We have previously reported that the glucocorticoid, dexamethasone, increases the expression of Complement Receptor Immunoglobulin (CRIg) in macrophages. Since the sex steroid hormones, progesterone, estradiol and testosterone also affect macrophages functions, it was of interest to see if these also modulated CRIg expression in macrophages and whether, the steroids effect these changes through the glucocorticoid receptor (GR). The data showed that only progesterone and dexamethasone increased CRIg expression in human monocyte-derived macrophages (MDM). The other two steroids had no effect on the expression of this receptor at both the mRNA and protein level. Western blot analysis revealed that this was the case for both spliced forms of CRIg, the long (L) and short (S) isoforms. However, all four steroids had no effect on the expression of CR1 (CD35) and CR3 (CD11b) but caused an increase in CR4 (CD11c) expression. Interestingly the effects of dexamethasone and progesterone correlated with their ability to induced expression of the GR and an increase in its nuclear translocation. The importance of the GR in regulating the steroid-induced increase in CRIg expression, was demonstrated by the finding that knocking down the levels of GR using shRNA, led to a loss of the dexamethasone-induced up regulation of CRIg expression. The increase in CRIg expression by dexamethasone and progesterone was also evident in cell surface expression of the receptor, assessed by flow cytometry. Our findings reveal that the steroids differ in their effects on macrophage CRIg expression and that they have different effects on the expression of the different complement receptors. The expression of CRIg including its up-regulation by steroids requires the GR.
dexamethasone, complement receptor immunoglobulin (CRIg), CR1, CR3, CR4, human macrophage, steroid hormones, glucocorticoid receptors
Steroid hormones are derived from cholesterol. They are transported through the bloodstream to various target cells where they carry out the regulation of a wide range of physiological processes. Sex steroid hormones, including progesterone, estrogen and testosterone are produced by ovaries, testes and adrenal cortex. While these are known for their influence on sexual development and reproduction, they have also been reported to modulate inflammation and immune functions [1]. Thus, reduction of sex steroid hormones promotes inflammation. Both estrogen and progesterone have been shown to drive macrophages towards an alternative activation type, regulating wound healing, angiogenesis and remodelling [1]. Progesterone can suppress LPS-induced IL-6, TNF-α and IL-1b production [2,3]. In vitro and in vivo studies suggested that testosterone displays immunosuppressive effects by causing a reduction in TLR 4 expression on macrophages [4]. Other effects of macrophage function, including phagocytosis, by these steroids have been reported [5-7]. Central to the anti-inflammatory function of steroids and glucocorticoid receptor (GR), is their ability to repress inflammatory genes; apart from the above include chemokines, inducible nitric oxide synthase 2, matrix metalloproteases 12 and 13. GR is the nuclear hormone receptor family of ligand activated transcription factors which bind to glucocorticoids (GC), steroid hormones. GC-GR binding is a powerful therapy for inflammatory diseases, controls systemic homeostasis and the body’s stress response through the hypothalamic-pituitary-adrenal axis [8]. GC can have anti-inflammatory effects and synthetic GC are commonly used anti-inflammation agents [8]. Previously it has been reported that the anti-inflammatory drug dexamethasone causes substantial increases in CRIg (B7 family-related protein V-set and Ig domain-containing 4; VSIG4) expression on macrophages [9,10] , and we recently postulated that the GC effects on CRIg expression occur through their binding to GR [11].
Macrophages are crucial cells of the innate immune system as well as the adaptive immune response [12,13]. The cells play a major role in the clearance of pathogens by complement-mediated as well as non-complement-mediated mechanisms [14,15]. CRIg is expressed exclusively on macrophages, where it promotes phagocytosis in a complement-dependent and -independent manner [16-18]. In addition CRIg has been shown to have anti-inflammatory and immunosuppressive properties, specifically anti-complement activity [19-21], suppression of T cell responses [22,23] and antibody production [24].
We have previously reported that the glucocorticoid, dexamethasone, increases the expression CRIg in macrophages. Since the sex steroid hormones, progesterone, estradiol and testosterone also affect macrophages functions [2-4], it was of interest to see if these also modulated CRIg expression in macrophages, and to compare their effects on all of complement receptors expressed by macrophages. It was also of interest to see if the steroids effect on CRIg expression was dependent on the GR. In this study, we demonstrate that while dexamethasone and progesterone increase the expression of CRIg in human macrophages, estrogen and testosterone had no effect. All steroids examined had no effect on either CR1 or CR3 expression but all four caused an increase in CR4 expression. Interestingly only dexamethasone and progesterone induced expression and translocation of the GR to the nucleus and their effects on CRIg expression were dependent on the GR.
Isolation and differentiation of human peripheral blood monocytes
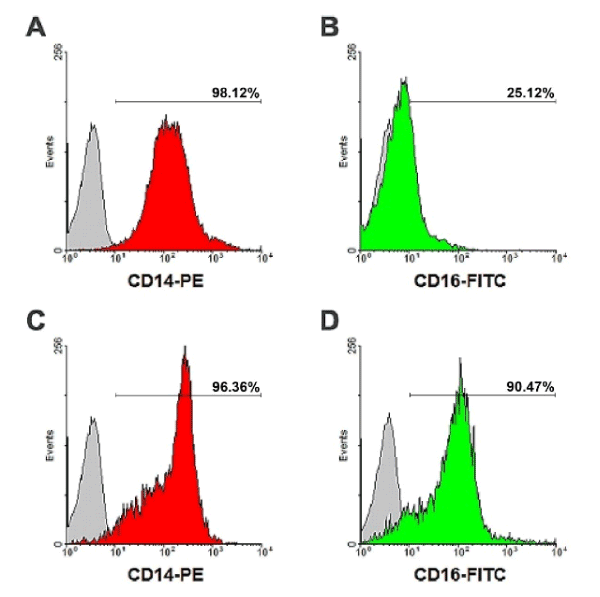
This study was approved by the Human Ethic Committee of Naresuan University, Phitsanulok, Thailand, under the certification number NUIRB-574/58. Human monocytes were isolated from the leukocytes-rich blood component of healthy donors obtained with permission from Naresuan University Hospital, Phitsanulok, Thailand. The blood component was diluted by the addition of an equal volume of Hank’s balanced salt solution (HBSS), layered above 1.077 g/ml Lymphoprep density gradient solution (Axis-Shield PoC AS, Oslo, Norway) and then centrifuged at 600 g for 30 min, 25°C for the isolation of peripheral blood mononuclear cells (PBMCs). Cells were washed with HBSS by centrifugation at 300 g for 5 min. PBMCs were diluted in RPMI-1640 medium (Thermo Fisher Scientific, Inc., New York, NY, USA), layered above 1.064 g/ml hyper-osmotic Percoll solution (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), and centrifuge at 400 g, 15 min, 25°C for the isolation of human monocytes. To remove platelets, the cell suspension was layered on 1.068 g/ml iso-osmotic Percoll solution followed by centrifugation at 300 g, 15 min, 25°C. The pellet containing predominantly monocytes was resuspended in RPMI-1640 and washed with HBSS, followed by centrifugation sequentially at 400, 300, 200 and 100 g. Isolated monocytes were cultured in RPMI-1640 medium supplemented with 10% FBS (Thermo Fisher Scientific). Cells were cultured at 37°C in humidified incubator with 5% CO2 air for 7 days to differentiate to monocyte-derived macrophages (MDM) [9,11]. The macrophages were detached from culture surface with detachment buffer, containing 0.4% lidocaine and 50 mM EDTA in HBSS. The cells were assessed for viability by the trypan blue exclusion assay. Flow cytometric analysis showed the cell population to be >99% CD14+ and >98% CD16+ cells (Supplementary Figure 1).
