [2]. .As a result, immunomodulatory therapies have been considered as potential candidates in the ongoing search for novel anti-epileptic drugs [2]. Steroids have been used [8-9] to treat medically refractory seizures on the basis of their known anti-inflammatory action mediated by modulation and suppression of the immune cascade [8-11]. In clinical practice steroids are used as a first-line treatment for infantile spasms and Landau-Kleffner syndrome, epilepsy with continuous spike-wave in a slow-wave sleep (CSWS) and they are also used early in the treatment of epileptic encephalopathies thought to have inflammatory or autoimmune etiology such as Rasmussen’s encephalitis [11].
Experimental models of neuroinflammation showed that neural damage and the onset of spontaneous recurrent seizures are modulated via complex interactions between innate and adaptive immunity [12]. Moreover, inflammation can also occur as a result of ongoing epileptic process as it has been shown in animal models. Seizure activity was shown to induce neuroinflammation in experimental animals in which recurrent seizures were perpetuated by chronic inflammation [12]. Epileptic seizures induce an inflammatory response within the brain through activation of resident immune cells, i.e. microglia and production of pro-inflammatory cytokines and related molecules [13,14]. The hypothesis based on experimental models, proposing that inflammatory mediators could be contributing to the onset and recurrence of seizures, as well as the presence of inflammatory markers documented in human epileptogenic specimen, points to targeting inflammation-related pathways as another way to control refractory seizures [15]. When we subdivided our patients according of the main seizure semiology, the statistical analysis showed a significant response to cortisone only for the group of focal tonic seizures at 30 days following the initiation of steroid therapy. This is in keeping with previous studies that showed a good response to steroid treatment in children with focal seizures, which were not traditionally believed to be inflammatory in nature [2,16].
Currently there are no studies on the efficacy of steroids in DRE secondary to genetic etiology. In our study, effectiveness of steroids in patients with DRE with genetic etiology was not demonstrated. However; considering a small number of patients with genetic epilepsies enrolled in the study, no definite conclusions could be made at this time and larger studies are needed.
The second aim of this study was to evaluate the efficiency of the 4 steroid treatment protocols used for seizure control in children with medically refractory epilepsy. Out of the four therapeutic used in our study, the protocols with best efficacy were the IV methylprednisolone 30 mg/Kg/day for 3 days (P1) and IV methylprednisolone 15 mg/Kg/day for 5 days (P2). With the limitation of small study sample, there did not appear to be a specific steroid treatment protocol, choice or dosing of corticosteroids, or duration of the therapy that has been shown to be superior for seizure control in children with intractable epilepsy.
A variety of protocols using corticosteroids in treating epilepsy have been reported. Almaabdi, et al. [17] published their centre experience on the use of intravenous methylprednisolone, 15–30 mg/kg/day for 3–4 days given monthly for 3–4 months in children with epilepsy refractory to multiple antiepileptic drug. Sevilla-Castillo, et al. [18], and Bakker [19] reported using methylprednisolone (15–30 mg/kg/day for 3–4 days) followed by a prednisone taper [11,12], while group led by You, et al. [20] used high dosage prednisone (1– 2 mg/kg/day) for 1–2 months, and Yamatogi, et al. [21] using ACTH (10–30 iu/day) given for 10–57 days.
This is a retrospective study focused on steroid treatment for the DRE. In the DRE, inflammatory mediators are likely to significantly contribute to the onset and recurrence of seizures. What our study adds, is providing supportive evidence on efficacy of steroids in children with drug resistant epilepsy. Use of steroids for refractory seizures appears to be safe, and it is associated with clinical reduction of seizures as well as EEG improvement.
There is currently no standard protocol for children with refractory seizures. Our study introduced four protocols used across major Italian pediatric neurology centers. We believe that our study strengthens the hypothesis of efficacy of steroids in the treatment of medically refractory epilepsy. Currently, larger and randomized clinical trials are needed to establish the therapeutic protocol.
The authors declared no conflicts of interest with respect to the research, authorship, and/or publication of this article
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
None of the Authors has received funding for the preparation of the present article.
This clinical research was approved by the ethic committee of the University of Catania.
