Abstract
Schizophrenia is a brain disconnection syndrome involving abnormal interactions between widespread networks. Genetic and non-genetic mechanisms are thought to interact affecting the developing brain resulting in a predisposition to suffer schizophrenia. We did a review of current data of research emphasizing on white matter abnormalities mainly punctual axonal disorganization in many pathways and reduction of total number of neurons in different parts of the brain, brainstem and/or cerebellum. Finally we consider: (i) hypoactivation of dorsolateral and dorsomedial prefrontal cortices from thalamic reticular and mediodorsal thalamus impairing the normal streaming of working memory and causing cognitive disorder, (ii) hyperactivation of hippocampal pyramidal neurons provoking hyperdopaminergic subcortical outbursts giving rise to positive symptoms like paranoid delusions and auditory visual hallucinations and (iii) inefficiency of salience networks linked to diminished population of Von Economo spindle neurons in anterior cingulate cortex and reduction of gray matter in anterior insula which can be linked with negative symptoms, aberrancies in default-mode network and self-face recognition disorder.
Keywords
Schizophrenia, Axonal dysconnectivity, Neuronal loss, Entorhinal-hippocampal system, Mediodorsal thalamic nuclei, Salience network
Introduction
Despite intensive research for over a hundred years, the exact causes of schizophrenia (SZ), remain unclear. Schizophrenia is a severe neuropsychiatric disorder with a lifetime prevalence of approximately 1% in most studied populations. It is a brain disconnection syndrome involving abnormal interactions between differentiated networks [1]. This causes disturbances in cognition, mood, reality testing, interpersonal relations, social abilities and workplace functionality [2].
Age of onset is frequently between 12 and 30 years, and is associated with multifactorial genetic risk, involving genes in chromosomes 1, 6, 8, 10, 13, 16, 17 and 22 among others.
Prefrontal Networks
Okubo et al. [3] found reduced radioligand binding to D1 neurons in the prefrontal cortex (PFC) of SZ patients, this reduction correlated with the severity of the negative symptoms and to poor performance in the Wisconsin Card Sorting Test. Thus, dysfunction of D1 in the PFC may contribute to the negative symptoms and cognitive deficits seen in SZ patients.
The currently dominant view is that SZ might be characterized by an imbalance between subcortical and cortical DA systems: subcortical mesolimbic DA projections being hyperactive through hyperstimulation of D2 receptors causing positive symptoms, while mesocortical DA projections to the PFC are hypoactive resulting in hypostimulation of D1 receptors, cognitive impairment and negative symptoms. Deficiency in mesocortical DA function would cause disinhibition of mesolimbic DA activity [4]. 5-HT2a antagonists may increase DA release in the PFC cortex by direct action at 5-HT2a receptors on DA nerve terminals [5–7]. 5-HT2a antagonists may also prevent the effect of decreased prefrontal glutamatergic activity that inhibits burst firing of ventral tegmental area (VTA) DA neurons and this would probably be one of the mechanisms by which 5-HT2A antagonists decrease negative symptoms [8].
Friston and Frith [9] have suggested a severe disruption in prefronto-temporal interactions known as “the disconnection hypothesis”. Although SZ is not characterized by gross abnormalities of white matter, it may involve a dysregulation of myelin-associated gene expression, coupled with reductions in oligodendrocyte numbers, and marked abnormalities in the ultrastructure of myelin sheaths. Since each oligodendrocyte (OLG) myelinates as many as 40 axon segments, changes in the number of OLG, and/or alterations in the integrity of myelin sheaths, and/or axoglial contacts can have a profound impact on signal propagation and integrity of neuronal circuits.
Kubicki et al. [10] using DTI information in SZ patients found fractional anisotropy (FA) decreased in SZ patients in the fornix, corpus callosum, also bilaterally in the cingulum bundle, bilaterally in the internal capsule, in the right inferior occipito-frontal fasciculus and in the left arcuate fasciculus. DTI data showed white matter abnormality is predominant in the anterior corpus callosum and fibers projecting to the medial frontal cortex. The medial frontal region identified by these imaging techniques corresponds to the anterior midline node of the default mode network (DMN), a brain system which is believed to support internally directed thought, a state of watchfulness, and/or the maintenance of the individual´s sense of self, all of the above are currently of interest in neuropsychiatric disorders [11]. DMN is most commonly shown to be active when an individual is not focused on the outside world and the brain is at wakeful rest, such as during mind-wandering, but it is also active when the individual is thinking about others, thinking about himself, remembering the past or planning for the future [12]. The hypoconnectivity with medial prefrontal/ACC change in SZ patients the normal pattern in DMN.
Wible et al. [13] using MRI found right prefrontal white matter significantly reduced in SZ patients and those with high negative symptom scores had significantly smaller bilateral white matter volumes. In addition, right prefrontal gray matter volume alterations were significantly correlated with reduced right hippocampal volume in the SZ patients but not in the control group. These results underscore the importance of temporal–prefrontal pathways in the symptomatology of schizophrenia, and they suggest an association between prefrontal abnormalities and negative symptoms. Other investigators reported white matter reduction to be more severe in orbitofrontal regions and these findings were associated with negative symptoms [14].
Alterations in brain function in SZ are evident during specific cognitive challenges (cognitive control network, CCN) and also found in functional MRI (fMRI) data obtained during resting state (DMN). In SZ patients, an inverse correlation of negative symptoms was found with right anterior PFC activity at rest. These findings suggest that aberrant DMN connectivity contributes to regional functional pathology in SZ bearing significance for core symptoms. Aberrant connections between anterior and lateral DMN have been observed [15], suggesting a pivotal role for DMN in SZ, however the dissociation pattern of functional connectivity (FC) in DMN subsystems remains poorly understood.
The reduction in functional connectivity between thalamus and PF cortex alters the working memory network (WMN) affecting the central executive complex [16] (Figure 1). Other investigators found correlations in SZ patients between WM performance and FA at the right anterior internal capsule [17] and left thalamus [18]. Atypical connectivity was associated with failure in introspective processing and limitations in theory of mind (ToM), as has been demonstrated in most SZ patients [19–21]. First degree relatives of these patients have been reported to present similar atypical connectivity to that observed in patients, although patients tend to exhibit more severe and widespread aberrances [22–24]. According to Marenco et al. patients with SZ had reduced thalamocortical connectivity than controls in the left PFC although having an increase in total connectivity with the motor cortex [25]. Neurons projecting to PFC in the MD were reduced in numbers en SZ [26,27,28] (Figure 1).
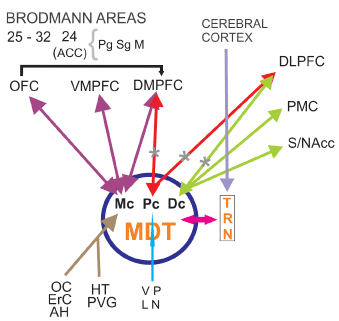
Figure 1: Frontocingulothalamic network (FCTN) Hypoactivation of DLPFC and DMPFC from TRN-MD thalamus. FCTN is involved in cognitive symptoms of SZ and disorganized psychosis. TRN thalamic reticular nucleus. MDT mediodorsal thalamic nucleus. MC: Magnocellular sector. PC: parvocellular sector. DC: Densocellular sector. OC: Olfactory Cortex. ErC: Entorhinal Cortex. AH: Amygdala-Hippocampus. HT: Hypothalamus. PVG: Periventricular Grey matter. VP: Ventral Pallidum. LN: Locus Niger. OFC: Orbitofrontal Cortex. VMPFC: Ventromedialprefrontal Cortex. DMFFC: Dorsomedialprefrontal Cortex. DLPFC: Dorsolateralprefrontal Cortex. PMC: Premotor Cortex. S/NAcc: Striatum/Nucleus accumbens. ACC: Anterior Cingulate Cortex. Pg: Perigenual sector. Sg: Supragenual sector. M: Middle sector
*Specific defects found in SZ patients, according to Popken et al. (2000) and Wagner et al. (2013) among others.
It has been pointed out that PF networks provide us the matrix in order to choose (free will?) between taking an action or not [29] and give anatomical substrate for active thought (tättige Gedanken [30], Kleist 1937), “the inner voice of thoughts”, found to be abnormal in SZ patients during auditory verbal hallucinations AVH [31,32].
