Post-translationally modified human β-amyloid (Aβ) is found to be a potent target for drug development. Implication of glycosylation, more particularly sialylation as identified with tyrosine at position ten (Y10) in the Aβ appears to play a role in pathogenesis of Alzheimer’s disease. Sialylation facilitates Aβ interaction, internalization, and transport to the brain.
It is therefore postulated that selective intervention with sialylation is a novel strategy to design drugs against Alzheimer’s disease and many other diseases where similar sialylation is involved. This commentary gives an overview of the impact of the observed sialylation on the structure of Aβ, which could play a role in the pathogenesis in Alzheimer’s disease.
Sialic acid derivatives with α 2-6 linkage and or α 2-8 specific linkage are hypothesized to be ideal drug candidates for AD.
Alzheimer’s disease, β-amyloid, sialic acid, glycosylation, sialylation, 2-6 linkage, α 2-8 specific linkage
The presented article is a commentary that is based on the observation that sialylated β-amyloid (Aβ) is found to be excess in the Alzheimer’s disease (AD), compared to the non-AD subjects. As sialylation may have a role in the pathogenesis of Aβ-induced AD symptoms, we postulate that selective intervention of sialylation of Aβ could be a novel strategy to design drugs against AD.
Alzheimer's Disease (AD) is a devastating neurodegenerative disease and is the sixth leading cause of death in the United States. Patients with AD live an average of eight years after their symptoms become noticeable, and survival then can be between 4 to 20 years, depending on age and other health conditions. The destruction and death of nerve cells causes memory failure, personality changes, problems carrying out daily activities, and other symptoms of AD. Two abnormal structures called plaques and tangles are prime suspects in damaging and killing nerve cells. Plaques are deposits of a protein fragment called Aβ that build up in the spaces between nerve cells. Tangles are twisted fibers of another protein called tau that build up inside cells. Although the specific contributions of plaques and tangles in AD are currently unknown, it is believed that they somehow play a critical role in blocking communication among nerve cells and disrupting processes that cells need to survive [1].
The neuropathological hallmarks of AD include extracellular Aβ deposits, intracellular neurofibrillary tangles (NFT), dystrophic neuritis and amyloid antipathy. A processing error of APP (Alzheimer’s precursor protein) and the defective clearance of Aβ generate a cascade of events including hyperphosphorylated tau (τ) mediated breakdown of microtubular assembly and resultant synaptic failure which results in AD. The exact aetiopathogenesis of AD is still obscure. The preeminent hypotheses of AD include amyloid cascade hypothesis and tau hyperphosphorylation [2].
AD is marked by consistent elevation in amyloid proteins thus leading researchers to target Aβ as the drug candidate. This is called the ‘amyloid hypothesis’ which is becoming increasingly less popular among clinicians and more controversial among researchers. In the past 2 decades, billions of dollars have been spent on multiple anti-Aβ antibody based therapeutics, but with limited success. However, some success was observed only on prodromal [patients with early phase or mild form of the disease] patient population which is being pursued by big pharma like Biogen. At the time this article was written, to the best of the author’s knowledge, Biogen’s anti - Aβ antibody has met with initial positive endpoints in clinical trials and a long-term trial is ongoing. Many failed antibodies had inherent immunological side effects. Therefore, an effective treatment for AD is still a challenging puzzle and an unmet need in the medical world. We, at Recombinant Technologies, have been involved in research and development of a non-immune based Aβ binding therapeutic called ‘Amytrap’ for the past decade [3]. We have been able to show, with little or no side effects, that Amytrap is able to reduce brain Aβ levels and improve memory parameters by about 25% in a clinically relevant mouse model of AD [4]. The API [active pharmacological ingredient] is a retro inverso peptide containing D-amino acids. The peptide is conjugated to albumin and polyethylene glycol for stable and slow release into the circulation upon subcutaneous injection. We do have intellectual property rights over this formulation and we continue to exploit the amyloid binding properties of our API via an extracorporeal apheresis column which can bind amyloid from the blood outside of the body (US Patent No.844,5438 B2). Overall, although the antibodies pose a threat through their immunological side effects such as micro hemorrhage, vasogenic edema etc., almost all the Aβ binders including Amytrap have demonstrated that there is a beneficial return to normalcy after at least 25% Aβ reduction. Therefore, the amyloid theory is still alive and worth pursuing. It is possible that modified amyloid such as glycosylated, phosphorylated and or sulfated amyloid might turn out to be the real culprit and thus could become a target for AD therapy. This is the nucleus for the current thinking that amyloid, while less popular, could very well be the target with one or more of these modifications. This new theory should satisfy both the critics and proponents of the amyloid hypothesis.
