Abstract
Biological aging is known as senescence, and gradually leads to death through aging and mature organ deterioration. Senescence plays an important role in growth and development. Under the normal physiological conditions, the action of senescence is similar to apoptosis, and has a positive protective response to stimulation. However, in some pathological processes, the senescence effect may be positive or negative. To clarify the role of senescence in a certain disease, the exploration of its special regulation mechanism is the key to search for the new targets of the clinical treatment of disease. Oxidative stress is caused by excessive ROS involved in intracellular reactions and leads to the accumulation of oxidative damage to induce the changes in molecules, cells and tissues. The senescence is inseparable from oxidative stress, so the relationship between them has already become a hot issue of research. In this review, it will be focused on the research progress of senescence and the correlation of senescence with oxidative stress and mitochondrial dysfunction.
Key words
aging, senescence, ROS, oxidative stress, mitochondrial dysfunction
Introduction
Senescence is a physiological phenomenon with a progressive decline in function of an organ or life with the increasing of the age, ultimately leading to death [1]. Cellular senescence is a permanent proliferation arrest by genomic instability or abnormal stimulation. And it is a protective or even tumor-suppressing physiological process [2,3]. Although senescent cell growth is quiescent, it still retains metabolic activity and changes in cellular and molecular levels, which can alter their microenvironment [4-7]. The characteristics of senescent cells include enlargement and flattened phenotype, large vacuoles observed under the microscope, occasional multi-nuclear phenomenon, and positive β-galactosidase staining reaction. It has shown that Sudan black can be used to detect lipofuscin in lysosomes as an indicator of the aging or senescence-related secretory phenotype (SASP) [4,8-11]. Cellular senescence can trigger tissue remodeling in the normal embryonic development and the tissue damage, and the subsequent process is clearance and regeneration. Therefore, temporary senescence is beneficial and can remove damaged cells, which is similar to apoptosis to some extent [12,13]. However, long-term senescence is harmful. The accumulation of senescent cells in some organs, usually in skin, liver, lungs, and spleen, will has an adverse effect on these organs [14]. At present, cellular senescence has been found in a variety of tissues and diseases, such as cancer (lung cancer, breast cancer, neuroblastoma, astrocytoma, colorectal cancer, etc.), fibrosis (idiopathic pulmonary fibrosis, renal fibrosis, etc.), chronic obstructive pulmonary disease COPD, pulmonary hypertension [4,15-19], etc.
Oxidative Stress refers to the excessive production of reactive oxygen species (ROS) exceeds its clearance in cells, which can result in the imbalance between the antioxidant and oxidative system [20]. It can be caused by smoking, obesity, ultraviolet radiation, drug abuse and so on. Excessive ROS participate in intracellular reactions to accumulate oxidative damage and the changes in molecules, cells, and tissues. It is characterized by increased ROS, decreased antioxidant capacity, and functional defects in antioxidant response [20]. Mitochondria are important sites for ROS production [21]. ROS are oxygenates produced by intracellular aerobic metabolism or exogenous oxidants. They are highly active molecules, including superoxide anion, hydroxyl and hydrogen peroxide [22]. Mitochondria are not only the main source of ROS, but also the main target of oxidative damage, which in turn can reduce the efficiency of mitochondria and lead to more ROS production [21]. Besides mitochondria, peroxisomes are also involved in ROS production and clearance [22]. ROS can be taken as an important biological medium and as a harmful medium. At physiologically low concentrations or at appropriate concentrations, ROS regulate protein phosphorylation, ion channels, and transcription factors through redox reactions [23]. ROS are involved in a variety of signaling cascades respond to growth factor stimulation and inflammatory signaling pathways, and involved in the regulation of a variety of cellular processes including differentiation, proliferation, senescence, apoptosis [22,23], etc. However, under pathological conditions, excessively produced ROS may affect DNA, RNA, fat, and protein to cause their interactions to result in functional damage or irreversible changes in the target substance [23,24]. In summary, ROS are currently considered to be major executors in cell damage. Oxidative stress can also sustain the damage to cells and even cause cancer [21,24].