Role of amygdala and entorhinal-hippocampal system
Amygdala
The Edinburgh High-Risk Study (EHRS) evaluated initially healthy adolescents with at least two affected relatives. This study found that the amygdala-hippocampal complex (AHC) volumes are reduced pre-morbidly but not to SZ levels, suggesting that further volume reductions may be associated with the onset of SZ. AHC volumes appear to be genetically mediated in families with a dominant pattern of transmission. Neuropsychological testing has also demonstrated pre-morbid impairments and symptom-related deterioration [33,34]. Krekzmanski et al. [35] examined 13 post-mortem brains of male SZ patients and found a 12.1-17.6 % (left-right) reduction in the volume of lateral nucleus of the amygdala and 15.9-16.2 (left-right) mean reduction in total neuron number at the same nucleus. According to Rasetti et al. [36] decreased amygdala reactivity suggests that SZ patients have a deficit in the recruitment and regulation of limbic areas implicated in the response to salient stimuli, specifically, fear related. Mier et al. [37] replicated previous findings of affected emotion recognition in SZ, these results provide further evidence for impaired social cognition in SZ and point to a central role of the amygdala in negative misperceptions of facial stimuli in SZ. Early postnatal lesions of the amygdala in rats lead to a SZ-like behaviour whereas such lesions in adults rats do not produce such changes [38]. Following these findings, a neurodevelopmental deficit in SZ affecting the basolateral amygdala was suggested [35]. The amygdala is involved in encoding valence and intensity of emotions [39]. The connections between PFC and hippocampus-amygdala are involved in emotional learning, modulation of memory, and emotional contribution to decision-making and social behaviour [40]. The amygdala is also involved in the organization of emotional tension discharge into consciousness, and the building of the nucleus of a conscious experience [41]. The amygdala is an essential point of contact between cognitive representations and emotional feelings [42]. Electric stimulation studies suggest it may be the site of parathymic experiences [43] which are frequent in schizophrenic patients [44].
Entorhinal-hippocampal system (EHS)
Bilateral hippocampal size reduction in SZ, has been described [45–48] as early as the first psychotic episode [49,50]. Morphometric neuroimaging, magnetic resonance spectroscopy, and functional neuroimaging, have provided evidence for abnormal hippocampal structure and functionality in SZ. Hippocampal volume reduction is now one of the most consistent structural abnormalities found in SZ; it can be found at onset of the illness and to a lesser degree in first-degree relatives of SZ patients. Decreased levels of N-acetyl-aspartate imply a cellular or axonal basis for the volume changes. Functional neuroimaging studies have demonstrated enhanced levels of hippocampal activity at rest, during the experience of auditory verbal hallucinations (AVH), and during the performance of memory retrieval tasks. These neuroimaging study results complete the evidence of post-mortem and behavioural studies, which have found specific regional hippocampal abnormalities and of memory function in SZ [51].
Patients with SZ undergoing fMRI during visual tasks showed reduced habituation of the hippocampus and visual cortex, and a lack of neural discrimination between old and new images in the hippocampus. Hippocampal discrimination correlated with memory performance, suggesting reduced habituation may contribute to the memory deficits commonly observed in SZ patients [52].
Compared to controls, the patients with SZ showed a significant decrease in the mean number of oligodendrocytes in the left and right CA4 area [53]. Reif et al. [54] have suggested diminished adult neurogenesis in the dentate gyrus in SZ, plausibly associated in some patients with changes in risk gene DISC-1 [55] and/or NRG1 [56]. Kolomeets et al. [57] reported reduction in the number of synapses of mossy fibers into CA3 pyramidal neurons in post-mortem SZ tissue. In addition, post-mortem examinations of SZ subjects revealed a marked decrease in the number of CA2 inhibitory neurons, while the rest of the hippocampus remained largely unaffected [58]. Experimental data showed activation of the subiculum promote release of DA in the VTA indicating the EHS region is a key structure involved in hyperdopaminergc states [59] (Figure 2).
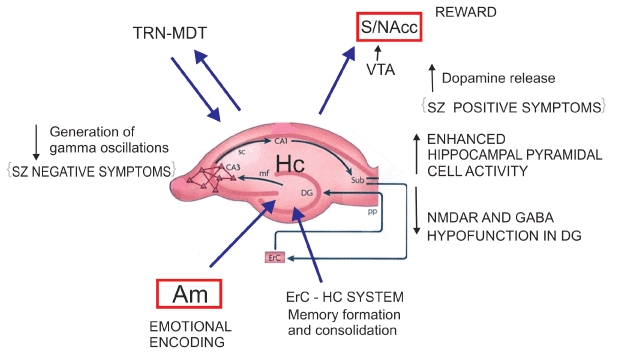
Figure 2: Entorhinal Hippocampal System (EHS) Neurochemical alterations at the EHS level enhancing their projections to VTA-NAcc and diminishing the generation of gamma oscillations at the thalamic level could explain SZ positive and negative symptoms (Lisman et al. (2008) and possibly paranoid psychosis derived from positive symptoms. CA1, CA3 hippocampal subfields. SC: Schaffer’s collateral bundle. Sub subiculum. ErC: Entorhinal Cortex. PP: Perforant Pathway. Dg: Dentate gyrus. mf: mossy fibers. TRN: Thalamic Reticular Nucleus. MDT: Mediodorsal Thalamic Nucleus. VTA: Ventral Tegmental Area. S/NAcc: Striatum/Nucleus accumbens. Am Amygadala. Hc Hippocampus.
Post-mortem molecular changes in the hippocampal formation in SZ patients suggest a selective reduction in glutamate transmission in the dentate gyrus and in its efferent fibers, the mossy pathway, provoking increased activity in disinhibited CA3 neurons and loss of dentate gyrus mnemonic functions, namely pattern separation. Through pattern separation, novel events are recognized as unique. Through pattern completion a conjunctive representation can be recalled capturing the co-occurrence of the multiple features that constitute a given experience, this is dependent on mechanisms located in CA3, CA1 and subiculum [60]. The abnormal changes in SZ patients may reduce discrimination between present and past experiences in memory. Thus CA3-mediated pattern completion could result in cognitive mistakes and memories with psychotic content. The relative shift from pattern separation to pattern completion can introduce inappropriate associations, illogical memories and paranoid susceptibility, retrieving a past event rather than encoding a new representation and the patients repeat the same patterns of delusions or hallucinations. In summary, the acquisition of new experiences could be disturbed in SZ patients as a consequence of structural and functional disturbances in some regions of the EHS.
Role of the thalamus
The thalamus provides a nodal link for multiple functional circuits that are impaired in SZ. Despite inconsistencies in the literature, a meta-analysis suggests that the volume of the thalamus, relative to that of the brain is decreased in SZ patients. Morphometric neuroimaging studies employing deformation, voxel-based and region of interest (ROI) methodologies suggest that the volume deficit preferentially involves the anterior, MD nuclei and the pulvinar [61]. In vivo studies using magnetic resonance imaging have demonstrated smaller thalamic volumes in SZ, as well as shape deformations, suggesting changes in those thalamic regions that are most densely connected to the portions of the brain responsible for executive function and sensory integration. These changes seem to be correlated with clinical symptomatology described in these patients [62]. Using stereological techniques in six pairs of individually matched brains from SZ patients and controls, Popken et al. have found a significant reduction in total neuron number in mediodorsal nucleus (MD). Because the subnuclei of MD have different connections and project to different areas of the frontal cortex, specific loss of neurons in MDpc and MDdc may have implications for the functional defects observed in SZ [26] (Figure 1) but its clear significance remains uncertain.
Glutamate receptor expression was lower at both transcriptional (NMDAR1, NMDAR2B, NMDAR2C, gluR1, gluR3, and KA2 subunit mRNAs) and posttranscriptional ([(3)H] ifenprodil and [(3)H]MDL105,519 binding to polyamine and glycine sites of the NMDA receptor) levels in the thalamus in patients with SZ than in comparison subjects. Differences were most prominent in nuclei with reciprocal projections to limbic regions consistent with diminished glutamatergic activity in the thalamus in SZ [63].
Wagner et al. during a fMRI study applied a Stroop task in an event-related design. In the fMRI analysis, the patients demonstrated a significantly decreased BOLD signal in the fronto-cingulo-thalamic network. A significantly decreased bilateral endogenous connectivity between the MD and the anterior cingulate cortex (ACC) was detected in patients in comparison to healthy controls (Figure 1). This was correlated with the Stroop interference score. A decrease in white matter volume was observed in the MD and frontal cortex of these patients [64].
Evidence suggests gamma oscillations are regulated by TRN influence on thalamocortical loops [65]. In this way, Pratt and Morris [66] emphasize the role of the thalamic reticular nucleus (TRN) in attentional disorders in SZ (see also Ferrarelli & Tononi [67], Steullet et al. [68]), considering the possibility of dynamic interactions with other thalamic nuclei and thalamocortical networks. TRN function is supposed to be linked with switching between environmental and internal stimuli and switching between DMN and CCN [69].