Expression of membrane surface markers was assessed by flow cytometric analysis. The cells were suspended in staining buffer solution in round-bottom test tubes at a concentration of 106 cells/ml and then centrifuged at 300 g, 8 min, 4°C. Cell supernatants were discarded, fluorescence-conjugated antibody added to a final concentration of 2 µg/106 cells in 100 µl, and the reaction was incubated at 4°C for 1h in the dark. Cells were washed twice with 2 ml of staining buffer and centrifuged at 300 g, 6 min, 4°C. The cell pellet was resuspended in 400 µl of staining buffer and analysed by flow cytometry using the FC 500 Flow Cytometer (Beckman Coulter, Inc., Indianapolis, IN, USA). Immunostaining was performed with PE-conjugated anti-CD14 antibody (HCD14) (BioLegend, Inc., San Diego, CA, USA) and FITC-conjugated anti-CD16 antibody (NKP15) (BD Biosciences, San Jose, CA, USA), respectively. To assess surface complement receptor expression the macrophages were stained with anti-Z39Ig/CRIg (6H8) (Santa Cruz Biotechnology, Inc., Dallas, TX) and PE-conjugated goat anti-mouse IgG antibody (Immuno Tools GmbH, Friesoythe, Germany), FITC-conjugated anti-human CR1/CD35 (UJ11) (Immuno Tools GmbH), PE-conjugated anti-human CR3/CD11b (D12) (BD Biosciences, San Jose, CA, USA), and FITC-conjugated anti-human CR4/CD11c antibodies (BU15) (Immuno Tools GmbH).
NR3C1/GR knock down
In order to knock-down expression of GR in human macrophages, DNA plasmid containing scrambled shRNA and NR3C1/GR shRNA were used. Plasmids were extracted from growth phase culture of E. coli (VectorBuilder (Guangzhou) Inc., Guangzhou, China) containing shRNAs using QIAGEN Plasmid Plus Midi Kit (QIAGEN, Hilden, Germany) with an optimized protocol.
The MDM were prepared and 1x 107 cells were added to microfuge tubes. These were centrifuged at 500 × g for 5 min, the supernatant discarded and the cell pellet was resuspended in 200 µl of Ingenio Electroporation Solution (Mirus Bio LLC., Madison, WI, USA) containing 20 µg/ml of shRNA plasmid. Then the cell suspension was transferred to 0.2 mm transfection cuvette, placed into Lonza-AmaxaNucleofector 2b device (Lonza Group AG, Basel, Switzerland), and electroporated with optimal pulse for human primary macrophages (Protocol Y-010). Viable shRNA-transfected cells were counted, transferred to 24-well plate and seeded as 2 × 105 cells/well, and allowed to rest for 48 h before use in experiments.
RNA isolation
Total RNA from macrophages was isolated with the TRIzol reagent (Thermo Fisher Scientific, Inc.,) containing phenol and guanidine isothiocyanate. Cultured macrophages were directly lysed inside the plate by the addition of 200 µl of TRIzol reagent. The homogenized sample was transferred to polypropylene microcentrifuge tube and 100 µl of chloroform were added to perform phase was separation. The reaction was vigorously mixed, incubated for 3 min at room temperature and centrifuged at 12000 g, 15 min, 4°C. The upper aqueous phase containing RNA was transferred to a new tube, 100 µl of isopropanol added to precipitate the RNA, left standing for 10 min at room temperature and centrifuged at 12000 g, 15 min, 4°C. The reaction supernatant was discarded, RNA pellet washed with 500 µl of 75% ethanol in RNase-free water followed by the centrifugation at 7500 g, 5 min, 4°C. The RNA pellet was dissolved by adding 50 µl of RNase-free water and incubation for 10 min at 55°C.
Measurement of complement receptor mRNA
The RNA was first converted to cDNA using Tetro cDNA Synthesis Kit (Bioline Reagents Limited, London, UK). The reaction contained 5 µl of 1 µg/ml RNA template, 1 µl of Oligo (dT)18 Primer, 1 µl of 10mM dNTP mix, 5 µl of 5x RT Buffer, 1 µl of RiboSafe RNase Inhibitor, 1 µl of 200 U/ µl Tetro Reverse Transcriptase, and 7 µl of RNase-free water. Reverse transcription was performed in the thermal cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) setting at 45°C for 30 min, and 85°C for 5 min.
Gene expression was evaluated by qPCR using SensiFAST SYBR No-ROX Kit (Bioline Reagents Limited, London, UK). The reaction was prepared by mixing the following reagents; 10 µl of 2x SensiFAST SYBR No-ROX Mix, 0.8 µl of 10 µM forward primer, 0.8 µl of 10 µM reverse primer, 5 µl of 1 ng/ml cDNA template, and 3.4 µl of RNase-free water. Oligonucleotide primers specific to genes used in this study are showed in Table 1 (Bio Basic Inc., Markham, ON, Canada) [25]. The qPCR conducted in CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA), set as 3 min of initial denaturation at 95°C, 45 cycles of 10 sec denaturation at 95°C followed by 30 sec of annealing and elongation at 60°C. RT-qPCR data was analysed by normalized gene expression using 2˗ΔΔCᴛ method [26].