In 1950s, Denham Harman et al. proposed the theory of free radicals in senescence [25]. The hypothesis was that the production of intracellular ROS and their toxic effects on various cellular components are the main determinants of longevity, which link the senescence with oxidative stress for the first time [25]. Since then, the theory of free radicals has been developed into the oxidative stress hypothesis [26]. It is believed that the reduction of the body's antioxidant components during senescence leads to a decrease in the body's ability to scavenge free radicals, which can causes the structural damage of biological macromolecules accumulated with age [26]. In addition, as mitochondria plays a key role in the regulation of bioenergetics, oxidant production and cell death, and play a central role in the development of cellular senescence, the theory has been extended to mitochondrial oxidative stress theory [27]. Although there has been extensive research, the theory of senescence free radicals/oxidative stress is still controversial. Therefore, this review summarizes the current research status of senescence and oxidative stress and focus on the effects of oxidative stress on mitochondria during senescence and the problem to be resolved.
Senescence classification and related mechanisms
Replicative senescence
Replicative senescence refers to the cell cycle arrest, which is expressed as an irreversible stop of proliferation, also known as the Hayflick limit, which can also be caused by increased telomere shortening and increased expression of cell cycle-dependent kinase inhibitors caused by massive replication [8]. When DNA double-strand breaks, it triggers DNA damage reaction (DDR), which leads to γ-H2AX-positive senescence-related DNA damage foci formation, activates ATM and ATR, phosphorylates p53, and then activates p21 [8,28]. In addition, both p16 and RB tumor suppressor have important roles in replicative senescence [29]. However, some studies have shown that p16 has little effect on aging in the mouse model, but it has a significant effect on aging in human [14].
Premature senescence
Premature senescence usually caused by many exogenous cellular stresses including oxidative stress, activation of oncogenes, DNA damage and chromatin abnormalities, which is non-autonomous. According to the different sources of exogenous stimulation, it can be divided into the following categories (Table 1).
Table 1. Classification of premature senescence.
Classification |
Incentive |
Cancer-related senescence |
Abnormal oncogene signaling activated |
Stress-induced senescence |
ROS increase induced by different stress |
Immunesenescence |
The immune system reshaped by aging |
Cancer-related senescence
Senescence is a highly potent, cell-independent tumor suppressor that blocks pre-cancerous cell proliferation and further reduces the risk of tumor formation through cell-independent paracrine SASP [30]. When the abnormal oncogene signaling is activated to prevent tumorigenesis, the cells often initiate the senescence processes [31]. This type of senescence is called oncogene-induced senescence (OIS), which is also a premature form [32]. This response is caused by hyperplasia and can also be caused by DNA hyper-replication, suggesting a same S-phase specific DNA damage response to replicative senescence [31]. Phosphorylation of p53 in OIS activates the ARF-p53 tumor suppressor signaling cascade and activates transcription of downstream target genes, such as CDKN1A, which encodes the protein p21 [31,32]. The induction of OIS is also related to other senescence effectors. Some related studies have shown that the absence of tumor suppressor can cause cellular senescence. For example, the absence of Rb1 tumor suppressor genes in vivo leads to senescence with OIS-like features [33]. Deletion of PTEN also induces senescence (known as PTEN-induced cell senescence, PICS) [8]. The characteristics of PICS are evident, but there is no OIS-like over-replication and DDR production [8]. At the same time, some studies have shown that the upregulation of INK4A and the accumulation of ETS2 in the absence of PTEN also contribute to senescence [34-36]. However, some factors secreted by senescent cells can promote the progression of tumors, and accelerate the proliferation and invasion of precancerous cells. Therefore, senescence in cancer has a dual dependence of time and environment [8].
Stress-induced senescence
After many different types of stress (such as oxidative stress), ROS levels increase significantly and the body's antioxidant capacity declines [37]. Studies have shown that due to mitochondrial dysfunction, older animals have more ROS produced than younger animals, and oxidative damage to DNA, protein, and fat is increased in older animals [37,38]. In the case of senescence caused by ROS, the administration of antioxidants can delay or even prevent senescence, thus indicating the relationship between oxidative stress and senescence [39]. Studies have confirmed that under high levels of ROS, p38 MAPK is activated by the RAS-RAF-MEK-ERK cascade, which leads to an increase in p53 transcriptional activity and up-regulation of p21 [40].