Reduced levels of spontaneous cortical gamma oscillations are typically detected in SZ patients and the degree of this reduction is correlated to negative symptoms [70] (Figure 2) also in association with sensory processing or performance of executive tasks [71–73]. Considering the relationship between oscillatory activity and cognition, the characteristic cognitive deficits of SZ may be directly connected with these impairments. Gamma band oscillations are linked with performance of working memory (WM) tasks [74–76], SZ patients show impairment in these tasks [77,78]. In PFC, reduced NMDAR signalling on fast-spiking GABAergic interneurons may disrupt gamma oscillations resulting in impaired WM and other cognitive abnormalities [79]. The alteration of high frequency oscillation and its effect on WM may account for the so called mental disaggregation.
Paralimbic and limbic cortices
Insula
A growing body of research suggests a crucial role for the insula in the neuropathology of this disease. There is consistent evidence that the insula´s gray matter volume is substantially decreased in SZ [80]. Patients with predominantly negative symptoms had significant volume reduction in right posterior insula compared to those with predominantly positive symptoms. Virupaksha et al. [81] conclude that insular volume deficits in antipsychotic-naive SZ patients, support intrinsic role for the insula in the pathogenesis of this disorder. Gong et al. reported reductions in the gray matter volume of the right anterior insula in SZ patients as compared to controls, describing their finding as “a neuroanatomical signature” [82].
Salience network comprised of anterior frontoinsular cortex and ACC have been identified as a system that enables the switch between dynamic brain states. Salience network dysfunction has been proposed as a mechanistic model for several core symptoms of SZ [83]. Functional and anatomical deficits in the insula, ACC and the connection between them have been found in early stages of the illness (Figure 3).
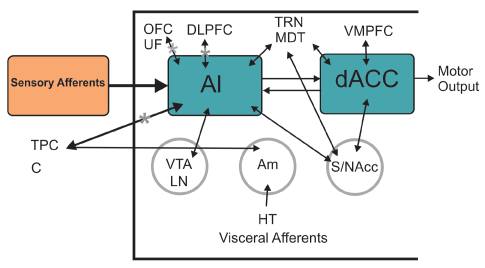
Figure 3: Salience Network (SN) Mainly comprised of AI (Anterior Insula) and dACC (dorsal anterior cingulate cortex) both structures reported to be dysfunctional in SZ patients. SN takes part in the distinction between external perceptions and inner feelings and thoughts switching between DMN and CCN and can be linked with negative symptoms, aberrancies in DMN and self-face recognition disorder. AI reflects affective and cognitive demands of self-awareness, meanwhile dACC interacts in response selection guiding overt behavior. HT: Hypothalamus. VTA: Ventral Tegmental Area. LN: Locus Niger. Am: Amygdala. S/NAcc: Striatum/Nucleus accumbens. TPC: Tempral Pole Complex. C: Cingulum. UF: Uncinate Fasciculus. MDT: Mediodorsal Thalamic Nucleus. TRN: Thalamic Reticular Nucleus. OFC: Orbitofrontal Cortex. VMPFC: Ventromedialprefrontal Cortex. DLPFC: Dorsolateralprefrontal Cortex. DMN: Default Mode Network. CCN: Cognitive Control Network (Modified from Menon V (2015) Salience Network. Brain Mapping. An: Encyclopedic Reference 2015(2): 597-611.)
Impaired insula function in SZ has been related to general deficits in self-awareness, introspection, empathy and the processing of affective experience. In this light, the finding of reduced insula responsiveness to disgusted faces might hint to a certain “social asymbolia” [80].
Anterior Cingulate Cortex (ACC)
The available MRI data suggests that gray matter reductions in the ACC precede psychosis onset in some groups of high-risk individuals -showing some sub-regional specificity in dorsorostral area Brodmann 32- and may be further reduced during illness progression. Post-mortem findings indicate these changes are accompanied by reductions in neuronal, synaptic, and dendritic density, as well as increased afferent input, suggesting the gray matter differences observed in MRI arise from alterations in both neuronal and non-neuronal tissue compartments [84]. Brune et al. have found Von Economo neurons (VEN) reduced density in the ACC in early onset schizophrenics [138]. Spindle neurons (VEN) is believed to be important for bodily feelings, self-control, empathy and social abilities. A significantly decreased bilateral endogenous connectivity between the MD thalamus and the ACC has been detected in SZ patients as compared to healthy controls [64] (Figure1). Neurochemical studies detected enhanced GABA binding in ACC of postmortem brain in SZ patients [85] and reduction in D2 binding in the same region [86].
Solid evidence that damage to ACC function is present in SZ has been found. Some studies have shown SZ patients present difficulties in dealing with conflicting spatial locations in a Stroop-like task and having abnormal error-related negativity [87,88]. Apathy and automatic obedience which are common in SZ patients are possibly related with impaired function in ACC. The ACC has been associated with response selection, conflict resolution and cognitive control [89].
Cerebellum and striatum
Cerebellum
Atrophy of the vermis is the most cited structural cerebellar abnormality in SZ [90]. Bibliographic evidence enables our presumption about the participation of cerebellar dysfunction in the pathogenesis of SZ, the so called “cognitive dysmetria” [91] based upon PET studies showing dysfunction of prefronto-thalamic cerebellar circuits. Andreasen et al. [92] established this concept as “difficulty in prioritizing, processing, coordinating, and responding to information”, in relationship with cognitive symptoms of SZ patients.
Investigators have reported a decreased linear density in Purkinje cells [93], coupled with a specific decrease in their size (8.3% reduction) [94] associated with decreased excitatory input from the granule cells [95]. From post-mortem studies, accumulating evidence supports GABAergic signalling decrease in the cerebellum of SZ patients secondary to a decreased expression of NMDA receptor subunit 2C which in turn provokes NMDA receptor dysfunction [96].
SZ patients present reduced correlations between modularity and microstructural integrity, as measured by FA in lobules I–IV and X. The aforementioned alterations were significantly correlated with the Positive and Negative Syndrome Scale symptom scores, suggesting altered network architecture in the cerebellum with reduced local microstructural connectivity in SZ patients [97]. Additionally, fiber tracts located between the cerebellar white matter and the thalamus exhibit a reduced FA in patients with SZ compared to controls. The FA values along the defined fiber tracts were not reduced overall but showed a reduction in the FA in the superior cerebellar peduncles projecting towards the red nucleus [98].
Although it has been established that cerebellum is related, albeit partially, in the genesis of cognitive symptoms of SZ, its role remains uncertain in relation to positive and negative symptoms.
Striatum
Despite numerous revisions and reformulations, dopamine (DA) striatal dysregulation may not be a primary dysfunction in SZ, but rather may represent a final common pathway [99]. According to a seminal paper from Laruelle et al, dysregulation of dopamine function revealed by the amphetamine challenge is present at onset of illness and in patients never previously exposed to neuroleptic medications; This dysregulation was observed in patients experiencing an episode of illness exacerbation, but not in patients studied during a remission phase [100]. The aetiology of such dysregulation is still unclear, but evidence support is related to alterations in other areas functionally connected and in other neurotransmitters systems, such as the frontal cortex and the glutamate, GABA and 5HT neurotransmission. Indeed, the striatum receives inputs from the hippocampus and the cerebral cortex, two areas involved in the pathology of SZ that directly and/or indirectly project back to the cortex and DA midbrain neurons. Striatal dopaminergic dysfunction can be also reciprocally linked to a cortical dopaminergic dysfunction [101]. Recent evidence indicates the hippocampus as a main source of altered modulatory influence on dopamine, possibly leading to excess striatal activity [102] (Figure 2). The striatal DA excess is attributed a glutamatergic corticostriatal deficit, although in catatonic and/or malignant neuroleptic syndrome the picture can be the reverse: increased striatal glutamatergic activity and impaired or blockade of DA terminals [103]. Some studies have reported higher DA levels in caudate nucleus tissue samples from patients with SZ [104] and NAcc [105] and increased number of D2-D3 receptors at the same regions [106].
On spiny GABAergic neurons in the striatum, D1 receptors facilitate Glu transmission but D2 receptors have the opposite effect. Increased density of striatal D2 receptors in SZ patients has been found in several postmortem studies [106–110]. This data is in line with increased vulnerability of SZ patients to DA enhancing drugs and this increase is believed to be psychotogenic. D2 receptor levels are increased in healthy monozygotic twins compared to dizygotic twins of patients with SZ and probably the caudate D2 receptor up-regulation is linked to a genetic risk factor for SZ [111].
