Abstract
Inflammation is a major contributor to beta cell destruction leading to diabetes. Generation of reactive oxygen species (ROS) by inflammatory signals facilitates beta cell dysfunction. Disruption of the ROS-generating enzyme NADPH oxidase-1 (NOX-1) confers protection to beta cells. Selective small molecule inhibitors of NOX-1 that confer protection to mouse or human beta cells (ML171 or GKT137831) have been systemically administered to NOD mice. A brief (4 week) administration of the NOX-1 inhibitors reduced the conversion of NOD mice to diabetes, relative to vehicle control. Histologic analysis of islet morphology showed mice administered the NOX-1 inhibitors had a predominant organization of leukocytes that was restricted to the peri-islet region, in contrast to leukocyte invasion of the islet that was predominantly seen in vehicle control mice. The data support the therapeutic potential of NOX-1 inhibition in diabetes and suggest a role for NOX-1 in the cross talk between inflammatory cells, beta cells and the integrity of the islet extracellular matrix.
Key words
inflammation, diabetes, beta cell function, NADPH oxidase-1, reactive oxygen species, small molecule.
Introduction
Diabetes is a complex and multifaceted disease. An increasing number of the world’s population is impacted by diabetes and its chronic complications with a predicted 50% increase in prevalence within the next three decades. The primary underlying etiology of diabetes is beta cell failure and loss of functional beta cell mass. Beta cells are sensitive to sustained elevation in reactive oxygen species (ROS) relative to other cells [1-3]. Inflammation contributes to beta cell dysfunction. Exposure of beta cells and primary islets to mediators of inflammation results in elevation of ROS and concomitant loss of beta cell function [4-7]. NADPH oxidase 1 (NOX-1), is a cellular source of ROS in beta cells that is induced by inflammatory cytokines [8-10].
Selective small molecule inhibition of NOX-1 blocks inflammatory cytokine-induced ROS in beta cell lines and islets [8]. Selective NOX-1 inhibition confers protection to beta cells from the damaging effects of inflammatory cytokines in homogeneous murine beta cell lines and mouse or human primary islets [8]. Over expression of NOX-1 results in beta cell dysfunction whereas knockdown of NOX-1 protects and preserves beta cells exposed to inflammatory factors [11]. NOX-1 inhibition is a candidate strategy to preserve beta cell function. In this study, proof-of-concept data in a diabetic animal model has been developed. The resultant data demonstrate conferred protection to diabetes development in NOD mice and support the therapeutic potential of NOX-1 inhibition in diabetes.
Methods
Ethics
Protocols and procedures were reviewed and approved by relevant institutional regulatory committees
Cell line
βTC-3 cells, a mouse β-cell line, were cultured at 37°C in a 5% CO2/humidified atmosphere using standard media (Life Technologies, Grand Island, NY) as previously described [12]. Human donor islets were obtained from non-diabetic donors (Prodo Laboratories Inc., Aliso Viehio, CA).
ROS measurement
Cells were treated with species specific cytokines (PIC: 5ng/mL IL-1β, 10ng/mL TNF-α, 100ng/mL IFN-γ; R&D Systems, Minneapolis, MN) for 4 hours. NOX-1 inhibitors (ML171 or GKT137831) were added 30 minutes before PICs. After treatment, ROS was measured as previously reported [10] using 10μM 6-carboxy-2,7’-dichloroduhydrofluorescein diacetate, di(acetoxymethyl ester); DCF-DA (Life Technologies).
Glucose-Stimulated Insulin Secretion (GSIS)
Human donor islets were assessed for GSIS as previously described in [8]. Insulin was measured by ELISA (Mercodia, Winston Salem, NC) following the manufacturer’s directions.
Compound plasma half life
Compound levels in plasma samples were determined using LC-MS/MS by Frontage Laboratories Inc., Secaucus, NJ. CD-1 mice were administered 30mg/Kg of NOX-1 inhibitor via intraperitoneal injection. For each compound, three mice were injected for each time point. Blood was collected into heparin tubes at 0.25, 0.5, 1,2 4, 8 ,12 hours post injection. Plasma samples were frozen and sent to Frontage Laboratories for analysis and calculation of half-life.