Immunesenescence
Senescence can continue to reshape the immune system. This process is called immunesenescence, which reflects the human senescence process or phenomenon in the immune system [41,42]. Human immunesenescence is a cellular and molecular process including the innate and adaptive immune reactions, to ultimately result in the entire immune deficiency [42]. Senescence is changing at different speeds, in different ways and towards different directions [43]. Many studies have shown that the impaired function of human monocytes and macrophages is associated with aging in innate immunity [41,44]. Besides innate immunity, acquired immunity is also targeted and remodeled in the aging process [41]. The key phenotypes and functions of peripheral blood T cells are changed, but the total T cell level is basically unchanged [41]. Earlier studies have confirmed that chronic stimulation is associated with thymus degeneration, slow proliferation of T cells and increased serum pro-inflammatory markers [43]. Human infection with cytomegalovirus can accelerate senescence by reducing the T cell receptor pool and promoting the proliferation of aged CD8+CD28-T cells [41]. These factors explain the relationship between inflammation and senescence, also known as "inflammatory senescence" [41]. Some other characteristic changes occur in the senescence of the immune system, especially T cells. The important characteristic changes comprise the reduced number and function of hematopoietic stem cells, degraded thymus, declined peripheral primary T cells, highly differentiated memory CD28-T cells, decreased CD4+/CD8+ ratio, and decreased IL-2 by T cell activation [42,43]. Administration of exogenous IL-2 reverses the defects in age-related T cell activation [45].
Oxidative stress
In the human body, more than 90% of oxygen is consumed by mitochondria, and 1-5% of the oxygen consumed is converted to superoxide by electron leakage from the mitochondrial electron transport chain [21]. The superoxide is produced spontaneously or catalyzed by superoxide dismutase (SOD) to be converted into hydrogen peroxide [46]. The hydrogen peroxide has membrane permeability and diffusibility, and it can be decomposed into water by glutathione peroxidase or thioredoxin peroxidase or catalase [46]. In the presence of divalent cations such as Fe2+ and Cu2+, Fenton's reaction occurs and produces more active hydroxyl radicals [46]. Mitochondria are not only the main source of ROS, but also the main target of oxidative damage which can reduce the efficiency of mitochondria and induce more ROS production [21]. Peroxisomes are also involved in ROS production and clearance. Peroxisomes have several oxygen-consuming metabolic functions [22]. Oxygen consumption in peroxisomes leads to the production of hydrogen peroxide, which can oxidize multiple molecules [22]. It is shown that peroxisome dysfunction affects mitochondrial function, but its specific mechanism remains unclear [47]. Except the cellular metabolic processes, ROS can also be produced by overstimulation of nicotinamide adenine dinucleotide phosphate oxidase or as a response to different environmental stimuli such as growth factors, inflammation, ionizing radiation, ultraviolet light [22], etc. ROS can both as an important biological medium and as a harmful medium. At physiologically low concentrations or at appropriate concentrations, ROS regulate protein phosphorylation, ion channels and transcription factors through redox reactions [23]. ROS is involved in a variety of signaling cascades respond to growth factor stimulation and inflammatory signaling pathways, and is also involved in the regulation of a variety of cellular processes including differentiation, proliferation, senescence, apoptosis [22,23]. Oxidative stress can participate in the regulation of cell membrane signal transduction, such as protein kinase C and MAP kinase activity, proliferation and differentiation [21,26]. Apoptosis induced by oxidative stress not only prepares the birth passage for childbirth but also enhances the defense function of the organism. Therefore, proper oxidative stress is physiologically beneficial [21]. However, under pathological conditions, excessive ROS may affect DNA, RNA, fat and protein, which causes their interactions to result in functional damage or irreversible changes of the targets [24]. In summary, ROS are currently considered to be the major executors in cell damage. Oxidative stress can also cause damage to cells and even sustain cancer [21, 24].
Senescence, oxidative stress and mitochondrial dysfunction
The classification of the mechanism between senescence and oxidative stress
At present, there are mainly four pathways of oxidative stress-induced cell senescence. One is the DNA damage response pathway which is that oxidative stress causes DNA damage to activate DDR reaction through activating p53 and up-regulating p21 expression to cause senescence [48]. The second is the nuclear factor κB (NF-κB) pathway. Excessive ROS by oxidative stress activates IκBs kinase, which phosphorylates IκB to activate NF-κB and make it transfer into the nucleus to stimulate interleukin-8 expression and increase p53 protein stability and then induce cellular senescence [49]. The third is the p38 MAPKs pathway which is activated by ROS, and it up-regulates p19 protein expression and limits self-renew to induce cellular senescence [50]. The fourth is the microRNA pathway. It has been reported that oxidative stress can affect the amount of microRNA and promote senescence [51].