The NAcc is a prime region of interest, as inputs from numerous brain areas altered in SZ are integrated here. McCollum et al. using electron microscopy, have studied the morphology of NAcc synaptic connections. The NAcc core and shell of 6 SZ subjects and 8 matched controls were compared in this pilot study. SZ subjects had a 19% increase in the density of asymmetric axospinous synapses (characteristic of excitatory inputs) in the core, but not in the shell. Both groups had similar densities of inhibitory inputs. These results indicate that the core receives increased excitatory input in SZ, potentially leading to dysfunctional dopamine neurotransmission on cortico-striatal-thalamic stimulus processing [112].
De Leeuw et al. [113] using DTI reported reduced FA in the tract connecting the left NAcc and left DLPFC, indicating a possible reduction of white matter integrity, commonly associated with SZ. As both, patients and unaffected siblings showed reduced FA, this may represent a vulnerability factor for SZ.
Krekzmanski et al. [35] examined 13 post-mortem brains of male SZ patients and found a 5-4.1% (left-right) reduction in the volume of the putamen and a 8.1-11.6% (left-right) mean reduction in total neuronal numbers at the same nucleus and 10.4-12% (left-right) in caudate nucleus. Buchsbaum et al. [114] have suggested that small putamen size at disease onset may be a predictor of outcome and the expansion of putamen size may be a physiological correlate of neuroleptic responsiveness.
Rajarethinam et al. [115] have observed smaller caudate nuclei in drug-naive individuals with SZ but larger in antipsychotic-treated patients and found volume reduction of right and left caudate by 8.9 and 8.1% respectively in MRI in offspring without psychosis of patients with SZ (n = 50) compared with controls, providing evidence that caudate volume reduction may be a trait-related abnormality in SZ.
Dopamine (DA) dysregulation plays a central role in the pathophysiology of SZ, as it relates most directly to the positive symptoms and to their treatment. Such dysregulation has been well characterized by in vivo imaging studies in humans and comprises two main components: a striatal excess and a cortical deficit.
Substantia Nigra (LN) and Ventral Tegmental Area (VTA)
LN is a mesencephalic nucleus responsible to produce dopamine and projection of dopaminergic neurons to the striatum. In a post-mortem study Williams et al. examined the cytoarchitecture of DA neurons in LN in SZ compared to matched controls. Astrocyte density was decreased in SZ as compared to controls. Significantly increased nuclear cross-sectional area and length and increased nucleolar volume in DA neurons were observed in SZ patients in comparison to controls, suggesting nuclear pleomorphic changes [116].
Imaging studies have consistently demonstrated SZ is associated with increased presynaptic activity of nigral DA neurons projecting to the striatum [117].
In event-related WM-fMRI studies, Yoon et al. reported task-evoked LN-caudate hyperconnectivity and PFC-striatal hypofunction in SZ patients. The magnitude of the LN-caudate hyperconnectivity was highly correlated with psychosis severity [118]. Decreased PFC-striatal functional connectivity leads to disinhibition of striatonigral cells which in turn can induce nigral overactivity and hyperfunction of D2 receptors in the striatum.
Ventral Tegmental Area
VTA is another midbrain DA nucleus supposed to be related to the pathogenesis of SZ. According to Bogerts et al. the number of DA neurons in VTA is diminished in SZ patients [119] and the remaining cells look smaller and dystrophic [120]. Yamashita et al. through a neuromelanin sensitive MRI study found that the intensity of the VTA signal in patients with SZ was significantly decreased compared to healthy controls. The VTA/LN ratio was also significantly lower in patients with SZ than in the control group. In addition, scores on the Scale for the Assessment of Positive Symptoms showed an independent negative correlation with VTA. Thus, selective signal attenuation in the VTA was correlated with positive symptoms, in patients with SZ [121]. In another study, SZ patients had significantly smaller midbrain measures compared with control subjects and midbrain size was significantly and inversely correlated with positive symptoms [122].
Alba-Ferrara and de Erausquin presented data revealing increased anisotropy in DA tracts in the midbrain of SZ patients and their unaffected relatives and discuss the possible biological underpinnings and physiological significance of this fact [123]. It could be thought that such neural overactivity may modulate synaptic maps resulting in deficient axonal pruning. Deficient axonal pruning may lead to redundant networks which may indicate decreased efficiency in information transmission.
Temporal Lobes
The association of SZ with the temporal lobes is generally accepted, in part by the occurrence of SZ-like psychosis associated with temporal lobe epilepsy and/or herpetic encephalitis. In this paper we have presented alterations described in relation to amygdala and/or EHS pathology, that seem to be milestones in the understanding of SZ.
Shenton et al. did a volumetric MRI with 3D reconstructions comparing SZ patients with normal controls, showing gray matter volume decreases in left anterior hippocampus/amygdala (19 percent), left parahippocampal gyrus (13 percent) and left superior temporal gyrus (15 percent). None of these regional volume decreases were paralleled by decreases in overall brain or temporal lobe volumes. The authors conclude that neuroanatomical pathology in schizophrenia does not consist of global atrophy but rather of strategically localized left-lateralized reductions in gray matter, especially in temporal lobe [29,124]. Additionally, a reduction in right temporal volume, has been found in deficit patients [125], a group of patients with enduring, primary negative symptoms.
Auditory verbal hallucinations (AVH) are frequently present symptoms in schizophrenia. While functional imaging studies have suggested the association of certain patterns of brain activity with positive symptoms, there has been only limited evidence from structural imaging or post-mortem studies. During such hallucinations Bentaleb et al. [126] have reported metabolic enhancement in the primary auditory cortex. Gaser et al. have investigated the relation of local brain structural deficits to severity of AVH using deformation-based morphometry to assess local gray and white matter deficits in MRI of SZ patients. They found severity of AVH to be significantly correlated with volume loss in the primary auditory cortex and left inferior supramarginal gyrus, as well as middle/inferior right prefrontal gyri. This demonstrates a pattern of distributed structural abnormalities specific for AVH and suggests alterations in frontotemporal network in relation to processing auditory information and language [31].
Chang et al. published a study combining MRI and fMRI on SZ first-episode patients comparing those who presented AVH and those who didn´t. All patients presented abnormalities in parahippocampus and striatum, but aberrant bilateral DMN in inferior frontal gyrus and cerebellum was only present in AVH patients, whereas alterations in superior temporal gyrus and precentral gyrus were specific to non-AVH patients. This study corroborates that SZ is characterized by aberrant dysconnectivity and suggests the localization of such abnormalities may be crucial to where AVH develops [32]. In a more recent study AVH patients were characterized by connective alterations in neural circuitry involving the anterior cingulate cortex, insular cortex (Figure 3) and various language-related regions [127].
Discussion
The current techniques for SZ research, like PET, morphometric and volumetric studies, fMRI with BOLD signals and/or FC, ROI techniques, DTI, etc. have changed the search for a unique brain area responsible for the occurrence of SZ symptoms, turning the attention to brain networks and their functional connectivity. Diffusion tensor imaging (DTI) studies in SZ, have demonstrated lower fractional anisotropic (FA) diffusion within white matter due to microstructural disorganization and axonal dysfunction secondary to either loss of coherence of white matter fiber tracts, changes in the number and/or density of interconnecting fibers or changes in myelination. These findings are in line with the Friston and Frith “disconnection” hypotheses [9]. It must be said that these networks can have evidenced aberrant behaviour in spite of normal anatomical structural appearance and/or lack of proven anatomical connections between them.
There are a considerable number of reports describing subtle decrease in number of cells or diminished volumes in different parts of the brain of SZ patients, sometimes prior to the debut of psychotic symptoms (Table 1) or in their first-grade relatives or siblings (Table 2,3). These facts show that the illness has a genetic component and/or pre/postnatal stressful events trigger the predisposition to suffer SZ in the adulthood. When the pruning of brain neurons reaches certain threshold, previously subnormal circuits become insufficient to generate normal drive and actions in order to respond to adult life demands. Regarding left or right hemisphere asymmetric data it is well established that both sides of the brain take part in the disease (Table 4).