NOD study
Female NOD/ShiLTJ (NOD) mice were purchased from Jackson Laboratories. All mice were maintained at the Indiana University School of Medicine Laboratory Animal Resource Center under pathogen-free conditions according to protocols approved by the institutional animal care and use committee. Mice were randomly assigned to each cohort (twenty three per group). Compounds were provided blind (coded). Mice were injected intraperitoneally daily from 6-10 weeks of age with 30mg/Kg (200 µl) NOX-1 inhibitor dissolved in 7:1:1:1 PEG200:cremophor:ethanol:water or vehicle alone. Glucose measurements were performed weekly using an AlphTrak glucometer (Abbott Laboratory, Abbott Park, IL). At the end of the respective studies, mice were euthanized, serum was collected, and pancreata harvested. In each group, mice were removed from the study early due to health issues; Control, one mouse; GKT137831, two mice; ML171, four mice. The health issues did not appear to be related directly to the compound(s) as the other mice remaining in the study that received compound presented no adverse events. Administration of treatments (vehicle and compound in vehicle) was by daily intraperitoneal injection, this is the likely source of health issues.
Histology
Pancreata were fixed in 10% formalin (Fisher Chemicals, Fair Lawn, NJ) and paraffin embedded with 7µM serial sections being prepared and stained with H & E (AML laboratories, Jacksonville, FL). For microscopic analysis, three non-overlapping pancreas sections were analyzed for each mouse. All islets present in these sections were captured and representative images are shown. Images were captured at 20x objective using a DP-70 camera with a BX-51 Olympus Microscope (Olympus, Center Valley, PA).
Data analysis
In vitro experiments were performed in triplicate, at minimum. Student’s t test or one-way ANOVA with post hoc testing (Prism 7.0; Graph-Pad Software, La Jolla, CA) were used to determine statistical significance (95% CI and p < 0.05).
Results and discussion
Current therapeutic options for diabetes are not curative and do not address beta cell failure, the underlying cause of diabetes. Inflammation and resultant elevation of proinflammatory cytokines has been identified as a significant contributor to loss of beta cell function; a mere short exposure of human islets to inflammatory cytokines ex vivo results in beta cell failure [13]. Inflammatory cytokines elevate reactive oxygen species (ROS) in beta cells that are highly prone to oxidative stress [1]. A source of this beta cell ROS is NADPH oxidase-1 (NOX-1) activity. The key role for NOX-1 in driving beta cell dysfunction has been demonstrated in study of chemical inhibitors and genetic deletion of NOX-1 that protect beta cells [8, 11]. Further, direct over expression of NOX-1 promotes a state of beta cell dysfunction analogous to inflammatory cytokine stimulation [11]. These data indicate that inhibition of NOX-1 is a candidate therapeutic strategy to preserve beta cell function in an ongoing inflammatory environment [9].
Inhibition of NOX-1 blocks ROS production in islets
The phenothiazine compound, ML171 is a NOX-1 inhibitor that provides significant protection to beta cells from the damaging effects of inflammatory cytokines [8]. This NOX-1 inhibitor blocked ROS production and preserved beta cell function when islets or beta cells were challenged with inflammatory cytokines. A second structural class, pyrazolopyridine dione, of molecules have been identified as potent dual specific inhibitors of NOX-1 and NOX-4 [14]. The activity of ML171 (phenothiazine) and GKT137831 (pyrazolopyridine dione) to inhibit beta cell dysfunction and ROS production was assessed (Figure 1). In the homogeneous beta cell line, βTC3, inflammatory cytokines (PIC) increased ROS by 105%. In contrast, inclusion of ML171 or GKT137831 with PICs blocked inflammation-induce ROS by 78% and 80% respectively (p<0.01). To assess beta cell function, glucose stimulated insulin secretion was measured in human donor islets (Figure 1). Beta cells are stimulated to secret insulin upon elevation of glucose from low to high (3 to 16.7mM glucose). With exposure to inflammatory cytokines, this functional response is absent resulting in a loss of glucose stimulated insulin secretion (GSIS). Addition of GKT137831 to human donor islets preserved beta cell function when islets are exposed to PIC, shown by the preserved increase in insulin secretion upon transition from low (L) to high (H) glucose (Figure 1). Equivalent preservation of GSIS in human donor islets with ML171 has previously been published [8]. The additional new results with GKT137831 show that small molecule inhibition of NOX-1 can preserve beta cell function. GKT137831 is a dual specific NOX-1/NOX-4 inhibitor and a role for NOX-4 has not been excluded, however compelling data has been accumulated to establish that NOX-1 deletion is necessary and sufficient in preserving beta cells [8,11].
