Immune checkpoint inhibitors and Chimeric Antigen Receptor (CAR) T-cell therapies have emerged as concomitant approaches to treat B-cell malignancies. Immune checkpoint blockade are monoclonal antibodies that override the evasion by tumors of self-destruction through inhibitory signaling. CAR T-cells are genetically modified to express CAR and, like monoclonal antibodies, are targeted therapies directed toward antigens on tumor cells. Initial studies of immune checkpoint inhibitors and CAR T-cell therapies report clinical data demonstrating safety and efficacy for both approaches and have constituted a paradigm shift in the treatment of cancer. Subsequent clinical data reveal safety and efficacy, and also associated toxicities for both approaches. Immune related adverse events can affect patients’ quality of lives adversely. Further studies may focus on mitigating adverse events and benefitting patients.
Immunotherapy, malignancies, B-cell, chimeric antigen receptor
“If we are at the dawn of a new day in immunotherapy for HM [Hematological Malignancies], we may begin to envision its morning,” an author reports in a 2015 issue of the journal Blood, referring to the immune checkpoint blockade approaches [1]. Anti-Programmed Death-1 (PD-1), its ligand Programmed Death-Ligand 1 (PD-L1), and anti-cytotoxic T-lymphocyte antigen 4 (anti-CTLA-4) inhibitors were being developed (and still are) at the time as immunological targets for hematological malignancies, and as the foundation for treatment of these blood cancers, and the author proclaims it a “paradigm-shift” for their clinical use. However, recently, this proclamation might be aptly described for chimeric antigen receptor (CAR) T-cell therapies, which may currently be eclipsing immune checkpoint blockade inhibitors.
This paper evaluates of both of these approaches for B-cell malignancies and focuses on diffuse large B-cell lymphoma (DLBCL), B-cell acute lymphocytic leukemia (B ALL), classical Hodgkin lymphoma (cHL), and chronic lymphocytic leukemia (CLL), and presents a critical appraisal of the clinical evidence supporting their use in the treatment of B-cell malignancies [2–4]. This review compares and contrasts their current roles for treating B-cell malignancies and the existing immune‐related toxicities [5] and discusses how both immune checkpoint inhibitors and adoptive cell therapies can produce strategies for the management of these malignancies [6]. Also, this paper addresses immune‐related adverse events. Both approaches have demonstrated a dramatic improvement in overall survival in patients with B-cell malignancies. Prolonged survival rates, longer remission and less refractory periods result, and each has benefits and limitations for patients in terms of symptomology and quality of life [7,8].
Approximately every three minutes one person in the United States (U.S.) is diagnosed with a blood cancer, of which B-cell malignancies comprise. An estimated combined total of almost 175,000 people in the U.S. were diagnosed with leukemia, lymphoma or myeloma in 2018. New cases of leukemia, lymphoma and myeloma are expected to account for 10 percent of the estimated two million new cancer cases diagnosed in the U.S. in 2018. An estimated 1.3 million people in the U.S. are either living with, or are in remission from, leukemia, lymphoma or myeloma. Approximately 22,000 people die of B cell malignancies each year in the U.S. Patients with some B cell malignancies cannot be cured by conventional chemotherapy and monoclonal antibodies. Allogeneic stem cell transplantation is limited by significant transplant related mortality and a shortage of suitable donors [9].
The hallmarks of cancer are constant proliferative signaling, evasion of growth suppressors, resistance to cell death, replicative immortality, induction of angiogenesis and activating invasion, and metastasis [10,11]. The dysregulated cellular processes in cancer cells are in tandem with accumulation of variable genetic alterations and consequent expression of tumor neoantigens which are not present in normal cells. In an ideal state, immune cells recognize these new antigens and kill the cancer cells [12,13].
Immune checkpoint inhibitors, such as anti-CTLA-4, PD-1, and PD-L1, and adoptive T-cell therapies, such as CAR T-cell therapy, have emerged at the forefront in the discovery of cancer immunotherapies [14,15]. A number of immunomodulatory agents that target immune system checkpoints, such as the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), a receptor that binds with certain molecules to tell the immune system to slow down, and the programmed cell death protein 1 (PD-1), a receptor involved with telling T-cells to die and reduce the death of regulatory T-cells, have received regulatory approval for the treatment of multiple cancers including malignant melanoma, non-small cell lung cancer, renal cell carcinoma, classical Hodgkin lymphoma, and recurrent or metastatic head and neck squamous cell carcinoma [16].
Additionally, the recent success of immune checkpoint inhibitors for the treatment of multiple different cancers has markedly increased interest in their investigation as combination regimens, often in conjunction with established systemic and local therapies [17,18]. Although the combination of immunooncology agents with molecularly targeted agents or radiotherapy offers the potential for synergistic antitumor immune responses, a valid concern is the increased risk of immune-mediated toxic effects [19].