Table 1. Alterations before or at the onset of psychotic symptoms in SZ patients
Deviant olfactory experiences can precede the onset of psychosis [130]. |
3rd and lateral ventricular enlargement in MRI studies [30]. |
Premorbid impairments in neuropsychological testing [33]. |
Gray matter reduction in ACC, showing a sub-regional specifity in dorsal and rostral areas [84]. |
Table 2. Alterations in first grade relatives or siblings of SZ patients
Eye tracking dysfunction in first grade relatives [135]. |
In MRI studies, reduced volumes in amygdala-hippocampal complex in relatives of SZ patients [33]. |
Caudate D2 receptors up-regulation in healthy monozygotic twins, PET study [111]. |
SZ patients and first-degree relatives showed significant deficit in their ability to correctly identify odorants [131]. |
Caudate volume reduction in 50 offspring’s of SZ patients without SZ; MRI study [115]. |
Temporoparietal P300 amplitude reduction and frontal P300 amplitude increase seem to be quantitative phenotypes associated with increased risk of schizophrenia [132]. |
Default-mode network dysfunction in healthy siblings of SZ patients [22–24]. |
Increased anisotropy in DA tracts in the midbrain of unaffected relatives [123]. |
Reduced FA in the tract connecting left NAcc and left DLPFC in unaffected siblings [113]. |
Table 3. Region of interest and specific findings in SZ patient, siblings or relatives
Region |
Finding |
Third and lateral ventricles |
Enlargement [130] |
Mediodorsal thalamic nuclei |
Reduction in total neuron number [26] |
Anterior cingulate cortex |
Gray matter reduction [84]
Spindle neurons reduction in the right side [138] |
Right insula |
Deficit in right anterior insula volume [82]
Deficit in right posterior insula volume [81] |
Left prefrontal cortex |
Lower thalamocortical connectivity [25] |
Right prefrontal cortex
(anterior)
(middle and inferior) |
Reduced white and gray matter [13]
Decreased activity at rest [15]
Volume loss correlated with severity of ADH [31] |
Parietal cortex (right superior) |
Increased rCBF in relation with positive symptoms |
Caudate nucleus |
Volume reduction [115]
Up-regulation D2 receptors [111] |
Midbrain |
Increased anisotropy in DA tracts [123] |
Hippocampus |
Volume decrease [33], idem left anterior section [124] |
Amygdala |
Volume decrease [33,124] |
Temporal lobe |
Volume decrease in left superior temporal gyrus [124]
Reduced volume in the right side [13,125] |
Table 4. Left (i) and right (ii) asymmetric findings in SZ patients
(i)
Volume decrease in left anterior hippocampus, amygdala, Para hippocampal and left superior temporal gyrus in MRI [124]. |
Using deformation-based morphometry (DBM) severity of AVH correlated with volume loss in left inferior supramarginal gyrus [31]. |
FA decreased in the left arquate fasciculus [10]. |
Left thalamic FA was correlated with spatial working memory deficits [18]. |
Through fMRI activation, lower thalamocortical connectivity in the left PFC [25]. |
Reduced FA in the tract connecting left NAcc and left DLPFC [113] |
(ii)
Right prefrontal white matter and right prefrontal gray matter and right hippocampal volume reduced in MRI studies [13]. |
Increased rCBF in right superior parietal cortex in relation to positive symptoms, a PET study [136]. |
From DBM data, severity of AVH correlated with volume loss in middle/inferior right prefrontal gyri [31]. |
FA decreased in the right inferior occipitofrontal fasciculus [10]. |
Reduced FA in the right superior longitudinal fasciculus in deficit subjects [137]. |
VEN density in the right ACC correlated with the age at onset, and inversely with the duration of the illness in early onset, post mortem study [138]. |
IN DTI, asymmetric microstructural change in the anterior limb of the right internal capsule, which correlates with cognitive impairment [17]. |
Reduced right temporal volume in deficit patients [125]. |
In fMRI, decreased right anterior prefrontal cortex activity at rest correlated with negative symptoms [15]. |
Deficit in right posterior insula volume in those with negative symptoms, 3T MRI study [81]. |
Through fMRI increased connectivity between right DLPFC and right MD thalamic nucleus was assessed [64]. |
Deficit in right anterior insula volume, MRI data [82]. |
Conclusion
As Price and Friston [128] have stated, SZ can be interpreted as a prefrontal cortex failure to control the temporal lobes activity, impairing volitive actions (negative) and causing psychotic hallucinations and delusions (positive symptoms). In neurochemical terms, the dysregulation of dopaminergic, serotonergic, GABAergic and/or glutamatergic neurons diminished the efficiency of context information and flexible updating of stored information (cognitive symptoms) [129]. Genetically determined glutamate receptor expression could be a common deficit in SZ patients in different areas like EHS, PF cortex, cerebellum, MD thalamus and ACC.
Any simple gene, dysgenesis, neurotransmitter or neuropathological locus nor any simple white matter tract or network can explain the disorder, so we will need complex approaches in order to restore mental health in SZ patients.
Acknowledgment
Sincere thanks to Dr. Juan Carlos Goldar for his inspiration
References
- Yu Q, Allen EA, Sui J, Arbabshirani MR, Pearlson G, et al. (2012) Brain connectivity networks in schizophrenia underlying resting state functional magnetic resonance imaging. Curr Top Med Chem 12: 2415-25. [Crossref]
- Roth BL, Meltzer HY (1995) The role of serotonin in schizophrenia, in Psychopharmacology: The Fourth Generation of Progress (Bloom FE and Kupfer DJ eds) pp 1215–1228, Raven Press, New York.
- Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, et al. (1997) Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385: 634-636. [Crossref]
- Weinberger DR (1987) Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 44: 660-669. [Crossref]
- Moghaddam B, Bunney BS (1990) Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem 54: 1755-1760. [Crossref]
- Pilowsky LS, Costa DC, Ell PJ, Verhoeff NP, Murray RM, et al. (1994) D2 dopamine receptor binding in the basal ganglia of antipsychotic-free schizophrenic patients. An 123I-IBZM single photon emission computerised tomography study. Br J Psychiatry 164: 16-26. [Crossref]
- Svensson TH, Mathe JM, Andersson JL, Nomikos GG, Hildebrand BE, et al. (1995) Mode of action of atypical neuroleptics in relation to the phencyclidine model of schizophrenia: role of 5-HT2 receptor and alpha 1-andrenoceptor antagonism. J Clin Psychopharmacol 15:11S-18S. [Crossref]
- Grenhoff J, Tung CS, Ugedo L, Svensson TH (1990) Effects of amperozide, a putative antipsychotic drug, on rat midbrain dopamine neurons recorded in vivo. Pharmacol Toxicol 66 Suppl 1: 29-33. [Crossref]
- Friston KJ, Frith CD (1995) Schizophrenia: a disconnection syndrome? Clin Neurosci 3: 89-97. [Crossref]
- Kubicki M, Park H, Westin CF, Nestor PG, Mulkern RV, et al. (2005) DTI and MTR abnormalities in schizophrenia: Analysis of white matter integrity. Neuroimage 26: 1109–1118. [Crossref]
- Pomarol-Clotet E, Canales-Rodríguez EJ, Salvador R, Sarró S, Gomar JJ, et al. (2010) Medial prefrontal cortex pathology in schizophrenia as revealed by convergent findings from multimodal imaging. Mol Psychiatry 15: 823-830. [Crossref]
- Buckner RL, Andrews-Hanna JR, Schacter DL (2008) The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci 1124: 1-38. [Crossref]
- Wible CG, Anderson J, Shenton ME, Kricun A, Hirayasu Y, et al. (2001) Prefrontal cortex, negative symptoms, and schizophrenia: an MRI study. Psychiatry Res 108: 65–78. [Crossref]
- Sanfilipo M, Lafargue T, Rusinek H, Arena L, Loneragan C, et al. (2000) Volumetric measure of the frontal and temporal lobe regions in schizophrenia: relationship to negative symptoms. Arch Gen Psychiatry 57:471–480. [Crossref]
- Mingoia G, Wagner G, Langbein K, Maitra R, Smesny S, et al. (2012) Default mode network activity in schizophrenia studied at resting state using probabilistic ICA. Schizophr Res 138: 143-149. [Crossref]
- Wang HL, Rau CL, Li YM, Chen YP, Yu R (2015) Disrupted thalamic resting-state functional networks in schizophrenia. Front Behav Neurosci 9: 45. [Crossref]
- Mamah D, Conturo TE, Harms MP, Akbudak E, Wang L, et al. (2010) Anterior thalamic radiation integrity in schizophrenia: a diffusion-tensor imaging study. Psychiatry Res 183: 144-150. [Crossref]
- Qiu A, Zhong J, Graham S, Chia MY, Sim K (2009) Combined analyses of thalamic volume, shape and white matter integrity in first-episode schizophrenia. Neuroimage 47: 1163-1171. [Crossref]
- Zhou Y, Liang M, Tian L, Wang K, Hao Y, et al. (2007) Functional disintegration in paranoid schizophrenia using resting-state fMRI. Schizophr Res 97: 194-205. [Crossref]
- Camchong J, MacDonald AW 3rd, Bell C, Mueller BA, Lim KO (2011) Altered functional and anatomical connectivity in schizophrenia. Schizophr Bull 37: 640-650. [Crossref]
- Liu H, Kaneko Y, Ouyang X, Li L, Hao Y, et al. (2012) Schizophrenic patients and their unaffected siblings share increased resting-state connectivity in the task-negative network but not its anticorrelated task-positive network. Schizophr Bull 38: 285-294. [Crossref]
- Van Buuren M, Vink M, Kahn RS. (2012) Default-mode network dysfunction and self-referential processing in healthy siblings of schizophrenia patients. Schizophr Res 142: 237–243. [Crossref]
- Chang X, Shen H, Wang L, Liu Z, Xin W, et al. (2014) Altered default mode and fronto-parietal network subsystems in patients with schizophrenia and their un affected siblings. Brain Res 1562: 87–99. [Crossref]
- Guo W, Su Q, Yao D, Jiang J, Zhang J, et al. (2014)Decreased regional activity of default-mode network in unaffected siblings of schizophrenia patients at rest. Eur Neuropsychopharmacol 24: 545–552. [Crossref]
- Marenco S, Stein JL, Savostyanova AA, Sambataro F, Tan HY, et al. (2012) Investigation of anatomical thalamo-cortical connectivity and FMRI activation in schizophrenia. Neuropsychopharmacology 37: 499-507. [Crossref]
- Popken GJ, Bunney Jr WE, Potkin SG, Jones EG (2000) Subnucleus-specific loss of neurons in medial thalamus of schizophrenics. Proc Natl Acad Sci USA 97: 9276–9280. [Crossref]
- Young KA, Manaye KF, Liang C, Hicks PB, German DC (2000) Reduced number of mediodorsal and anterior thalamic neurons in schizophrenia. Biol Psychiatry 47: 944-953. [Crossref]
- Byne W, Buchsbaum MS, Mattiace LA, Hazlett EA, Kemether E, et al. (2002) Postmortem assessment of thalamic nuclear volumes in subjects with schizophrenia. Am J Psychiatry 159: 59-65. [Crossref]
- Goldar JC (1990) The frontal lobe and psychoses. Acta Psiquiatr Psicol Am Lat 36: 37-47. [Crossref]
- Kleist K (1937) Bericht über die Gehirnpathologie in ihrer Bedeutung für Neurologie und Psychiatrie. Zeitschr. f. d. ges. Neurol. u.Psychiatrie 1937: 158-193.