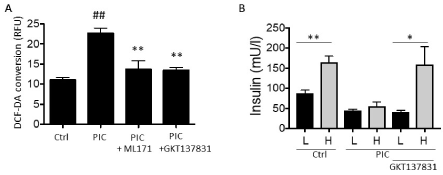
Figure 1. Inhibitors of NOX-1 block ROS generation and GSIS uncoupling induced by inflammatory cytokines. A, generation of ROS (measured by conversion of DCF-DA) in βTC3 beta cells treated with inflammatory cytokines (PIC) in the absence or presence of NOX-1 inhibitors ML171 or GKT137831. B, Glucose stimulated Insulin Secretion response in human donor islets exposed to low (L) 3mM glucose or high (H) 16.7 mM glucose treated with inflammatory cytokines in the absence or presence GKT137831.Data is shown relative to control (Ctrl) response and is from triplicate experiments. *, **/## = p<0.05, 0.01 resp.; # = comparison to control
These data highlight the key role of NOX-1 in mediating beta cell dysfunction when islets are exposed to inflammatory factors. Importantly, the study introduces the therapeutic potential for NOX-1 inhibition in diabetes. To advance these studies evaluation of compounds in proof-of-concept in vivo study has been assessed.
Systemic administration of NOX-1 inhibitors to NOD mice disrupts development of hyperglycemia
NOD mice are the archetypal mouse model for type one diabetes. The model is a genetic inbred strain where female mice have a predisposition to develop inflammation mediated beta cell destruction and hyperglycemia [15]. The NOD mouse model, which has close similarities to human type 1 diabetes, is heavily driven by genetic predisposition but also influenced by environmental factors. For example, not all female mice within a colony will develop diabetes. Rates of diabetic conversion between research institutions can vary significantly both in age at development of hyperglycemia and percent of colony conversion. This institutional variation occurs even with NOD mice identically sourced from vendor and shipped to different institutes (data not shown). For this report, young NOD mice were sourced from Jackson Labs and studies were performed at Indiana University Center for Diabetes and Metabolic Diseases where female NOD mice have a robust and reliable conversion to hyperglycemia (70 – 85%) commencing at age week 12.
Exploration of a range of excipients and combinations thereof determined stable solubility of compounds at 1.5mg/ml in PEG200: Cremaphore EL; Ethanol; ddH20 (7:1:1:1). BL6 mice were administered compound acutely with escalating compound dose up to 30mg/Kg and no adverse effects observed. Mice were subsequently administered compound daily for 5 days at maximal dose (chronic exposure) and monitored for two subsequent weeks with no adverse effects being observed (not shown). No reported data was available on systemic administration of ML171. Compound GKT137831 has been reported in systemic studies and proven to be well tolerated in oral administration [16]. Compounds were studied for pharmacokinetic properties with the chosen intraperitoneal administration route for this study. The calculated half-life for ML171 and GKT137831 were 2.58 hr and 4.79 hr respectively, both achieving a sustained plasma concentration higher than the compound IC50 for two hours or greater following bolus 30mg/Kg compound administration.
For pre-clinical analysis, NOD mice were administered compound or vehicle daily starting at week six and continuing for four weeks. Twenty-three mice were randomly assigned to each of the cohorts. Three mice from each cohort were harvested at week ten. Commencing at week ten blood glucose levels for each mouse were monitored weekly until study endpoint, this being either when mice were classified as diabetic (defined as, two consecutive blood glucose readings at ≥250mg/dl) or at age twenty-five weeks.
Shown is the incidence of diabetes in each of the groups (Figure 2): Control (vehicle only) and compound treated groups. Cohorts treated with NOX-1 inhibitors (GKT137831 and ML171) with daily intraperitoneal injection from age 6 weeks for four weeks show a clear separation in incidence of diabetes relative to the control cohort that received the equivalent administration of vehicle only.