Adoptive T-cell therapies such as CAR T-cell therapies have been developed as approaches to specifically treat hematological malignancies. CAR T cell therapies – that have been genetically engineered for a new antigen-specificity – have displayed significant success in treating patients with hematologic malignancies, leading to three recent U.S. Food and Drug Administration approvals [20,21].
The development of immune checkpoint blockade treatments:
The treatment of cancer has largely relied on killing tumor cells with nonspecific cytotoxic therapies and radiotherapy [2]. This approach, however, has limitations including severe systemic toxicities, bystander effects on normal cells, recurrence of drug‐resistant tumor cells, and the inability to target micro-metastases or subclinical disease [2].
Immune checkpoint inhibition evolved as a way to exploit the cancer cell’s inhibitory pathway and self-destruct upon stimulation or activation. For proper T cell activation, two separate signals are required (Figure 1) [10,22]. The first signal is mediated by antigen-dependent T cell receptor (TCR) binding to the major histocompatibility complex (MHC) molecule of an antigen-presenting cell (APC). The second signal is antigen-independent, co-stimulatory, or co-inhibitory signal delivered by the APCs. The second signal modulates TCR signaling and determines the T cell’s fate [10,22].
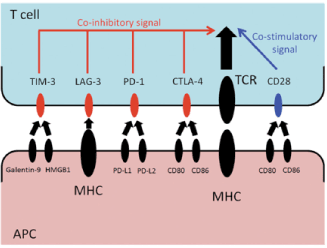
Figure 1. Immune Checkpoint Inhibition Mechanisms. The antigen presenting cell presents the inhibitory signal through the major histocompatibility complex, which blocks the T-cell from killing tumor cells. Immune checkpoint inhibitors, such as anti-CTLA4 and anti-PD-1, override this inhibition [10]
Cancer cells can exploit the programmed death-1 (PD-1) immune checkpoint pathway to avoid immune surveillance by modulating T-lymphocyte activity. In part, this may occur through overexpression of PD-1 and PD-1 pathway ligands (PD-L1 and PD-L2) in the tumor microenvironment. PD-1 blockade has produced significant antitumor activity in solid tumors, and similar evidence has emerged in hematologic malignancies [23].
The development of immune checkpoint inhibition, whose mechanisms are described above, is one of the most striking innovation in the clinical development of immunotherapy. Monoclonal antibodies (mAbs) restore and augment the antitumor immune activities of cytotoxic T cells by mainly blocking immune checkpoint molecules on T cells or their ligands on antigen-presenting and tumor cells [24]. Based on preclinical data, many clinical trials have demonstrated the acceptable safety profiles and efficacies of mAb in various cancers. The first-in-class approved immune checkpoint inhibitor was ipilunimab, which is a fully humanized mAb that blocks the immunosuppressive signal by cytotoxic T-lymphocyte antigen-4. Thereafter, nivolumab was also approved for use in the treatment of advanced renal cell cancer in August 2016, of Hodgkin's lymphoma in December 2016, and of head and neck cancer in March 2017 in Japan [24].
It is now recognized that established tumors have numerous mechanisms of suppressing the antitumor immune response including production of inhibitory cytokines, recruitment of immunosuppressive immune cells, and upregulation of coinhibitory receptors known as immune checkpoints. La-Beck et al. conducted one of the first reviews on the immune checkpoint inhibitors. In highlighting similarities and differences among the three immune checkpoint inhibitors approved by the U.S. Food and Drug Administration—ipilimumab, pembrolizumab, and nivolumab—they facilitate therapeutic decision making [2,25]. However, there is limited clinical success amongst pediatric patients [26,27].
Immune checkpoint blockade inhibitors: The landscape of treatments from recent clinical trials
Based on the success of PD-1 blocking mAbs in the treatment of solid tumors, two phase 1 studies were initiated testing those antibodies in a broad array of HMs. The first study (NCT01592370) tested the safety and single-agent activity of the anti-PD1 mAb nivolumab in patients with relapsed or refractory (R/R) multiple myeloma (MM), non-HL (NHL), and classical HL [3]. The second study (NCT01953692, KEYNOTE-013) tested another PD-1 mAb, pembrolizumab, in R/R myelodysplastic syndromes (MDS), MM, NHL, and HL [1]. Preliminary results from both studies have now been reported. Both studies enrolled patients with multiply R/R HL, with a median of 4 to 5 lines of prior systemic therapy; most had had prior brentuximab and prior autologous stem cell transplantation (ASCT) [1,28,29].