- Gaser C, Nenadic I, Volz HP, Büchel C, Sauer H (2004) Neuroanatomy of "hearing voices": a frontotemporal brain structural abnormality associated with auditory hallucinations in schizophrenia. Cereb Cortex 14: 91-96. [Crossref]
- Chang X, Xi YB, Cui LB, Wang HN, Sun JB, et al. (2015) Distinct inter-hemispheric dysconnectivity in schizophrenia patients with and without auditory verbal hallucinations. Sci Rep 5: 11218. [Crossref]
- Lawrie S, Whalley HC, Job D, Johnstone HC (2003) Structural and Functional Abnormalities of the Amygdala in Schizophrenia. Ann N Y Acad Sci 985: 445-460. [Crossref]
- Johnstone EC, Ebmeier KP, Miller P, Owens DG, Lawrie SM (2005) Predicting schizophrenia: findings from the Edinburgh High-Risk Study. Br J Psychiatry 186: 18-25. [Crossref]
- Kreczmanski P, Heinsen H, Mantua V, Woltersdorf F, Masson T, et al. (2007) Volume, neuron density and total neuron number in five subcortical regions in schizophrenia. Brain 130: 678-692. [Crossref]
- Rasetti R, Mattay VS, Wiedholz LM, Kolachana BS, Hariri AR, et al. (2009) Evidence that altered amygdala activity in schizophrenia is related to clinical state and not genetic risk. Am J Psychiatry 166: 216-225. [Crossref]
- Mier D, Lis S, Zygrodnik K, Sauer C, Ulferts J, et al. (2014) Evidence for altered amygdala activation in schizophrenia in an adaptive emotion recognition task. Psychiatry Res 221: 195-203. [Crossref]
- Wolterink G, Daenen LE, Dubbeldam S, Gerrits MA, van Rijn R, et al. (2001) Early amygdala damage in the rat as a model for neurodevelopmental psychopathological disorders. Eur Neuropsychopharmacol 11: 51–59. [Crossref]
- Benarroch EE (2015) The amygdala. Functional organization and involvemente in neurologic disorders. Neurology 84: 313-324. [Crossref]
- Salzman CD, Fusi S (2010) Emotion, cognition, and mental state representation in amygdala and prefrontal cortex. Annu Rev Neurosci 33: 173-202. [Crossref]
- Halgren E (1981) The amygdala contribution to emotion and memory: current studies in humans. In The Amygdaloid Complex INSERM Symposium 20 (Ben-AriY ed) pp 395-408, Elsevier, North Holland Biomedical Press, Amsterdam.
2021 Copyright OAT. All rights reserv
- Halgren E (1992) Emotional neurophysiology of the amygdala within the context of human cognition. In The amygdala: Neurobiological aspects of emotion, memory and mental dysfunction (Aggleton JP ed) pp 191-228, Willey-Liss, New York.
- King HE (1961) Psychological effects of excitation in the limbic system. In Electrical Stimulation of the Brain (Sheer DE ed) pp 477-486, University of Texas Press: Austin, TX USA.
- Ure JA (1981) Schizophrenia: Biochemical, Anatomical and Physiological aspects. Eumens 1981, 1:42-47.
- Suddath RL, Casanova MF, Goldberg TE, Daniel DG, Kelsoe JR Jr, et al. (1989) Temporal lobe pathology in schizophrenia: a quantitative magnetic resonance imaging study. Am J Psychiatry 146: 464-472. [Crossref]
- Bogerts B, Ashtari M, Degreef G, Alvir JM, Bilder RM, et al. (1990) Reduced temporal limbic structure volumes on magnetic resonance images in first episode schizophrenia. Psychiatry Res 35: 1-13. [Crossref]
- Bilder RM, Bogerts B, Ashtari M, Wu H, Alvir JM, et al. (1995) Anterior hippocampal volume reductions predict frontal lobe dysfunction in first episode schizophrenia. Schizophr Res 17: 47–58. [Crossref]
- Becker T, Elmer K, Schneider F, Schneider M, Grodd W, et al. (1996) Confirmation of reduced temporal limbic structure volume on MRI in male patients with schizophrenia. Psychiatry Res 67: 135–143. [Crossref]
- Szeszko PR, Goldberg E, Gunduz-Bruce H, Ashtari M, Robinson D, et al. (2003) Smaller anterior hippocampal formation volume in antipsychotic-naive patients with first-episode schizophrenia. Am J Psychiatry 160: 2190–2197. [Crossref]
- Narr KL, Thompson PM, Szeszko PR, Robinson D, Jang S, et al. (2004) Regional specificity of hippocampal volume reductions in first-episode schizophrenia. Neuroimage 21: 1563–1575. [Crossref]
- Heckers S (2001) Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus 11: 520-528. [Crossref]
- Williams LE, Blackford JU, Luksik A, Gauthier I, Heckers S (2013) Reduced habituation in patients with schizophrenia. Schizophr Res 151: 124-132. [Crossref]
- Schmitt A, Steyskal C, Bernstein HG, Schneider-Axmann T, Parlapani E, et al. (2009) Stereologic investigation of the posterior part of the hippocampus in schizophrenia. Acta Neuropathol 117: 395-407. [Crossref]
- Reif A, Schmitt A, Fritzen S, Lesch KP (2007) Neurogenesis and schizophrenia dividing neurons in a divided mind? Eur Arch Psychiatry Clin Neurosci 257: 290–299. [Crossref]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, et al. (2007) Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130: 1146–1158. [Crossref]
- Barros CS, Calabrese B, Chamero P, Roberts AJ, Korzus E, et al. (2009) Impaired maturation of dendritic spines without disorganization of cortical cell layers in mice lacking NRG1/ErbB signaling in the CNS. Proc Natl Acad Sci USA 106: 4507–4512. [Crossref]
- Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA (2005) Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a post-mortem morphometric study. Synapse 57: 47–55. [Crossref]
- Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, et al. (2016) Age-Dependent Specific Changes in Area CA2 of the Hippocampus and Social Memory Deficit in a Mouse Model of the 22q11.2 Deletion Syndrome. Neuron 89: 163-176. [Crossref]
- Floresco SB, Todd CL, Grace AA (2001) Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci 21: 4915-4922. [Crossref]
- Tamminga CA, Stan AD, Wagner AD (2010) The hippocampal formation in schizophrenia. Am J Psychiatry 167: 1178-1193. [Crossref]
- Byne W, Hazlett EA, Buchsbaum MS, Kemether E (2009) The thalamus and schizophrenia: current status of research. Acta Neuropathol 117: 347-368. [Crossref]
- Cronenwett WJ, Csernansky J (2010) Thalamic pathology in schizophrenia. Curr Top Behav Neurosci 4: 509-528. [Crossref]
- Ibrahim HM, Hogg AJ Jr, Healy DJ, Haroutunian V, Davis KL, et al. (2000) Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am J Psychiatry 157: 1811-1823. [Crossref]
- Wagner G, Koch K, Schachtzabel C, Schultz CC, Gaser C, et al. (2013) Structural basis of the fronto-thalamic dysconnectivity in schizophrenia: A combined DCM-VBM study. Neuroimage Clin 3: 95-105. [Crossref]
- Pinault D (2004) The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev 46: 1-31. [Crossref]