Table 1. Oligonucleotide primers specific to genes used in this study
Gene |
Sequence (5' → 3') |
Tm
(°C) |
Amplicon Size
(Base Pair) |
ACTB: |
Actin beta [Homo sapiens], Reference Gene |
|
F: |
CATGTACGTTGCTATCCAGGC |
60.8 |
250 |
|
R: |
CTCCTTAATGTCACGCACGAT |
60.2 |
VSIG4: |
V-set and immunoglobulin domain containing 4 [Homo sapiens], CRIg |
|
F: |
TCCTGGAAGTGCCAGAGAGT |
60.2 |
112 |
|
R: |
TGTACCAGCCACTTCACCAA |
59.2 |
CR1: |
Complement C3b/C4b receptor 1 (Knops blood group) [Homo sapiens] |
|
F: |
CACGAAGCCGCCAATTTGTC |
62.5 |
204 |
|
R: |
CCCACTTGATCGTCATTGCTG |
61.3 |
ITGAM: |
Integrin subunit alpha M [Homo sapiens], CR3 |
|
F: |
GCCTTGACCTTATGTCATGGG |
60.4 |
185 |
|
R: |
CCTGTGCTGTAGTCGCACT |
61.6 |
ITGAX: |
Integrin subunit alpha X [Homo sapiens], CR4 |
|
F: |
AGAGCTGTGATAAGCCAGTTCC |
61.7 |
95 |
|
R: |
AATTCCTCGAAAGTGAAGTGTGT |
60.1 |
NR3C1: |
Nuclear receptor subfamily 3 group C member 1 [Homo sapiens], GR |
|
F: |
ATAGCTCTGTTCCAGACTCAACT |
60.5 |
111 |
|
R: |
TCCTGAAACCTGGTATTGCCT |
60.8 |
Subcellular fractionation and protein extraction
Fractionation of cytoplasmic protein and nuclear protein from experimental macrophages was performed using Nuclear Extraction kit, according to the manufacturer’s instructions (Catalog number: SK-0001, Lot number: 12041501) (Signosis, Inc., Santa Clara, CA, USA) [27] with DTT solution and Halt Protease/Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Inc.,). The cells were washed with HBSS and then harvested by treating with detachment buffer (HBSS buffer containing 50 mM EDTA and 0.04% lidocaine hydrochloride). Cells were transfer to 1.5 ml microcentrifuge tube and washed twice with HBSS by centrifugation at 500 g for 5 min. The cell supernatant was discarded and the cell pellet was solubilized by adding 100 µl of lysis Buffer I on an ice box, for 10 min with shaking. Cells were centrifuge at 12000 g for 5 min at 4°C. Supernatants containing cytoplasmic proteins were collected. The remaining cells pellet was then lysed with 100 µl of lysis Buffer II on an icebox, for 2 h with shaking. Cell lysates were centrifuge at 12000 g for 5 min at 4°C and supernatant containing nuclear protein was collected for further analysis.
Total protein from macrophages was extracted using the cell lysis buffer (150 mM NaCl, 50 mM Tris, 1.0% NP-40) with DTT solution, and Halt Protease/Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Inc.,). Cell culture supernatants from experimental wells were discarded and cells washed twice with cold HBSS. Cold RIPA buffer was added to the cells at 100 µl/well, and then kept on ice for 5 min. Cells were detached using a cell scrapper and the cell lysate was transferred to 1.5 ml microcentrifuge tube. The lysate was centrifuged at 14000 g for 15 min to remove cell debris, and then the protein-containing extract was collected for further analysis.
Quantification of the protein concentration was performed by Bradford coomassie-binding, colorimetric method. Using 96-well plate, 5 µl of diluted sample or series of protein standards were added to the wells. Then 250 µl of coomassie reagent was added to each well and mixed with a plate shaker at RT for 30 sec. The reaction mixture was further incubated at RT for 10 min, and then the absorbance at 595 nm was measured using a microplate reader (PerkinElmer Inc., Waltham, MA, USA).
Western blot analysis
In order to prepare proteins from experimental samples for separation by polyacrylamide gel electrophoresis, 25 µl of 1000 µg/ml protein extract was mixed with an equal volume of 2X Leammli loading buffer, and heated at 95°C for 5 min to denature the protein. Samples were then mixed and centrifuged at 16000 g for 1 min to separate cell debris and the precipitated proteins. For the separation of proteins by SDS-PAGE, 40 µl of each protein sample was loaded to the well of mini 12% SDS-PAGE gel, along with Spectra Multicolor Broad Range Protein Ladder (Thermo Fisher Scientific, Inc.,). Gels were placed in Mini-PROTEAN Tetra cell (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and then run in Tris/Glycine/SDS buffer for 10 min at constant 100 V, followed by 35 min at constant 200 V. SDS-PAGE gel and PVDF membranes were placed in Mini Trans-Blot Module (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and transferred in Towbin buffer with SDS for 1 h at constant 100 V.
For western blot analysis, PVDF blotting membranes were blocked with 2.5% polyvinyl alcohol in TBST (0.05% Tween-20 in tris-buffered saline solution) at RT for 1h with shaking. The membrane was then incubated overnight in primary antibody at 4°C with shaking. Monoclonal primary antibodies used in this study were mouse anti-β-actin (C4), mouse anti-human Z39Ig/CRIg (6H8), mouse anti-human GR (G-5) (Santa Cruz Biotechnology, Inc.,), anti-CD11b (LT11), anti-CD11c (BU15), and anti-CD35 (UJ11) (Immuno Tools GmbH, Friesoythe, Germany). Membrane was washed 4 times with TBST at RT, each for 5 min with shaking. The membrane was then incubated in HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody (Thermo Fisher Scientific, Inc.,) at RT for 1 h with shaking.
For the detection of protein bands by enhanced chemiluminescence (ECL), the PVDF blotting membrane was washed 4 times with TBST at RT, each for 5 min with shaking. The membrane was soaked in SuperSignal West Pico ECL substrate (Thermo Fisher Scientific, Inc.,) for 5 min, and placed in ChemiDoc XRS+ Imaging System (Bio-Rad Laboratories, Inc.,). Chemiluminescence signal of blotted membrane was acquired, bands of proteins were detected and normalized by Image Studio Lite software (LI-COR Corporate, Lincoln, NE, USA).
Statistical analysis
Three-independent experiments were performed in this study. One-way ANOVA and Bonferroni multiple comparisons test was used to analyse data using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). Confident intervals of 99% (p=0.01) were used in all statistical analysis.
Effect of steroid hormones on complement receptors expression
Human macrophages were treated with two different concentrations of the steroids, based on previous reports (United States Biological, Salem, MA, USA), progesterone (hydroxyprogesterone caproate) (Bayer Pharma AG, Berlin, Germany), estradiol (estradiol valerate) (Bayer Pharma AG, Berlin, Germany), or testosterone (testosterone enantate) (Bayer Pharma AG, Berlin, Germany). Doses of steroid hormones used in this study were calculated from therapeutic doses of steroid hormones [28-30]. Macrophages were plated at a density of 2×105 cells/well in 24-well plate and treated with dexamethasone (12.50 ng/ml or 150 ng/ml), progesterone (4166.67 ng/ml or 8333.33 ng/ml), estradiol (166.67 ng/ml or 333.33 ng/ml), and testosterone (3333.33 ng/ml or 6666.67 ng/ml) for 24 h. Complement receptor mRNA and protein expression was measured by RT-qPCR and western blot, respectively.