APP is best known as the precursor molecule whose proteolysis generates Aβ, a polypeptide containing 37 to 42 amino acid residues whose amyloid fibrillar form is the primary component of amyloid plaques found in the brains of AD patients. APP including Aβ undergoes extensive post-translational modifications including glycosylation, phosphorylation, sialylation, and tyrosine sulfation, as well as many types of proteolytic processing to generate peptide fragments with post-translational modifications [5].
Glycosylation is one of the post-translational modifications that occur in the Aβ portion in APP [6]. A significant correlation between disease states and glycosylation pattern on human proteins has been discussed in the literature [7]. The post-translational modifications including glycosylation are shown in Figure 1.

Figure 1. Amino acids of non-amyloidogenic and amyloidogenic amyloids with observed post-translational modifications in Alzheimer’s Disease subjects
Glycosylation patterns, including the glyosidic linkage, are considerably altered during disease states. Depending upon the disease state, a specific set of glycosylation enzymes is either up- or down-regulated, which results in the synthesis of the same protein with different types of oligosaccharide and/or with different types of glyosidic linkages [8]. In AD subjects, Aβ, tyrosine at position 10 (Y10) is O-glycosylated. The different glycoforms such as mono-, di-, and tri-sialylated glycoforms are predominantly found to be associated with the site [9].
Biological functions of the N- and O-linked glycosylation are not fully understood. Differences in glycosylation and particularly sialylation create multiple isoforms of Aβ in humans [9]. Aβ isoforms in AD subjects are significantly different and the level of sialylation is found to be significantly higher (~2.5 times) in AD subjects compared to healthy humans [9]. A striking feature observed in cerebrospinal fluid (CSF) of AD subjects is that the amino acid tyrosine in the tenth position on Aβ (Y10) is O-glycosylated with mono-, bi-, and tri-sialic acids as shown below in Figure 2.
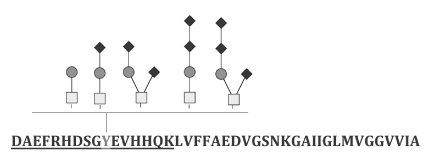
Figure 2. Tyrosine glycosylation in β- amyloid with mono-, di-, and tri-sialylated glycoforms containing α 2, 3, α 2, 6 and α 2, 8- linkage
Interestingly, they are predominantly in the bi-sialylated glycoform. Though the glyosidic linkage of the bi-sialic acids chain associated with Y10 remains unknown, the bi-sialylated terminals are likely to be linked to each other through α-2,8 linkage. In human brain proteins, α-2,8 linked sialic acids have been found predominantly. For instance, neural cell adhesion molecule (NCAM), and synaptic cell adhesion molecule 1 (SynCAM1) are displayed with α-2,8 linked poly sialic acids (PSA; 3, 4) [10]. In AD subjects, the sialylated structures including lactone formation are analogous to the terminal sialylated structures found on brain gangliosides GD1c, GT1a, and GQ1b [6]. It is worth mentioning that Aβ has a sialic acid binding site which facilitates Aβ interaction with cell surface gangliosides or glycoproteins that contain sialic acids [11]. Aβ binds to gangliosides on the neuronal membrane forming a ganglioside-Aβ moiety, which further acts as an endogenous seed for amyloid fibril formation [12].
Careful review of the literature supports the hypothesis that sialic acid derivatives could act as potential therapeutic candidates in AD. Evidence for this comes from the observation that the sialic acid binding immunoglobin like protein, CD33, is one of the AD risk alleles in humans [13-15]. CD33 is a transmembrane protein, and is expressed in human microglial cells. Microglia are a type of glial cells located throughout the brain and spinal cord and act as the first and main form of active immune defense and are constantly scavenging the central nervous systems for plaques, damaged neurons and synapses and infectious agents. In AD patients, CD33 is over expressed in microglial cells present in and around the Aβ plaques [16]. On the other hand, the AD mice deficient in CD33 exhibited reduction in plaques [16]. Additionally, CD33 -/- microglia also show an enhanced capacity to internalize Aβ while the overexpression of CD33 impaired Aβ update [16]. Siglec 11 is a CD33 related human sialic acid binding Ig-like lectin (siglec) family protein and exclusively overexpressed in human brain [17]. Siglec 11 recognizes α-2,8 linked sialic acids and acts as negative immunomodulator and induce immunosuppression [18]. It is through this receptor that we believe sialic acid analogues will be interacting to counter the negative effects of amyloid.