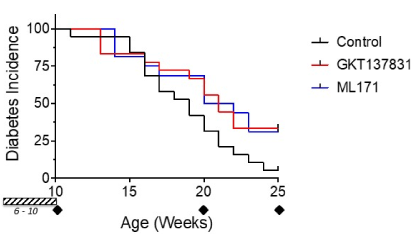
Figure 2.Inhibition of NOX-1 reduces diabetes conversion in NOD mice. Diabetes incidence relative to age of mouse is shown for each cohort (n=20 enrolled for each group). An additional three mice per group where studied at week 10. Mice were administered compound daily for four weeks (hashed block week 6-10) at 30mg/Kg. The representative ages analyzed and presented in this study are marked (diamonds)
These data show that a short four-week prophylactic exposure to inhibitors of NOX-1 reduce diabetes conversion in NOD mice. To provide histological evidence to support this pre-clinical outcome, pancreata from mice along the course of the study were studied.
Systemic administration of NOX-1 inhibitors confers protection to islets from inflammatory cells
Analysis of pancreas has been performed for mice from each cohort at ten (10), twenty (20) and twenty-five (25) weeks by histology.
At ten weeks of age, islets were examined for evidence of compound related effects on islet morphology and structure (Figure 3). Secondly, evidence for the development of early inflammation was sought to support the normal development of the type one diabetic model. The hypothesis being tested is protection to beta cells in ongoing inflammation by inhibition of NOX-1. Evidence of inflammation starting at week ten would serve to rule out ‘off-target’ effects of compounds on the immune activation; a central process in the development of this diabetic model. Islet morphology (top panel) was not changed by compound administration (Figure 3). Evidence of early inflammation is present as evidenced by invading lymphocytes (open arrows, bottom panel) Figure 3. The presence of inflammatory cells is indicative that the immune component underlying this NOD model has not been disrupted by the administration of compounds.
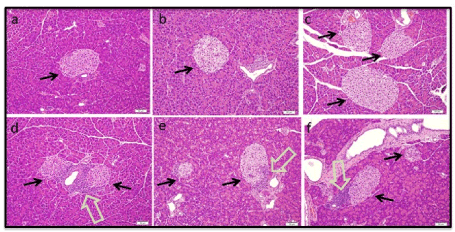
Figure 3. Histology of pre-diabetic (10 week) NOD mice. Images of Hematoxylin and Eosin stained pancreas sections for vehicle control mice (a,d) and mice administered GKT137831 (b, e) or ML171 (c, f). Location of islets are marked (black arrows). Early inflammatory cell infiltrate is identified in representative sections (d-f) and marked (open arrows). Images are representative of all islets captured in analysis of three non-overlapping pancreas sections for each mouse (n=3 per group)
Mice were harvested in the study at onset of diabetes. Consequently, all mice analyzed earlier than week 25 had developed diabetes (Figure 2). These mice would be expected to have significant inflammatory infiltrate with presentation of a number of condensed islets, due to loss of beta cells. Mice in the study first started to convert to diabetes starting at age 11 weeks and conversions continued until the study endpoint. To minimize variability due to age, this report will focus on mice that developed diabetes at age 20 weeks and those that were non-diabetic at 25 weeks (study endpoint).
At twenty weeks of age, pancreas sections contained inflammatory infiltrate and thus support for an immune-mediated onset of diabetes. With the progression of beta cell destruction, the overall size of islets condenses, creating smaller islets. Ongoing beta cell attack is associated with significant immune cell involvement. In all three cohorts the diabetic mice at age week 20 presented with condensed islet structures typical of beta cell destruction, shown on upper panels of Figure 4, marked with open green arrows. Present also were islets with an active immune infiltrate, bottom panel Figure 4. It is notable that the presentation of islets in the compound treated group was with a peri-islet (or halo) arrangement of inflammatory cells. This is in contrast to the control (vehicle treated) mice where active inflammation for control islets engulfed (invaded) the majority of the islet.