With regard to senescence, the most accepted theory is that oxidative stress is an important driver for senescence [37, 46]. Generally, ROS produced by oxygen metabolism can induce damage in cells or organisms [52]. Oxidative stress plays an important role in senescence by transforming protein conformation, changing catalytic activity, altering protein-protein interactions and protein-DNA interactions, affecting protein transport, and altering some signaling mechanisms to activate NF-κB and Smad3 [53]. Studies have shown that under the conditions of cellular stress or DNA damage, senescence will be initiated to prevent the proliferation of DNA abnormal cells and the formation of tumors [54]. At the same time, autophagy is initiated to remove residual organelles in the cells, eventually to induce apoptosis to maintain the tissue and body homeostasis [54].
The relationship between senescence and mitochondrial dysfunction
Mitochondrion, an important organelle in eukaryotic cells, determines cell fate and death and participates in cell signaling [21]. Mitochondrial dysfunction disrupts the function of cells/tissues/organs, which can causes lesions and even some age-related diseases [27]. Mitochondrial membrane phospholipids are sensitive to ROS-induced lipid peroxidation, because it has many unsaturated fatty acids on it [55]. The reports about the increased lipid peroxidation with age-related mitochondrial membrane double bonds and fat suggest that ROS damage to mitochondria is related to the senescence process [55]. There is a mitochondrial protein quality control progress in mitochondria, which is accumulated with oxidative damage [56]. When it exceeds its tolerance, the protein quality control mechanisms of other organelles are intervened to deal with the accumulation of mismatched proteins [56]. However, when the damage accumulation is beyond the ability of all quality control mechanisms, the damaged organelles are separated from other mitochondrial networks and then cleared out by mitochondrial autophagy [21]. Mitochondrial kinetics and mitochondrial autophagy regulate protein homeostasis and mitochondrial function accompanied by protein translocation and protein quality control [55]. The process is disrupted to cause mitochondrial stress and even senescence. Mitochondrial proteins are the first attacked by oxidative stress [27]. The proteins damaged by oxidative stress may be inactivated to lose their tertiary structure or to form toxic substances in organelles. Therefore, mitochondrial protein quality control system is very important [56]. The accumulation of mitochondrial DNA mutations induced by oxidative stress is associated with a progressive mitochondrial dysfunction, and promotes age-related physiological function decline [21]. The relevant studies indicate that the mitochondrial ROS in long-lived mammals and birds is lower than that in short-lived species, accompanied by less mitochondrial membrane unsaturated fatty acid, less oxidative mitochondrial DNA damage and less lipid oxidation resistance [57].
Peroxisome dysfunction may be related with Mitochondria dysfunction to give rise to cellular senescence
The relationship between mitochondria oxidative stress and cell death has been well discussed, but the potential role of peroxisomes in cell death pathways is emphasized in the recent studies that have shown that peroxisomes and mitochondria cooperate at different levels to maintain multiple metabolic and signaling pathways and share important components of some organelle division mechanisms, and that peroxisomes also have redox sensitivity [22]. Peroxisome plays an important role in lipid metabolism and often catalyzes reduction reactions [22]. Peroxisomes are important sites for ROS production and degradation, and maintain cellular oxidative balance and proper membrane lipid components to combat oxidative stress [47]. Disturbance of peroxisome metabolism makes cells more sensitive to oxidative stress [47]. Peroxisome dysfunction leads to impaired mitochondrial oxidative phosphorylation, increased number of abnormal mitochondrial structures, and altered activity and expression of the respiratory chain complex. After overexpression of catalase, mitochondrial redox balance and function are restored [47]. In summary, changes in peroxisome metabolism can seriously affect mitochondrial function, but the specific relationship between them remains to be further studied [47].
Conclusion
Senescence plays an important role in various physiological and pathological processes. Senescence research maybe a potential breakthrough in the treatment of diseases [8,58]. It is reported that oxygen exercise, calorie restriction, balanced diet and other means can reduce the level of oxidative stress and maintain the balance of oxidation-antioxidant system in the body, which will delay the occurrence of age-related diseases as much as possible and the process of the body aging and prolong the life [59]. However, oxidative stress is the key to cellular senescence. As mentioned as above, the accumulation of external stimuli induced the production of ROS and reduced the ability of the body's antioxidant components to scavenge free radicals to cause the age-related diseases and shorten the life span [26]. Abuse of oxidants may interfere with the normal physiological signaling pathways in the body and increase the risks of senescence-related diseases [60].