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, et al. (2010) Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51: 1069-1077. [Crossref]
- Wilson S (2015) Task Force members. In response: Indications and expectations for neuropsychological assessment in routine epilepsy care: Report of the ILAE Neuropsychology Task Force, Diagnostic Methods Commission, 2013-2017. Epilepsia 56:1316-7. [Crossref]
- Sinclair DB (2003) Prednisone therapy in pediatric epilepsy. PediatrNeurol 28: 194-198. [Crossref]
- Marescaux C, Hirsch E, Finck S, Maquet P, Schlumberger E, et al. (1990) Landau-Kleffner syndrome: a pharmacologic study of five cases. Epilepsia 31: 768–777. [Crossref]
- Riikonen R (1983). Infantile spasms: some new theoretical aspects. Epilepsia 24: 159-168. [Crossref]
- Butler T, Ichise M, Teich AF, Gerard E, Osborne J, et al. (2013) Imaging inflammation in a patient with epilepsy due to focal cortical dysplasia. J Neuroimaging 23: 129-131. [Crossref]
- Fisher RS, Blum DE, DiVentura B, Vannest J, Hixson JD, et al. (2012) Seizure diaries for clinical research and practice: limitations and future prospects. Epilepsy Behav 24: 304-310. [Crossref]
- Prasad AN, Stafstrom CF, Holmes GL (1996) Alternative epilepsy therapies: the ketogenic diet, immunoglobulins, and steroids. Epilepsia 37: 81-95. [Crossref]
- Gupta R, Appleton R (2005) Corticosteroids in the managements of the paediatric epilepsies. Arch Dis Child 90: 379-384. [Crossref]
- Yoshikawa H, Yamazaki S, Abe T, Oda Y (2000) Liposteroid therapy for refractory seizures in children. J Child Neurol 15: 702-704. [Crossref]
- Kneen R, Appleton RE (2006) Alternative approaches to conventional antiepileptic drugs in the management of paediatric epilepsy. Arch Dis Child 91: 936-941. [Crossref]
- Vezzani A, Balosso S, Ravizza T (2008) The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun 22: 797-803. [Crossref]
- Vezzani A, Granata T (2005) Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46: 1724-1743. [Crossref]
- Vitaliti G, Pavone P, Mahmood F, Nunnari G, Falsaperla R, et al. (2014) Targeting inflammation as a therapeutic strategy for drug-resistant epilepsies: an update of new immune-modulating approaches. Hum Vaccin Immunother 10: 868-875. [Crossref]
- Roberts DJ, Goralski KB (2008) A critical overview of the influence of inflammation and infection on P-glycoprotein expression and activity in the brain. Expert. Opin. DrugMetab. Toxicol 4: 11-20. [Crossref]
- Marchi N, Granata T, Freri E, Ciusani E, Ragona F et al. (2011) Efficacy of anti-inflammatory ther- apyin a model of acute seizures and in a population of pediatric drug resistant epileptics. PLoSONE 6: e18200. [Crossref]
- Almaabdi KH, Alshehri RO, Althubiti AA, Alsharef ZH, Mulla SN, et al. (2014) Intravenous methylprednisolone for intractable childhood epilepsy. Pediar Neurol 50: 334-336. [Crossref]
- Sevilla-Castillo RA, Palacios GC, Ramirez-Campos J, Mora-Puga M, Diaz-Bustos R (2009) Methylprednisolone for the treatment of children with refractory epilepsy. Neuropediatrics 40: 265-268. [Crossref]
- Bakker DP, Catsman-Berrevoets CE, Neuteboom RF (2015) Effectiveness of a hybrid corticosteroid treatment regimen on refractory childhood seizures and a review of other corticosteroid treatments. Eur J PaediatrNeurol 19: 553–560. [Crossref]
- You SJ, Jung DE, Kim HD, Lee HS, Kang HC (2008) Efficacy and prognosis of a short course of prednisolone therapy for pediatric epilepsy. Eur J Paediatr Neurol 12: 314-320. [Crossref]
- Yamatogi Y, Ohtsuka Y, Ishida T, Ichiba N, Ishida S, et al. (1979) Treatment of the lennox syndrome with ACTH: a clinical and electroencephalographic study. Brain Dev 1: 267-276. [Crossref]