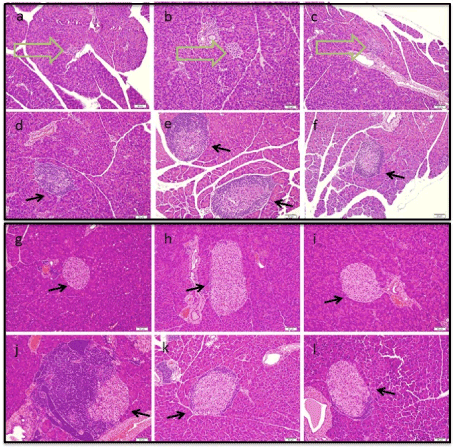
Figure 4. Histology of diabetic (20 week) and non-diabetic (25 week) NOD mice. Images of Hemotoxylin and Eosin stained pancreas sections for vehicle control mice (a,d,g,j) and mice administered GKT137831 (b,e,h,k) or ML171 (c,f,i,l). Panels show representative images from diabetic mice at 20 weeks (a-f) and non-diabetic mice at 25 weeks (g-l). Location of islets are marked (black arrows). Condensed islets in diabetic mice are identified in representative sections (a-c) and marked (open arrows). Images are representative of all islets captured in analysis of three non-overlapping pancreas sections for each mouse
At twenty-five weeks of age, islets were examined from mice at the end of the study. These mice had not reached the criteria for diabetes onset (blood glucose ≥250 mg/dl). Examination of pancreas sections showed presentation of preserved ‘normal’ islets, as represented in the top panel of Figure 4. At 25 weeks islets associated with early inflammation were also observed shown in the bottom panel of Figure 4, no condensed islets were observed. Islet inflammation in the control islets was much more involved than that observed in the mice treated with compounds. Inflammatory cells observed in the compound treated mice were in the peri-islet presentation. One mouse remained non-diabetic in the control group having a blood glucose reading of 129. Five mice were non-diabetic in each of the compound treated groups. The blood glucose ranges were 95 – 210 mg/dl for GKT137831, n=5 and 124 – 228 mg/dl for ML171, n=5.
In summary, the results from this study help to support proof-of concept evaluation for the utility of NOX-1 inhibition in diabetes. Extensive in vitro and ex vivo data demonstrate the importance of NOX-1 activation in mediating inflammation-driven beta cell dysfunction [8,9,11,17,18]. Systemic administration of NOX-1 inhibitors is tolerated in mice and achieves a plasma concentration above the in vitro IC50 for the compounds. A brief exposure of NOD mice to compounds in the pre-diabetic stage resulted in reduction in conversion to diabetes. This preclinical observation is supported in histological evaluation of the pancreas. The data warrant additional study with more sustained exposure to the NOX-1 inhibitors. Importantly, compound administration had no effect on overall islet presentation or morphology or the initiation of the inflammatory mechanism that underlies this diabetic model. It was a consideration that systemic administration of a NOX-1 inhibitor could have ‘off-target’ action and disrupt immune function. The presence of inflammatory infiltrate seen in all study groups at week 10 supports the underlying mechanism of the model is preserved and serves to rule out this trivial explanation for the preclinical outcome.
Islets from mice at week 20 and 25 treated with NOX-1 inhibitors were strikingly different in their presentation relative to the vehicle control group. While inflammatory cells were present in mice administered NOX-1 inhibitors, they predominated in a peri-islet region in the mice treated with compound in contrast to vehicle controls where an inflammatory infiltrate of the entire islet was present. Islets are encapsulated in an extracellular matrix (ECM) consisting of basement membrane and interstitial matrix. In the pathogenesis of type 1 diabetes, discrete steps can be summarized as extravasation of leukocytes from capillary vessel surrounding the islet, migration and accumulation of immune cells to the peri-islet region, breakdown of ECM of the islet capsule, invasion of leukocytes into the islet and finally beta cell destruction [19]. Korpos and Sorokin et al have demonstrated a pre-requisite loss of ECM components in the peri-islet capsule for resultant islet invasion by leukocytes and onset of diabetes [20]. In response to inflammation, beta cells actively respond by upregulating pathways and secreting factors [6, 21]. This crosstalk may be an important link in loss of ECM integrity [19,22,23]. Inhibition of NOX-1 appears to disrupt the crosstalk that results in a breakdown of peri-islet ECM thereby confining inflammatory leukocytes to the peri-islet region and reducing diabetes conversion. Loss of peri-islet ECM integrity directly correlates with diabetes incidence, highlighting the therapeutic potential for NOX-1 inhibition in diabetes.
Acknowledgements
This study was supported by funding from Commonwealth Health Research Board (274-03-15) and Commonwealth Research Commercialization Fund (MF16-019-LS). This work utilized core services supported by NIH grant P30 DK097512 to the Indiana University School of Medicine.