The MDM were treated for 24 h with the steroids and then analysed for expression of CRIg mRNA using RT-PCR. The data was normalized gene expression using 2˗ΔΔCᴛ method. The normalized gene expression data showed that only dexamethasone and progesterone significantly increased the expression of CRIg mRNA (p<0.01) (Figure 1). Both low and high concentrations of dexamethasone and progesterone up-regulated the expression of CRIg mRNA to 1.60, 1.66, 1.48, and 1.47-fold compared to untreated control cells, respectively (p<0.01). Interestingly, the steroids had no significant effect on the expression of the other complement receptors, CR1/CD35, CR3 except for CR4.
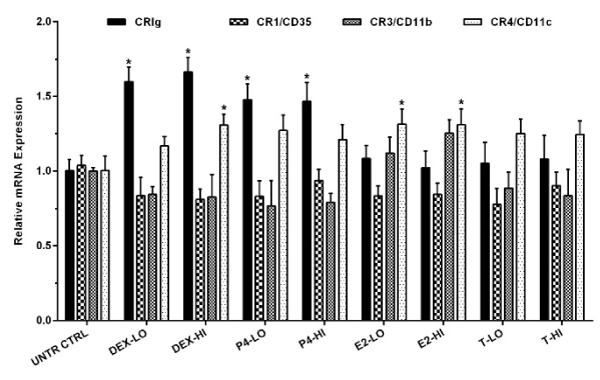
Figure 1. Effect of steroids on the expression of complement receptor mRNA in MDM
MDM were treated with either high (HI) or low (LO) concentrations of steroid hormones for 24 h. The level of complement receptor mRNA was measured by RT-qPCR. Data was analysed by normalized gene expression and presented as mean ± SD of 3 experiments. Statistical analyses: Receptor expression in treated versus non-treated control cells; * p<0.01 Abbreviations: UNTR CTRL, Untreated Control; DEX, Dexamethasone; P4, Progesterone; E2, Estradiol; T, Testosterone.
The macrophages were examined for expression of total CRIg protein by subjecting the cell lysates to western blotting. Protein samples were denatured, diluted to 1000 µg/ml and 50 µl was added to the wells. Western blot analysis showed the presence of both the long (L) and short (S) (Figure 2) forms of CRIg. Supplementary Figure 2 shows typical gel runs with molecular weight references range shown. The results presented in Figure 2 show that only dexamethasone and progesterone caused a significant increase in expression of CRIg. Neither estrogen nor testosterone had any effect on CRIg expression. Low and high concentrations of dexamethasone and progesterone significantly increased the expression of CRIg protein to 1.58, 1.44, 1.39, and 1.57-fold compared to the untreated control MDM, respectively (p<0.01) for the L isoform. Similar effects were seen for the S form (Figure 2). The steroids had no effect on the expression of CR1/CD35 and CR3/CD11b, and only a very small but significant increase in the latter receptor was caused by estradiol (Figure 2). However, the expression of CR4/CD11c protein was significantly increased in MDM treated with any one of these steroid hormones.
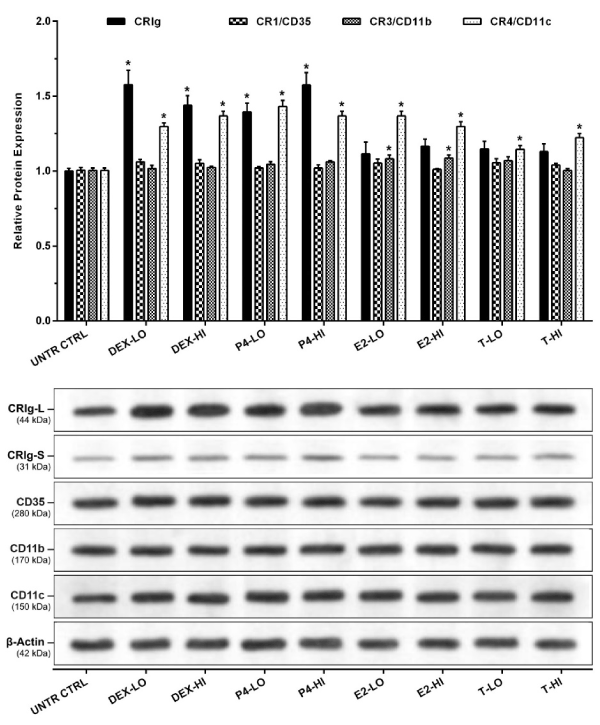
Figure 2. Effects of steroid hormones on the expression of total complement receptor protein
The MDM were treated with the steroid hormones for 24 h. Expression of complement receptors was determined by western blot analysis. Data was normalized and presented as mean ± SD of 3 experiments (top panel). Representative imaging of chemiluminescent from western blots analysis are shown (bottom panel). Molecular weights were established from the molecular weight references range (Supplementary Figure 2). Statistical analyses: Receptor expression in treated versus non-treated control cells; * p<0.01.
Effect on expression and translocation of the glucocorticoid receptor
MDM were treated with steroid hormones as described above. Normalized expression analysis of GR gene showed that low and high concentrations of the dexamethasone and progesterone up-regulated gene expression to 2.42, 2.65, 1.76, and 2.25-fold of untreated controls MDM (p<0.01) (Figure 3A). Denatured protein samples were loaded (50 µl/well) at a concentration of 1000 µg/ml for total GR and 2000 µg/ml for cytoplasmic and nuclear GR. Western blot analysis was used to assess changes in the amount of total, cytoplasmic, and nuclear GR expressed (Figure 3B, 3C and 3D). Molecular weight reference range is shown in Supplementary Figure 3. Low and high concentrations of dexamethasone significantly increased the expression of total GR to 1.49 and 1.61-fold (Figure 3B) of untreated control MDM (p<0.01). Dexamethasone and progesterone, at low and high concentrations caused a significant decrease in cytoplasmic GR content to 0.69, 0.62, 0.82, and 0.73-fold (Figure 3C) of untreated controls (p<0.01). In comparison these caused significant increases in nuclear GR content by 1.49, 1.62, 1.33, and 1.40-fold (Figure 3D) of untreated control cells, respectively (p<0.01).
When the results were expressed as the nuclear: cytoplasmic GR content ratio, dexamethasone and progesterone-treated MDM displayed an increase in this ratio (Figure 3E). Low and high concentrations of dexamethasone and progesterone, significantly increased nuclear: cytoplasmic GR ratio from 1.50 in untreated control MDM to 3.24, 3.95, 2.43, and 2.89 in treated cells, respectively (p<0.01).