Sailylated Aβ appears to be a fitting target for AD treatment. Aβ has been modified at Y10 with sialylation [9]. When compared to healthy humans, AD subjects show more mono-, di, and tri-sialylation with lactone modification. Figure 2 shows O- glycosylation linked to Y10 with no, mono-, bi-, and tri-sialic acids. Selective interference of such modification is expected to reduce the cross-linking and fibril formation that are the hallmark of AD and thus provides a novel strategy to develop new drugs against Aβ and ameliorate AD and many other diseases [19]. The in-silico analysis with sialyllactose, a sialic acid analogue reaffirms this postulation. Therefore, sialyllactose, its derivatives and other sialic acid containing derivatives provide suitable drug candidates to treat AD.
In the 3D structure, Aβ N-terminal region (1-16), as well as amino acids region 26-30, are found to be flexible regions. These flexible regions are involved in inter and intra-amyloid interaction. Figure 3 shows intra-amyloid interactions. Without being held to theory, it is postulated that sialic acid present at Y10 in combination with or without S8 phosphorylation facilitates Aβ interaction, internalization, and transport into the brain.

Figure 3. Y10 mediated loop formation and cross-linking between intra- Aβ interactions
The P13 region of the Aβ with amino acids (1-17) is not implicated in AD pathogenesis, whereas the Aβ isoforms N-terminal region comprising Y10 may be associated with AD pathogenesis. Thus, the Aβ N-terminal amino acid region 1-16 is considered critical for disease pathogenesis, and therefore is identified as potent target for drug development. Post-translational modifications such as sialylation and phosphorylation create negative charge and influence the pathological properties of Aβ. The negative charge may facilitate Aβ transport across the blood brain barrier.
There appears a sialic acid binding site in Aβ and the N-terminal region 1-11 including Y10 is critical for sialic acid binding. Thus, selective interference of sialic acid binding is expected to interfere with the Aβ interaction, internalization, and transport into the brain. For example, sialyllactose, a sialic acid containing oligosaccharide selectively binds Aβ and will interfere with Y10 mediated transport and plaque formation. Figure 4 shows α2-3 and α2-6 linked sialyllactose. In silico study confirms that these two oligosaccharides are possible drug candidates and can selectively interfere with loop formation and inter- and intra-amyloid cross-linking and subsequent plaque formation. Thus, α 2-3 and α 2-6 linked sialyllactose and n-acetylneuraminic acid (NeuAc) are considered as potential drug candidates.
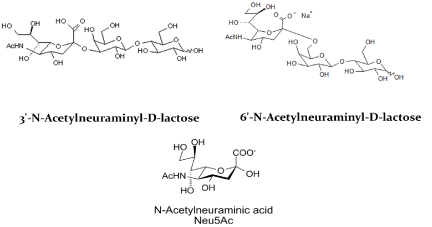
Figure 4. α 2-3 and α 2-6 linked sialic acid drug candidates
The sialic acid linkage determines the positive or negative biological properties in humans. For instance, α2-3 linked sialic acid is involved in inflammation while the α2-6 linked sialic acid is involved in anti-inflammation [20]. Thus, without being held to theory, it is believed that the most favorable drug candidate is α2-6 linked sialic acid analogues and or their derivatives. In AD subjects, there appear bi-and tri-sialic acids with or without lactone formation. Sialic acids are linked to the core saccharide or to the terminal sialic acid with α2-8 linkage. Since the AD subjects generate Aβ with α2-8 linkage, the drug candidates may be designed with such α2-8 linkage to effectively inhibit glycosylated Aβs in AD. Additionally, lactone formation occurs likely due to acidification of sialic acid at inflammation sites. The drug candidate with lactone will also be potent in interfering with glycosylated Aβ (Figure 5).