- Pratt JA, Morris BJ (2015) The thalamic reticular nucleus: a functional hub for thalamocortical network dysfunction in schizophrenia and a target for drug discovery. J Psychopharmacol 29: 127-137. [Crossref]
- Ferrarelli F, Tononi G (2011) The thalamic reticular nucleus and schizophrenia. Schizophr Bull 37: 306-315. [Crossref]
- Steullet P, Cabungcal JH, Bukhari SA, Ardelt MI, Pantazopoulos H, et al. (2017) The thalamic reticular nucleus in schizophrenia and bipolar disorder: role of parvalbumin-expressing neuron networks and oxidative stress. Mol Psychiatry. [Crossref]
- Halassa MM, Chen Z, Wimmer RD, Brunetti PM, Zhao S, et al. (2014) State-dependent architecture of thalamic reticular subnetworks. Cell 158: 808-821. [Crossref]
- Lee KH, Williams LM, Breakspear M, Gordon E (2003) Synchronous gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain Res Brain Res Rev 41: 57-78. [Crossref]
- Rutter L, Carver FW, Holroyd T, Nadar SR, Mitchell-Francis J, et al. (2009) Magnetoencephalographic gamma power reduction in patients with schizophrenia during resting condition. Hum Brain Mapp 30: 3254-3264. [Crossref]
- Sun L, Castellanos N, Grützner C, Koethe D, Rivolta D, et al. (2013) Evidence for dysregulated high frequency oscillations during sensory processing in medication-naïve, first episode schizophrenia. Schizophr Res 150: 519-525. [Crossref]
- Uhlhaas PJ, Singer W (2010) Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 11: 100-113. [Crossref]
- Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, et al. (2003) Gamma oscillations correlate with working memory load in humans. Cereb Cortex 13: 1369-1374. [Crossref]
- Roux F, Uhlhaas PJ (2014) Working memory and neural oscillations: α-γ versus θ-γ codes for distinct WM information? Trends Cogn Sci 18: 16-25. [Crossref]
- Yamamoto J, Suh J, Takeuchi D, Tonegawa S(2014) Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell 157: 845-857. [Crossref]
- Bor J, Brunelin J, Sappey-Marinier D, Ibarrola D, d'Amato T, et al. (2011) Thalamus abnormalities during working memory in schizophrenia. An fMRI study. Schizophr Res 125: 49-53. [Crossref]
- Perlstein WM, Carter CS, Noll DC, Cohen JD (2001) Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry 158: 1105-1113. [Crossref]
- Winterer G, Weinberger DR (2004) Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci 27: 683-690. [Crossref]
- Lindner C, Dannlowski U, Walhöfer K, Rödiger M, Maisch B, et al. (2014) Social alienation in schizophrenia patients: association with insula responsiveness to facial expressions of disgust. PLoS One 9: e85014. [Crossref]
- Virupaksha HS, Kalmady SV, Shivakumar V, Arasappa R, Venkatasubramanian G, et al. (2012) Volume and asymmetry abnormalities of insula in antipsychotic-naive schizophrenia: A 3-Tesla magnetic resonance imaging study. Indian J Psychol Med 34: 133-139. [Crossref]
- Gong Q, Dazzan P, Scarpazza C, Kasai K, Hu X, et al. (2015) A Neuroanatomical Signature for Schizophrenia Across Different Ethnic Groups. Schizophr Bull 41: 1266-1275. [Crossref]
- Palaniyappan L, Balain V, Radua J, Liddle PF (2012) Structural correlates of auditory hallucinations in schizophrenia: a meta-analysis. Schizophr Res 137: 169–173. [Crossref]
- Fornito A, Yücel M, Dean B, Wood SJ, Pantelis C (2009) Anatomical abnormalities of the anterior cingulate cortex in schizophrenia: Bridging the gap between neuroimaging and neuropathology. Schizophr Bull 35: 973–993. [Crossref]
- Benes FM, Vincent SL, Alsterberg G, Bird ED, SanGiovanni JP (1992) Increased GABAA receptor binding in superficial layers of cingulate cortex in schizophrenics. J Neurosci 12: 924-929. [Crossref]
- Suhara T, Okubo Y, Yasuno F, Sudo Y, Inoue M, et al. (2002) Decreased dopamine D2 receptor binding in the anterior cingulate cortex in schizophrenia. Arch Gen Psychiatry 59: 25-30. [Crossref]
- Posner MI, Di Girolamo GJ (1998) Executive attention: Conflict, target detection, and cognitive control. In The attentive brain. (Parasuraman R, ed) pp 401-423, MIT Press, Cambridge, MA.
- Holroyd CB, Nieuwenhuis S, Yeung N, Nystrom L, Mars RB, et al. (2004) Dorsal anterior cingulate cortex shows fMRI response to internal and external error signals. Nat Neurosci 7: 497–498. [Crossref]
- Menon V (2015) Salience network. Brain Mapping: An Encyclopedic Reference 2:597-611.
- Bottmer C, Bachmann S, Pantel J, Essiq M, Amann M, et al. (2005) Reduced cerebellar volume and neurological soft signs in first-episode schizophrenia. Psychiatry Res 140: 239–250. [Crossref]
- Andreasen NC, O'Leary DS, Cizadlo T, Arndt S, Rezai K, et al. Schizophrenia and cognitive dysmetria: A positronemission tomography study of dysfunctional prefrontalthalamic-cerebellar circuitry. Proc Natl Acad Sci U S A 93: 9985-9990. [Crossref]
- Andreasen NC, Paradiso S, O’Leary DS (1998) “Cognitive Dysmetria" as an Integrative Theory of Schizophrenia: A Dysfunction in CorticalSubcortical-Cerebellar Circuitry? Schizophr Bull 24: 203-218. [Crossref]
- Reyes MG, Gordon A (1981) Cerebellar vermis in schizophrenia. Lancet 2: 700-701. [Crossref]
- Tran KD, Smutzer GS, Doty RL, Arnold SE (1998) Reduced Purkinje cell size in the cerebellar vermis of elderly patients with schizophrenia. Am J Psychiatry 155:1288–1290. [Crossref]
- Andreasen NC, Pierson R (2008) The role of the cerebellum in schizophrenia. Biol Psychiatry 64: 81-88. [Crossref]
- Yeganeh-Doost P, Gruber O, Falkai P, Schmitt A (2011) The role of the cerebellum in schizophrenia: from cognition to molecular pathways. Clinics (Sao Paulo) 66 Suppl 1: 71-77. [Crossref]
- Kim DJ, Kent JS, Bolbecker AR, Sporns O, Cheng H, et al. (2014) Disrupted modular architecture of cerebellum in schizophrenia: a graph theoretic analysis. Schizophr Bull 40: 1216-1226. [Crossref]
- Magnotta VA1, Adix ML, Caprahan A, Lim K, Gollub R, et al. (2008) Investigating connectivity between the cerebellum and thalamus in schizophrenia using diffusion tensor tractography: a pilot study. Psychiatry Res 163: 193-200. [Crossref]
- Howes OD, Kapur S (2009) The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull 35: 549-562. [Crossref]
- Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R (1999) Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry 46: 56-72. [Crossref]
- Brunelin J, Fecteau S, Suaud-Chagny MF (2013) Abnormal striatal dopamine transmission in schizophrenia. Curr Med Chem 20: 397-404. [Crossref]
- Abi-Dargham, A, Grace, AA (2010) Dopamine and Schizophrenia, in Schizophrenia, Third Edition (Weinberger DR and Harrison PJ eds) Wiley-Blackwell, Oxford, UK.