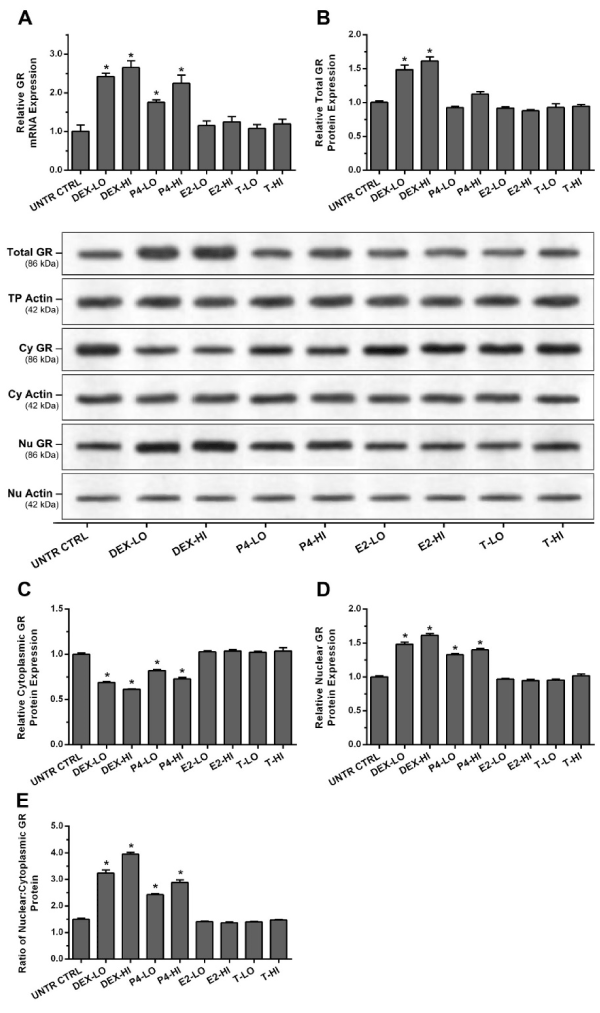
Figure 3. Steroid hormones regulate expression and nuclear translocation of glucocorticoid receptor (GR) in human macrophages
MDM were treated with the steroid hormones for 24 h. Expression of GR was measured by RT-qPCR and western blot Translocation of GR was analysed by measuring changes in the ratio of nuclear: cytoplasmic GR content using western blot. (A) shows the level of GR mRNA, (B) total GR protein levels and representative imaging of chemiluminescent from western blots, (C) cytoplasmic GR protein levels and (D) nuclear GR protein levels. (E) shows the ratio of the nuclear GR protein levels to cytoplasmic levels. Data was normalized and presented as mean ± SD of 3 experiments. Statistical analyses: * p<0.01 compared to untreated control cells.
The role of the gr in glucocorticoid-induced crig expressions
To examine whether the effects of the steroids worked through for GR receptor, we examined the effects of dexamethasone on GR deficient MDM. The macrophages were transfected with either scramble shRNA or NR3C1/GR shRNA by electroporation. Expression of the GR and complement receptors then analysed by gene expression and western blot analysis. Normalized gene expression data showed a significant decrease of GR and CRIg mRNA in NR3C1 shRNA-transfected macrophages by 0.37 and 0.55-fold of control transfected cells (p< 0.01) (Figure 4A). Expression of other complement receptors including CR1, CR3, and CR4 was not influenced by GR knock down.
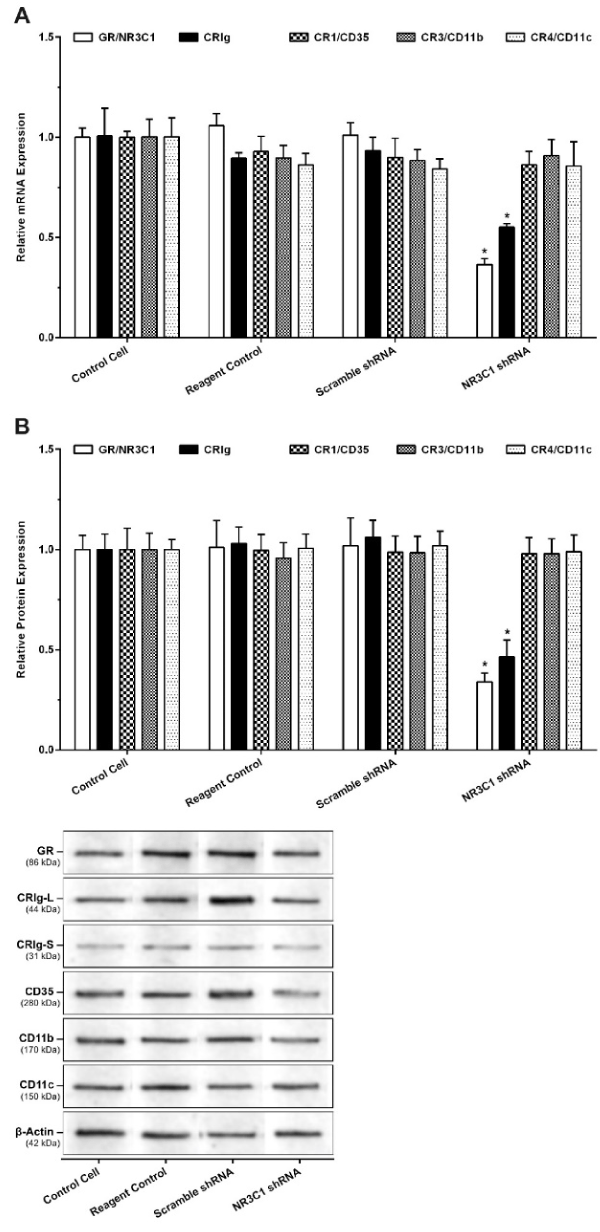
Figure 4. Effect of NR3C1/GR Gene Silencing on the Expression of Glucocorticoid Receptor mRNA (A) and protein (B)
MDM were transfected with plasmid containing either scramble shRNA or NR3C1/GR shRNA by electroporation. Expression of GR and CR mRNA was measured by RT-PCR and protein by western blots. Data was normalized and presented as mean ± SD of 3 experiments. Statistical analyses: * p<0.01 compared to non-transfected control.
For western blots, protein samples were denatured, diluted to 1000 µg/ml and 50 µl was added to each well. The results showed that there was a significant decrease in GR and CRIg proteins in NR3C1/GR shRNA transfected macrophages to 0.36 and 0.46-fold of control cells (p<0.01) (Figure 4B). No significant effect was found for the expression of other complement receptors. Thus, it is interesting that when the NR3C1/GR shRNA transfected cells were treated with either dexamethasone or progesterone there was no significant increase in CRIg expression (Figure 5).