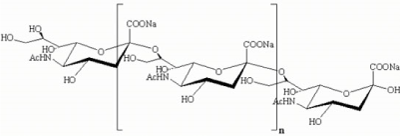
Figure 5. α 2-8 linked Sialic acid drug candidate. (Polysialic acid)
It is very plausible that sialylated Aβ, such as O- glycosylation linked to Y10 as described previously could be effective targets to developing candidates that could inhibit Aβ -induced cytotoxicity. Our laboratory is currently screening and testing several such compounds for AD treatment.
None
Part of the research analysis was supported by an SBIR phase 2 grant R44 AG0 50336, from National Institute of Aging, National Institutes of Health, USA. The author Dr. Pazhani Sundaram thanks NIH for the above grant award. The publication cost is supported by the above grant in which Dr. P.S is the principal investigator.
Help rendered by Dr. Madan Anant, in preparing this manuscript is gratefully acknowledged.
- Takahashi RH, Nagao T, Gouras GK (2017) Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer's disease. Pathol Int 67: 185-193. [Crossref]
- Mondragón-Rodríguez S, Perry G, Zhu X, Boehm J (2012) Amyloid Beta and tau proteins as therapeutic targets for Alzheimer's disease treatment: rethinking the current strategy. Int J Alzheimers Dis 630182. [Crossref]
- Sundaram RK, Kasinathan C, Stein S, Sundaram P (2008) Detoxification depot for beta-amyloid peptides. Curr Alzheimer Res 5: 26-32. [Crossref]
- Sundaram RK, Kasinathan C, Stein S, Sundaram P (2012) Novel Detox Gel Depot sequesters β-Amyloid Peptides in a mouse model of Alzheimer's Disease. Int J Pept Res Ther 18: 99-106. [Crossref]
- Kummer MP, Heneka MT2 (2014) Truncated and modified amyloid-beta species. Alzheimers Res Ther 6: 28. [Crossref]
- Schedin-Weiss S, Winblad B, Tjernberg LO (2014) The role of protein glycosylation in Alzheimer disease. FEBS J 281: 46-62. [Crossref]
- Ohtsubo K, Marth JD (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126: 855-867. [Crossref]
- Lauc G, Pezer M, Rudan I, Campbell H (2016) Mechanisms of disease: The human N-glycome. Biochim Biophys Acta 1860: 1574-1582. [Crossref]
- Halim A, Brinkmalm G, Rüetschi U, Westman-Brinkmalm A, Portelius E, et al. (2011) Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc Natl Acad Sci U S A 108:11848-11853. [Crossref]
- Janas T, Janas T (2011) Membrane oligo- and polysialic acids. Biochim Biophys Acta 1808: 2923-2932. [Crossref]
- Williamson MP, Suzuki Y, Bourne NT, Asakura T (2006) Binding of amyloid beta-peptide to ganglioside micelles is dependent on histidine-13. Biochem J 397: 483-490. [Crossref]
2021 Copyright OAT. All rights reserv
- Ariga T, Wakade C, Yu RK (2011) The pathological roles of ganglioside metabolism in Alzheimer's disease: effects of gangliosides on neurogenesis. Int J Alzheimers Dis 2011: 193618. [Crossref]
- Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, et al. (2008) Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet 83: 623-632. [Crossref]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, et al. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 43: 429-435. [Crossref]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 43: 436-441. [Crossref]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, et al. (2013) Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 78: 631-643. [Crossref]
- Wang X, Mitra N, Cruz P, Deng L; NISC Comparative Sequencing Program, Varki N, et al. (2012) Evolution of siglec-11 and siglec-16 genes in hominins. Mol Biol Evol 29: 2073-2086. [Crossref]
- Angata T, Kerr SC, Greaves DR, Varki NM, Crocker PR, et al. (2002) Cloning and characterization of human Siglec-11. A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J Biol Chem 277: 24466-24474. [Crossref]
- Büll C, Heise T, Adema GJ, Boltje TJ (2016) Sialic Acid Mimetics to Target the Sialic Acid-Siglec Axis. Trends Biochem Sci 41: 519-531. [Crossref]
- Yasukawa Z, Sato C, Kitajima K (2005) Inflammation-dependent changes in alpha2,3-, alpha2,6-, and alpha2,8-sialic acid glycotopes on serum glycoproteins in mice. Glycobiology 15: 827-837. [Crossref]