In summary, the effect of oxidative stress on senescence is related to the function and structure of peroxisomes and mitochondria, but oxidative stress may cause senescence finally through mitochondrial dysfunction as shown as Figure 1.
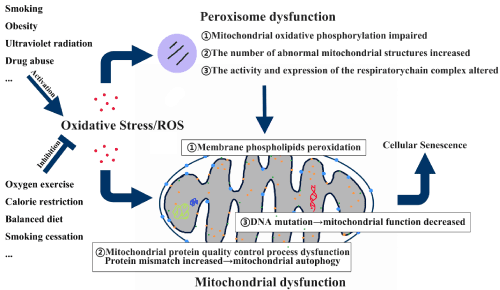
Figure 1. Oxidative stress results in cellular senescence through mitochondrial dysfunction.
Acknowledgment
This work was supported by National Natural Science Foundation of China [No. 81503058] and Natural Science foundation of Liaoning Province [No. 2014021065].
References
- Allan GJ, Beattie J, Flint DJ (2008) Epithelial injury induces an innate repair mechanism linked to cellular senescence and fibrosis involving IGF-binding protein-5. J Endocrinol 199: 155-164. [Crossref]
- Shih CT, Chang YF, Chen YT, Ma CP, Chen HW, et al. (2017) The PPAR?-SETD8 axis constitutes an epigenetic, p53-independent checkpoint on p21-mediated cellular senescence. Aging Cell 16: 797-813. [Crossref]
- Walters HE DS, Cox LS (2016) Reversal of phenotypes of cellular senescence by pan-mTOR. Aging (Albany NY) 8: 231-244. [Crossref]
- Zhou F, Onizawa S, Nagai A, Aoshiba K (2011) Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res 12: 78. [Crossref]
- Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, et al. (2016) Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig 54: 397-406. [Crossref]
- Kim CO, Huh AJ, Han SH, Kim JM (2012) Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch Gerontol Geriatr 54: e35-e41. [Crossref]
- Gorgoulis VG, Halazonetis TD (2010) Oncogene-induced senescence: the bright and dark side of the response. Curr Opin Cell Biol 22: 816-827. [Crossref]
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP (2011) Pro-senescence therapy for cancer treatment. Nat Rev Cancer 11: 503-511. [Crossref]
- Disayabutr S, Kim EK, Cha SI, Green G, Naikawadi RP, et al. (2016) miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLoS One 11: e0158367. [Crossref]
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, et al. (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23: 798-803. [Crossref]
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, et al. (2005) Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 8: 19-30. [Crossref]
- Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, et al. (2103) Programmed cell senescence during mammalian embryonic development. Cell 155: 1104-1118. [Crossref]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, et al. (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119-130. [Crossref]
- Muñoz-Espín D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482-496. [Crossref]
- Gao N, Wang Y, Zhen2021 Copyright OAT. All rights reservlig;2-Microglobulin participates in development of lung emphysema by inducing lung epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol 312: L669-L677. [Crossref]
- Davalli P, Mitic T, Caporali A, Lauriola A, D'Arca D (2016) ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longev: 3565127. [Crossref]
- Dong D, Cai GY, Ning YC, Wang JC, Lv Y, et al. (2017) Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling. Oncotarget 2017 8: 16109-16121. [Crossref]
- Feng C, Liu H, Yang M, Zhang Y, Huang B, et al. (2016) Disc cell senescence in intervertebral disc degeneration: Causes and molecular pathways. Cell Cycle 15: 1674-1684. [Crossref]
- Kida Y, Goligorsky MS (2016) Sirtuins, Cell Senescence, and Vascular Aging. Can J Cardiol 32: 634-641. [Crossref]
- Ashok BT, Ali R (1999) The aging paradox: free radical theory of aging. Exp Gerontol 34: 293-303. [Crossref]
- Kong Y, Trabucco SE, Zhang H (2014) Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip Top Gerontol 39: 86-107. [Crossref]
- Bonekamp NA, Völkl A, Fahimi HD, Schrader M (2009) Reactive oxygen species and peroxisomes: struggling for balance. Biofactors 35: 346-355. [Crossref]
- Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 8: 245-313. [Crossref]
- Marnett LJ (2000) Oxyradicals and DNA damage. Carcinogenesis 21: 361-370. [Crossref]
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298-300. [Crossref]
- Sies H, Cadenas E (1985) Oxidative stress: damage to intact cells and organs. Philos Trans R Soc Lond B Biol Sci 311: 617-631. [Crossref]
- Shinmura K (2013) Effects of caloric restriction on cardiac oxidative stress and mitochondrial bioenergetics: potential role of cardiac sirtuins. Oxid Med Cell Longev: 528935. [Crossref]
- d'Adda dFF, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, et al. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194-8. [Crossref]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, et al. (1998) Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396: 84-88. [Crossref]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24: 2463-2479. [Crossref]
- Di MR, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, et al. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638-642. [Crossref]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593-602. [Crossref]
- Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, et al. (2009) Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 15: 255-269. [Crossref]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, et al. (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725-730.
- Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, et al. (2010) A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest 120: 681-693. [Crossref]
- Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, et al. (2009) Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal 2: ra2. [Crossref]
- Dasari A, Bartholomew JN, Volonte D, Galbiati F (2000) Oxidative stress indu ces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res 66: 10805-10814. [Crossref]
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, et al. (1999) Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 274: 7936-7940. [Crossref]
- Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, et al. (2002) Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J 21: 2180-2188. [Crossref]
- Sun P, Yoshizuka N, New L, Moser BA, Li Y, et al. (2007) PRAK is essential for ras-induced senescence and tumor suppression. Cell 128: 295-308. [Crossref]
- Bauer ME, Wieck A, Petersen LE, Baptista TS (2015) Neuroendocrine and viral correlates of premature immunosenescence. Ann N Y Acad Sci 1351: 11-21. [Crossref]
- Pawelec G (2006) Immunity and ageing in man. Exp Gerontol 41: 1239-1242. [Crossref]
- Jorg J. Goronzy CMW (2013) Understanding immune senescence to improve vaccine responses. Nat Immunol 14: 428-436. [Crossref]
- Shaw AC, Goldstein DR, Montgomery RR (2013) Age-dependent dysregulation of innate immunity. Nat Rev Immunol 13: 875-887. [Crossref]
- Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, et al. (2008) T cell subset-specific susceptibility to aging. Clin Immunol 127: 107-18. [Crossref]
- Kirkinezos IG, Moraes CT (2001) Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol 12: 449-457. [Crossref]
- Nordgren M, Fransen M (2014) Peroxisomal metabolism and oxidative stress. Biochimie 98: 56-62. [Crossref]
- Rai P, Onder TT, Young JJ, McFaline JL, Pang B, et al. (2009) Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc Natl Acad Sci U S A 106: 169-174. [Crossref]
- Lee MY, Wang Y, Vanhoutte PM (2010) Senescence of cultured porcine coronary arterial endothelial cells is associated with accelerated oxidative stress and activation of NFkB. J Vasc Res 47: 287-298. [Crossref]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, et al. (2006) Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12: 446-451. [Crossref]
- Li G, Luna C, Qiu J, Epstein DL, Gonzalez P (2009) Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev 130: 731-741. [Crossref]
- Barnes PJ (2017) Senescence in COPD and Its Comorbidities. Annu Rev Physiol 79: 517-539. [Crossref]
- Faner R, Rojas M, Macnee W, Agustí A (2012) Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186: 306-313. [Crossref]
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, et al. (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126: 121-134. [Crossref]
- Schlame M, Rua D, Greenberg ML (2000) The biosynthesis and functional role of cardiolipin. Prog Lipid Res 39: 257-288. [Crossref]
- Lionaki E, Tavernarakis N (2013) Oxidative stress and mitochondrial protein quality control in aging. J Proteomics 92: 181-194. [Crossref]
- Pamplona R, Barja G, Portero-Otín M (2002) Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: a homeoviscous-longevity adaptation. Ann N Y Acad Sci 959: 475-490. [Crossref]
- Naesens M (2011) Replicative senescence in kidney aging, renal disease, and renal transplantation. Discov Med 11: 65-75. [Crossref]
- El AM, Angulo J, Rodríguez-Mañas L (2013) Oxidative stress and vascular inflammation in aging. Free Radic Biol Med 65: 380-401. [Crossref]
- Ludovico P, Burhans WC (2014) Reactive oxygen species, ageing and the hormesis police. FEMS Yeast Res 14: 33-39. [Crossref]