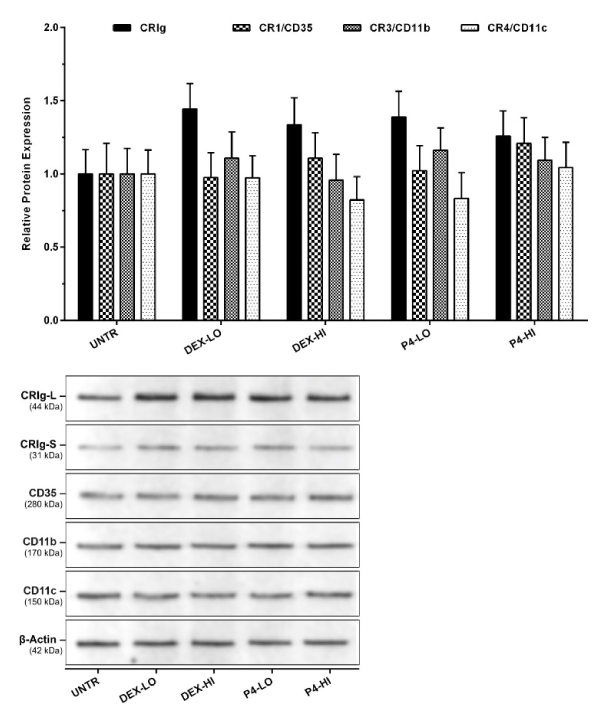
Figure 5. Effect of dexamethasone and progesterone on the expression of complement receptor proteins in GR gene-silenced macrophages
MDM were transfected with plasmid containing NR3C1/GR shRNA for the GR knockdown. The cells were treated with the steroids and then complement receptor expression measured by western blot analyses of cell lysates. Data was normalized and presented as mean ± SD of 3 experiments. Bottom panel shows a representative western blot of the effect of steroids on GR deficient macrophages.
Effects of steroids on cell surface expression of crig
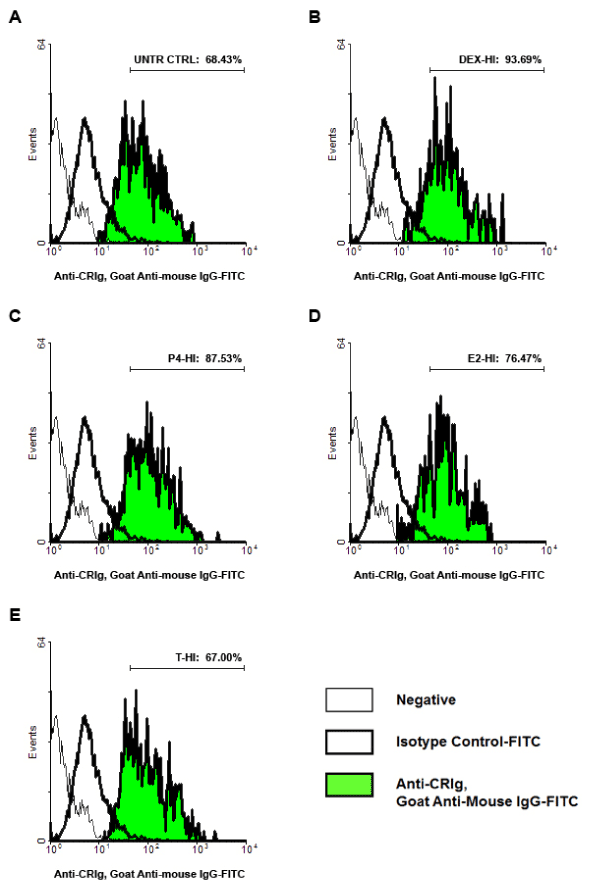
Since the functional properties of CRIg depend on expression on the cell surface, MDM surface complement receptors expression was examined by flow cytometry. The data showed that dexamethasone and progesterone increased expression of CRIg, from 68.43% in untreated control cells to 99.58, 93.69, 89.89, and 87.53%, respectively (p<0.01) (Figure 6). Expression of CR1/CD35 was significantly decreased in macrophages treated with low concentration of dexamethasone and progesterone from 73.92% in untreated control cells to 65.44 and 67.94%, respectively (p<0.01). Expression of CR3/CD11b was significantly decreased in cells treated with dexamethasone from 86.54% in untreated control cells to 76.88 and 78.83%, respectively for low and high concentrations (p<0.01). Expression of CR4/CD11c was significantly decreased in MDM treated with dexamethasone, progesterone and high concentrations of estradiol, and low concentration of testosterone from 89.95% in untreated control cells to 81.40, 83.18, 82.61, 84.05, 83.20, and 84.04%, respectively (p<0.01) (Figure 6).
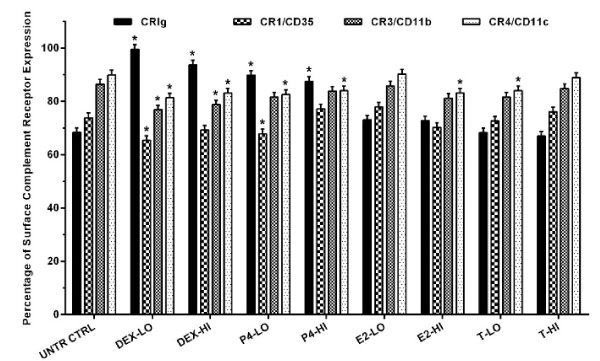
Figure 6. The effect of steroid hormones on the expression of complement receptors on the cell surface
MDM were treated with steroid hormones for 24 h. Complement receptors expression was examined by flow cytometry. Data were analysed and presented as mean ± SD of 3 experiments. Statistical analyses: Different between untreated cells versus steroid treated cells * p<0.01.
The data demonstrate that steroid hormones differentially regulate the expression of CRIg in macrophages. Thus, while progesterone like dexamethasone caused a marked increase in expression of this receptor in MDMs, both estrogen and testosterone had no effect. These steroids were active on macrophages since they caused an increase in expression of CD11c. While it is not clear why there are such differences, the structures depicted in Figure 7 show a closer similarity between dexamethasone and progesterone compared to estrogen and testosterone [31]. We have previously postulated that dexamethasone acts via GR to increase the expression of CRIg at the transcriptional level [11]. The steroid hormones have the same basic structure but differ in functional groups (Figure 7) [31]. Because CRIg has anti-inflammatory and immunosuppressive properties [22,23], it is likely that dexamethasone and progesterone cause their anti-inflammatory effects through this mechanism. All four steroids had no effect on CR3 expression but caused an increase in CR4 expression. Moreover, from the transcription factor binding predictions there are response elements of steroid hormone (glucocorticoid, progesterone, estrogen and androgen) on the promoter of all CRs genes. Interestingly CR1 and CR3 promoters contain NGRE (negative binding sites for glucocorticoid receptor). This may be a reason as to why glucocorticoids had no effect on CR1 and CR3 expression.