References
- Lenzen S (2008) Oxidative stress: The vulnerable beta-cell. Biochem Soc Trans 36: 343-347. [Crossref]
- Modak MA, Datar SP, Bhonde RR, Ghaskadbi SS (2007) Differential susceptibility of chick and mouse islets to streptozotocin and its co-relation with islet antioxidant status. Journal of comparative physiology. B Biochemical systemic and environmental physiology 177: 247-257.
- Tiedge M, Lortz S, Drinkgern J, Lenzen S (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46: 1733-1742.
- Michalska MG, Wolf R, Walther, Newsholme P (2010) Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta-cell dysfunction. Bioscience reports 30: 445-453.
- Morgan D, Oliveira-Emilio HR, Keane D, Hirata AE, Santos da Rocha MS (2007) Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 50: 359-369.
- Taylor-Fishwick DA, Weaver JR, Grzesik W, Chakrabarti S, Green-Mitchell S, et al. (2013) Production and function of IL-12 in islets and beta cells. Diabetologia 56: 126-135. [Crossref]
- Weaver JR, Nadler JL, Taylor-Fishwick DA (2015) Interleukin-12 (IL-12)/STAT4 axis is an important element for beta-cell dysfunction induced by inflammatory cytokines. PLoS One 10: e0142735.
- Weaver JR, Grzesik W, Taylor-Fishwick DA (2015) Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia 58: 113-121.
- Taylor-Fishwick DA (2013) NOX, NOX who is there? the contribution of NADPH oxidase one to beta cell dysfunction. Front Endocrinol (Lausanne) 4: 40.
- Weaver JR, Holman TR, Imai Y, Jadhav A, Kenyon V, et al. (2012) Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol Cell Endocrinol 358: 88-95.
- Weaver J, Taylor-Fishwick DA (2017) Relationship of NADPH oxidase-1 expression to beta cell dysfunction induced by inflammatory cytokines. Biochem Biophys Res Commun 485: 290-294.
- Cozar Castellano I, Harb G, Selk K, Takane K, Vasavada R, et al. (2008) Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes 57: 3056-3068.
- Taylor Fishwick DA, Weaver J, Glenn L, Kuhn N, Rai G, et al. (2015) Selective inhibition of 12-lipoxygenase protects islets and beta cells from inflammatory cytokine-mediated beta cell dysfunction. Diabetologia 58: 549-557.
- Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, et al. (2010) First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. Journal of medicinal chemistry 53: 7715-7730.
- Serreze DV, Chapman HD, Varnum DS, Gerling I, Leiter EH, et al. (1997) Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol 158: 3978-3986. [Crossref]
- Gray SP, Jha JC, Kennedy K, van Bommel E, Chew P, et al. (2017) Jandeleit-Dahm, Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 60: 927-937.
- Weaver J, Taylor-Fishwick DA (2015) Role of NADPH Oxidase in Beta Cell Dysfunction, in Islets of Langerhans S. Islam (Eds). Springer: Dordrecht, Heidelberg, New York, London. pp. 923-954.
- Weaver JR, Taylor-Fishwick DA (2013) Regulation of NOX-1 expression in beta cells: a positive feedback loop involving the Src-kinase signaling pathway. Mol Cell Endocrinol 369: 35-41.
- Bogdani M, Korpos E, Simeonovic CJ, Parish CR, Sorokin L, et al. (2014) Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr Diab Rep 14: 552. [Crossref]
- Korpos E, Kadri N, Kappelhoff R, Wegner J, Overall CM, et al. (2013) The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes 62: 531-542.
- Ocana GJ, Perez L, Guindon L, Deffit SN, Evans-Molina C, et al. (2017) Inflammatory stress of pancreatic beta cells drives release of extracellular heat-shock protein 90alpha. Immunology 151: 198-210.
- Jezek P, Jaburek M, Plecita-Hlavata L (2019) Contribution of oxidative stress and impaired biogenesis of pancreatic beta-cells to type 2 diabetes. Antioxid Redox Signal.
- Kuehn C, Vermette P, Fulop T (2014) Cross talk between the extracellular matrix and the immune system in the context of endocrine pancreatic islet transplantation. Pathol Biol 62: 67-78.