- Ure JA (2016) Emotional Motor System: neuropsychological and neuropsychiatrical correlations. Rev Argent Neuropsicol 28:39-51.
- Owen F, Cross AJ, Crow TJ, Longden A, Poulter M, et al. (1978) Increased dopamine-receptor sensitivity in schizophrenia. Lancet 2: 223-226. [Crossref]
- Mackay AV, Iversen LL, Rossor M, Spokes E, Bird E, et al. (1982) Increased brain dopamine and dopamine receptors in schizophrenia. Arch Gen Psychiatry 39: 991-997. [Crossref]
- Cross AJ, Crow TJ, Ferrier IN, Johnstone EC, McCreadie RM, et al. (1983) Dopamine receptors changes in schizophrenia in relation to the disease process and movement disorder. J Neural Transm Suppl 18: 265-272. [Crossref]
- Dean B, Pavey G, Opeskin K (1997) [3H]raclopride binding to brain tissue from subjects with schizophrenia: methodological aspects. Neuropharmacology 36: 779-786. [Crossref]
- Hess EJ, Bracha HS, Kleinman JE, Creese I (1987) Dopamine receptor subtype imbalance in schizophrenia. Life Sci 40: 1487-1497. [Crossref]
- Joyce JN, Lexow N, Bird E, Winokur A (1988) Organization of dopamine D1 and D2 receptors in human striatum: receptor autoradiographic studies in Huntington's disease and schizophrenia. Synapse 2:546-557. [Crossref]
- Knable MB, Hyde TM, Herman MM, Carter JM, Bigelow L, et al. (1994) Quantitative autoradiography of dopamine-D1 receptors, D2 receptors, and dopamine uptake sites in postmortem striatal specimens from schizophrenic patients. Biol Psychiatry 36: 827-835. [Crossref]
- Hirvonen J, van Erp TG, Huttunen J, Aalto S, Nagren K, et al. (2005) Increased caudate dopamine D2 receptor availability as a genetic marker for schizophrenia. Arch Gen Psychiatry 62: 371-378. [Crossref]
- McCollum LA, Walker CK, Roche JK, Roberts RC (2015) Elevated Excitatory Input to the Nucleus Accumbens in Schizophrenia: A Postmortem Ultrastructural Study. Schizophr Bull 41: 1123-1132. [Crossref]
- de Leeuw M, Bohlken MM, Mandl RC, Kahn RS, Vink M (2015) Reduced fronto–striatal white matter integrity in schizophrenia patients and unaffected siblings: a DTI study. NPJ Schizophr 1:15001. [Crossref]
- Buchsbaum MS, Shihabuddin L, Brickman AM, Miozzo R, Prikryl R, et al. (2003) Caudate and putamen volumes in good and poor outcome patients with schizophrenia. Schizophr Res 64: 53-62. [Crossref]
- Rajarethinam R, Upadhyaya A, Tsou P, Upadhyaya M, Keshavan MS (2007) Caudate volume in offspring of patients with schizophrenia. Br J Psychiatry 191: 258-259. [Crossref]
- Williams MR, Galvin K, O'Sullivan B, MacDonald CD, Ching EW, et al. (2014) Neuropathological changes in the substantia nigra in schizophrenia but not depression. Eur Arch Psychiatry Clin Neurosci 264: 285-96. [Crossref]
- Weinberger DR, Laruelle M (2001) Neurochemical and neuropharmacological imaging in schizophrenia. In Neuropharmacology – The Fifth Generation of Progress (Davis KL, Charney DS, Coyle JT, Nemeroff C eds) pp 833-856, Lippincott, Williams, & Wilkins, Philadelphia, Pennsylvania.
- Yoon JH, Minzenberg MJ, Raouf S, D’Esposito M, Carter CS (2013) Impaired prefrontal-basal ganglia functional connectivity and substantia nigra hyperactivity in schizophrenia. Biol Psychiatry 74: 122-129. [Crossref]
- Bogerts B, Hantsch J, Herzer M (1983) A morphometric study of the dopamine-containing cell groups in the mesencephalon of normals, Parkinson patients and schizophrenics. Biol Psychiatry 18: 951-969. [Crossref]
- Kolomeets NS, Uranova NA (1999) Synaptic contacts in schizophrenia: studies using immunocytochemical identification of dopaminergic neurons. Neurosci Behav Physiol 29: 217-221. [Crossref]
- Yamashita F, Sasaki M, Fukumoto K, Otsuka K, Uwano I, et al. (2016) Detection of changes in the ventral tegmental area of patients with schizophrenia using neuromelanin-sensitive MRI. Neuroreport 27: 289-294. [Crossref]
- Nopoulos PC, Ceilley JW, Gailis EA, Andreasen NC (2001) An MRI study of midbrain morphology in patients with schizophrenia: relationship to psychosis, neuroleptics, and cerebellar neural circuitry. Biol Psychiatry 49:13–19. [Crossref]
- Alba-Ferrara LM, de Erausquin GA (2013) What does anisotropy measure? Insights from increased and decreased anisotropy in selective fiber tracts in schizophrenia. Front Integr Neurosci 11:9. [Crossref]
- Shenton ME, Kikinis R, Jolesz FA, Pollak SD, LeMay M, et al. (1992) Abnormalities of the left temporal lobe and thought disorder in schizophrenia. A quantitative magnetic resonance imaging study. N Engl J Med 327: 604-612. [Crossref]
- Galderisi S. Brain imaging correlates of schizophrenia negative symptoms. 15th World Congress of Psychiatry-Abstract Book 2011, pp 205-206.
- Bentaleb LA, Beauregard M, Liddle P, Stip E (2002) Cerebral activity associated with auditory verbal hallucinations: a functional magnetic resonance imaging case study. J Psychiatry Neurosci 27: 110–115. [Crossref]
- Chang X, Collin G, Xi Y, Cui L, Scholtens LH, et al. (2017) Resting-state functional connectivity in medication-naïve schizophrenia patients with and without auditory verbal hallucinations: A preliminary report. Schizophr Res 188: 75-81. [Crossref]
- Price CJ, Friston KJ (2001) Functional neuroimaging of neuropsychologically impaired patients. In Handbook of Functional Neuroimaging of Cognition (Cabeza R and Kingstone A eds) pp 379-399, A Bradford Book, The MIT Press, Cambridge, MA.
- Braver TS, Barch DM, Cohen JD (1999) Cognition and control in schizophrenia: a computational model of dopamine and prefrontal function. Biol Psychiatry 46: 312-328. [Crossref]
- Kwapil TR, Chapman JP, Chapman LJ, Miller MB (1996) Deviant olfactory experiences as indicators of risk for psychosis. Schizophr Bull 22: 371-382. [Crossref]
- Roalf DR, Turetsky BI, Owzar K, Balderston CC, Johnson SC, et al. (2006) Unirhinal olfactory function in schizophrenia patients and first-degree relatives. J Neuropsychiatry Clin Neurosci 18:389–396. [Crossref]
- Winterer G, Egan MF, Raedler T, Sanchez C, Jones DW, et al. (2003) P300 and genetic risk for schizophrenia. Arch Gen Psychiatry 60: 1158-1167. [Crossref]
- Kubicki M, Westin CF, Maier SE, Frumin M, Nestor PG, et al. (2002) Uncinate fasciculus findings in schizophrenia: a magnetic resonance diffusion tensor imaging study. Am J Psychiatry 159: 813-820. [Crossref]
- Fannon D, Tennakoon L, O'Ceallaigh S, Doku V, Soni W, et al. (2000) Third ventricle enlargement and developmental delay in first-episode psychosis: preliminary findings. Br J Psychiatry 177: 354-359. [Crossref]
- Ure JA, D’Onofrio H (1999) Ocular movements in psychiatric practice. Part One. Alcmeon 7: 333-362.
- Franck N, O'Leary S, Flaum M, Hichwa RD, Andreasen NC (2002) Cerebral blood flow changes associated with Schneiderian first-rank symptoms in Schizophrenia. J Neuropsychiatry Clin Neurosci 14: 277–282. [Crossref]
- Rowland LM, Spieker EA, Francis A, Barker PB, Carpenter WT, et al. (2009) White matter alterations in deficit schizophrenia. Neuropsychopharmacology 34: 1514–1522. [Crossref]
- Brüne M, Schöbel A, Karau R, Benali A, Faustmann PM, et al. (2010) Von Economo neuron density in the anterior cingulate cortex is reduced in early onset schizophrenia. Acta Neuropathol 119: 771-778. [Crossref]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, et al. (2008) Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 31: 234-242. [Crossref]