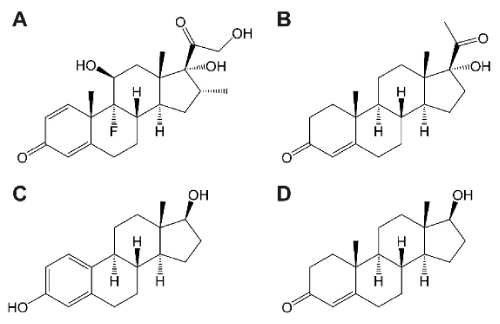
Figure 7. Structure of steroid hormones used in this study
All steroid hormones are generated from cholesterol and are then enzymatically modified by steroid oxidoreductases. Steroid hormones have the same cholesterol backbone structure but different functional groups. (A), glucocorticoid (dexamethasone); (B), progesterone (hydroxyprogesterone) (C), estrogen (estradiol) and (D), androgen (testosterone).
The steroid effects on expression of CRIg protein were also evident at the CRIg mRNA level. This suggests that the regulatory mechanisms are likely to be at the level of gene transcription. Regulation of target gene transcription by these hormones requires binding of the hormones to their associated receptors, nuclear translocation and homodimerization of the hormone-receptor complexes, and then binding of hormone receptor dimer to hormone response element at DNA binding sites of the target gene promoter [32,33]. Glucocorticoids control inflammatory responses by primarily binding to GR. GR directly transactivates anti-inflammatory genes (ANXA1, IL10, NFKBIA), and transrepresses NF-κB-dependent pro-inflammatory genes (TNF, IL6, IL8) [32,33]. Progesterone functions mainly in cell proliferation and secretion of matrix metalloproteinase during pregnancy. Progesterone primarily binds to the progesterone receptor (PR/NR3C3), which specifically regulates expression of genes such as Tnfsf11/RANKL, MMP2, and MMP9 [34,35]. PR also plays a role in cell growth, differentiation, proliferation, and development, through the non-genomic activation of Src/MAPK (ERK1, ERK2) signaling pathway [36,37]. Interestingly, binding of progesterone to hormone receptors is not limited to its cognate PR but includes GR, as an alternative receptor [2,38].
Interestingly both dexamethasone and progesterone but not estrogen and testosterone increased total GR expression in macrophages. This was manifested by decreased expression in the cytoplasm and an increase in the nucleus, indicating that of the steroids promote the nuclear translocation of GR. Dexamethasone and progesterone increased nuclear: cytoplasmic GR ratio. The results showed that dexamethasone was most effective in inducing nuclear translocation of GR in macrophages. Others have shown that progesterone increases expression and nuclear translocation of PR isoform A in human umbilical vein endothelial cells [39], and estradiol to increase expression of both ER (estrogen receptor)-α and ER-β [36,37]. Testosterone also increased expression and nuclear translocation of AR (androgen receptor) [40]. The mechanism of glucocorticoid induced regulation of CRIg expression probably relates to the translocation of GR, and functions of GR as transcriptional regulator [32,41,42]. GR is normally expresses in cytoplasmic compartment of cells. Binding of glucocorticoid to GR changes conformation of GR complex to become hyperphosphorylated enabling it to dissociate from its accessory heat shock protein 90 (HSP90). Glucocorticoid-bounded GR then translocate to nucleus through importin nucleocytoplasmic transporter. GR forms homodimer in the nucleus and binds to glucocorticoid response elements at gene promoters. Activities of GR include direct transactivation, tethered transactivation, direct transrepression, and tethered transrepression [32,41,42]. Our study also investigated the expression of complement receptors in MDM transfected with NR3C1 shRNA. MDM deficient in GR were found to also express significantly lower levels of CRIg but normal levels of other complement receptors. This suggests that the GR receptor controls CRIg expression on macrophages, in line with our previously reported concept [11] and intracrine production and action of steroids [43]. We also provide evidence that in GR deficient MDM both dexamethasone and progesterone failed to significantly increase CRIg expression, showing that their effects are via the GR.
The data demonstrated that only the effects of dexamethasone and progesterone culminated in an increase in cell surface expression of CRIg. Thus although there was an increase in the levels of total CD11c protein induced by the four steroids, this was not reflected in a corresponding increase in cell surface expression of this receptor. Such effects are likely to translate to functional interactions with complement opsonized microbial pathogens. Our study makes prominent the effect of these steroids on the spectrum of complement receptors present on macrophages. These are differentially regulated by the steroids, indeed with only dexamethasone and progesterone regulating CRIg expression, a process involving the GR. However, since regulation of CR3 and CR4 can also be at a functional level our work did not assess this aspect of the complement receptors.
It is most likely that engagement of the GR leads to inhibition of production of TNF. We have previously shown that TNF inhibits CRIg expression in macrophages by activating PKCα [11]. Because CRIg has been shown to have anti-inflammatory and immunosuppressive properties [22,23], it is likely that some of the anti-inflammatory effects of dexamethasone and progesterone involve this mechanism. However, other types of anti-inflammatory drugs, which also alter expression of CRIg [9,43], are likely to involve different mechanisms.
The data demonstrate that of the steroid hormones tested only dexamethasone and progesterone increased the expression of CRIg in human macrophages and that all four steroids had no effect on the expression of CR1 and CR3, but these all increased expression of CR4. The effects of dexamethasone and progesterone on CRIg correlated with their ability to increase the nuclear translocation of GR and that their action was mediated via this receptor.
The authors declare no conflicts of interest.
Designed the experiments: N.K, B.M and K.U. Execution of experiments: N.K. Data analysis and interpretation: N.K, P.P, Y.T, S.P, A.F and K.U. Wrote and critical reading of manuscript: N.K, P.P, Y.T, A.F and K.U. Final approval of the version to be published: N.K, P.P, Y.T, S.P, A.F. and K.U.
This study received financial support from the Thailand Research Fund (RSA6080042). A.F.’s work is supported by the National Health and Medical Research Council of Australia.
We thank members of our faculty who provided technical assistance for this work. Nateelak Kooltheat was in receipt of postgraduate scholarship from Faculty of Allied Health Sciences, Naresuan University. The research received financial support from Franco-Thai cooperation program in higher education and research year 2016 – 2017 for laboratory visiting and training at Centre d’Immunologie de Marseille-Luminy and Centre d’Immunophenomique, Aix-Marseille University, France.
View Supplementary Data
- Routley CE, Ashcroft GS (2009) Effect of estrogen and progesterone on macrophage activation during wound healing. Wound Repair Regen 17: 42-50.
- Lei K, Chen L, Georgiou EX, Sooranna SR, Khanjani S, et al. (2012) Progesterone acts via the nuclear glucocorticoid receptor to suppress IL-1ß-induced COX-2 expression in human term myometrial cells. PLoS One 7: e50167. [Crossref]
- Su L, Sun Y, Ma F, Lu P, Huang H, et al. (2009) Progesterone inhibits Toll-like receptor 4-mediated innate immune response in macrophages by suppressing NF-kappaB activation and enhancing SOCS1 expression. Immunol Lett 125:151-5.
- Rettew JA, Huet-Hudson YM, Marriott I (2008) Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reprod 78: 432-437.
- Curuvija I, Stanojevic S, Arsenovic-Ranin N, Blagojevic V, Dimitrijevic M, et al. (2017) Sex differences in macrophage functions in middle-aged rats: Relevance of estradiol level and macrophage estrogen receptor expression. Inflammation 40: 1087-101.
- Devadas K, Biswas S, Ragupathy V, Lee S, Dayton A, et al. (2018) Modulation of HIV replication in monocyte derived macrophages (MDM) by steroid hormones. PLoS One 13: e0191916.
- Kim BY, Son Y, Lee J, Choi J, Kim CD, et al. (2017) Dexamethasone inhibits activation of monocytes/macrophages in a milieu rich in 27-oxygenated cholesterol. PLoS One12: e0189643.
- Greulich F, Hemmer MC, Rollins DA, Rogatsky I, Uhlenhaut NH (2016) There goes the neighborhood: Assembly of transcriptional complexes during the regulation of metabolism and inflammation by the glucocorticoid receptor. Steroids 114: 7-15.
- Gorgani NN, Thathaisong U, Mukaro VR, Poungpair O, Tirimacco A, et al. (2011) Regulation of CRIg expression and phagocytosis in human macrophages by arachidonate, dexamethasone, and cytokines. Am J Pathol (2011) 179:1310-8. [Crossref]
- Munawara U, Small AG, Quach A, Gorgani NN, Abbott CA, et al. (2017) Cytokines regulate complement receptor immunoglobulin expression and phagocytosis of Candida albicans in human macrophages: A control point in anti-microbial immunity. Sci Rep 7: 4050.
- Ma Y, Usuwanthim K, Munawara U, Quach A, Gorgani NN, et al. (2015) Protein kinase calpha regulates the expression of complement receptor Ig in human monocyte-derived macrophages. J Immunol 194: 2855-61.
- Chawla A (2010) Control of macrophage activation and function by PPARs. Circ Res 106: 1559-1569. [Crossref]
- Davies LC, Jenkins SJ, Allen JE, Taylor PR (2013) Tissue-resident macrophages. Nat Immunol 14: 986-995. [Crossref]
- Freeman SA, Grinstein S (2014) Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol Rev 262: 193-215. [Crossref]
- Underhill DM, Goodridge HS (2012) Information processing during phagocytosis. Nat Rev Immunol 12: 492-502. [Crossref]
- Broadley SP, Plaumann A, Coletti R, Lehmann C, Wanisch A, et al. (2016) Dual-track clearance of circulating bacteriabalances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe 20: 36-48.
- Helmy KY, Katschke KJ, Jr. Gorgani NN, Kljavin NM, Elliott JM, et al. (2006) CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124: 915-27. [Crossref]
- Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, et al. (2016) CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe 20: 99-106.
- He JQ, Wiesmann C, van Lookeren Campagne M (2008) A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol Immunol 45: 4041-4047. [Crossref]
- Katschke KJ Jr., Helmy KY, Steffek M, Xi H, Yin J, et al. (2007) A novel inhibitor of the alternative pathway of complement reverses inflammation and bone destruction in experimental arthritis. J Exp Med 204: 1319-1325. [Crossref]
- Wiesmann C, Katschke KJ, Yin J, Helmy KY, Steffek M, et al. (2006) Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature 444: 217-220.
- Jung K, Kang M, Park C, Hyun Choi Y, Jeon Y, et al. (2012) Protective role of V-set and immunoglobulin domain-containing 4 expressed on kupffer cells during immune-mediated liver injury by inducing tolerance of liver T- and natural killer T-cells. Hepatology 56: 1838-1848.
- Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI, et al. (2006) VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest 116: 2817-2826. [Crossref]
- Jung K, Seo SK, Choi I (2015) Endogenous VSIG4 negatively regulates the helper T cell-mediated antibody response. Immunol Lett 165: 78-83.
- Spandidos A, Wang X, Wang H, Seed B (2010) PrimerBank: A resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 38: D792-D799. [Crossref]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408. [Crossref]
- Wu X, Li J, Zhu M, Fletcher JA, Hodi FS (2012) Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-kappaB pathways. Mol Cancer Ther 11:1905-14. [Crossref]
- U.S. Food and Drug Administration (2017) Approved Drug Products with Therapeutic Equivalence Evaluations. Silver Spring: U.S. Food and Drug Administration.
- World Health Organization (2007) WHO Drug Information. Geneva: World Health Organization.
- World Health Organization (2015) WHO Model Lists of Essential Medicines. Geneva: World Health Organization.
- Hanukoglu I (1992) Steroidogenic enzymes: Structure, function, and role in regulation of steroid hormone biosynthesis. J Steroid Biochem Mol Biol 43: 779-804.
- Moutsatsou P, Kassi E, Papavassiliou AG (2012) Glucocorticoid receptor signaling in bone cells. Trends Mol Med 18: 348-359.
- Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9: 429-39.
- Goldman S, Shalev E (2006) Difference in progesterone-receptor isoforms ratio between early and late first-trimester human trophoblast is associated with differential cell invasion and matrix metalloproteinase 2 expression. Biol Reprod 74: 13-22.
- Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, et al. (2013) Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med 5:182ra55.
- Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, et al. (2007) The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol 21: 359-375.
- Leonhardt SA, Boonyaratanakornkit V, Edwards DP (2003) Progesterone receptor transcription and non-transcription signaling mechanisms. Steroids 68: 761-70.
- Engler JB, Kursawe N, Solano ME, Patas K, Wehrmann S, et al. (2017) Glucocorticoid receptor in T cells mediates protection from autoimmunity in pregnancy. Proc Natl Acad Sci U S A 114: E181-E90. [Crossref]
- Hsu SP, Yang HC, Kuo CT, Wen HC, Chen LC, et al. (2015) Progesterone receptor-NFkappaB complex formation is required for progesterone-induced NFkappaB nuclear translocation and binding onto the p53 promoter. Endocrinology 156: 291-300.
- Karaca M, Liu Y, Zhang Z, De Silva D, Parker JS, et al. (2015) Mutation of androgen receptor N-terminal phosphorylation site Tyr-267 leads to inhibition of nuclear translocation and DNA binding. PLoS One 10: e0126270. [Crossref]
- Kadmiel M, Cidlowski JA (2013) Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 34: 518-530. [Crossref]
- Yang YH, Morand E, Leech M (2013) Annexin A1: Potential for glucocorticoid sparing in RA. Nat Rev Rheumatol 9: 595-603.
- Rubinow KB (2018) An intracrine view of sex steroids, immunity, and metabolic regulation. Mol Metab 15:92-103.