Despite this, single-agent PD-1 blockade yielded overall response rates (ORR) of 87% (with a complete response (CR) rate of 17%) and 65% (CR rate of 21%) with nivolumab and pembrolizumab, respectively [1,30]. Although the median follow-up is still short, many of the responses appear durable, with some patients now in continued remission for over a year. Another important finding in both studies was the favourable safety profile in patients with HM, as was also demonstrated by prior studies of another anti–PD-1 mAb, pidlizumab [1]. This is a salient result, as many patients on those studies had previously received other agents with potential lung toxicity, including radiation, high-dose carmustine, and brentuximab vedotin, raising the concern that PD-1 blockade could lead to a high incidence of pneumonitis in this patient population. In fact, the incidence of pneumonitis in those trials, although not negligible, did not appear excessive, with 13 cases (including 3 severe and 1 fatal case) among 134 patients [31]. Overall, the safety profile of PD-1 blockade in HM appeared similar to that in patients with solid tumors; the rate of severe (grade 3) drug-related adverse events was around 20% in both trials, and only 2 life-threatening (grade 4) and 1 fatal treatment-related toxicities were reported among 134 patients. PD-1 blockade therefore appears to be a tolerable treatment in HM [32].
In a recent preliminary Phase Ib study, nivolumab showed efficacy and tolerability in patients with relapsed and refractory B-cell lymphoma. In phase I, open-label, dose-escalation, cohort-expansion study, patients with relapsed or refractory B-cell lymphoma received the anti–PD-1 monoclonal antibody nivolumab at doses of 1 or 3 mg/kg every 2 weeks [23,32]. The study aimed to evaluate the safety and efficacy of nivolumab and to assess PD-L1/PD-L2 locus integrity and protein expression. Eighty-one patients were treated, and drug-related adverse events occurred in 51 (63%) patients. Objective response rates were 40%, 36%, 15%, and 40% among patients with follicular lymphoma, diffuse large B-cell lymphoma, mycosis fungoides, and peripheral T-cell lymphoma, respectively. Nivolumab was well tolerated and exhibited antitumor activity in extensively pretreated patients with relapsed or refractory B- cell lymphomas [23,32].
Loss of donor-mediated immune antitumor activity after allogeneic hematopoietic stem-cell transplantation (HSCT) permits relapse of hematologic cancers. According to Davids, et al, immune checkpoint blockade established by targeting cytotoxic T-lymphocyte-associated protein 4 with ipilimumab could restore antitumor reactivity through a graft-versus-tumor effect. A phase 1/1b multicenter, investigator-initiated study was conducted to determine the safety and efficacy of ipilimumab in patients with relapsed hematologic cancer after allogeneic HSCT [33]. A total of 28 patients were enrolled. Immune-related adverse events, including one death, were observed in 6 patients (21%), and graft-versus-host disease (GVHD) that precluded further administration of ipilimumab was observed in 4 patients (14%). Among 22 patients who received a dose of 10 mg per kilogram, 5 (23%) had a complete response, 2 (9%) had a partial response, and 6 (27%) had decreased tumor burden [33]. Complete responses occurred in 4 patients with extramedullary acute myeloid leukemia and 1 patient with the myelodysplastic syndrome developing into acute myeloid leukemia. Four patients had a durable response for more than 1 year. Early-phase data showed that administration of ipilimumab was feasible in patients with recurrent hematologic cancers after allogeneic HSCT, although immune-mediated toxic effects and GVHD occurred [33].
A phase II study evaluated pidilizumab monotherapy in patients with recurrent DLBCL who had received auto-HSCT [34]. The rationale was to enhance effector T cell function through PD-1 blockade after auto- HSCT to eliminate possible residual disease. Patients started by receiving pidilizumab intravenously at a dose of 1.5 mg/kg every 42 days for three cycles from 30 to 90 days after auto-HSCT. A total of 72 patients were enrolled and 60 patients finished all 3 cycles. A total of 66 patients were evaluable. The overall progression free survival (PFS) was 72% at 16 months (90% CI: 60%-82%) exceeding the historical data of 69%. The median time to initial response was 30 weeks (range: 6–69 weeks). Among the 35 patients who had residual disease by CT-imaging at time of post-auto-HSCT restaging, the overall response rate (ORR) was 51%, including 12 (34%) complete response (CR) and 6 (17%) partial response (PR). However, the ORR fell to 33% for the 9 patients with positive PET scan at restaging. The most frequently seen grade 3/4 adverse events (AEs) were neutropenia (19% of patients) and thrombocytopenia (8%). No autoimmune toxicity, infusion reactions or treatment-related mortality was observed. Treatment with pidilizumab has led to a substantial expansion of PD-L1 bearing activated helper T cells within 24 h, suggesting an on-target effect. Eleven patients with relapsed or refractory DLBCL were enrolled in the aforementioned phase I study of nivolumab. Four patients (36%) responded to the therapy with 1 CR while the PFS rate at 24 weeks was 24% [34].
A Phase II trials were registered on clinicaltrials.gov for nivolumab in hematologic malignancies: A single-arm, open-labelled, Phase II study of patients with relapsed/refractory DLBCL that failed AHCT or two prior standard regimens in patients that are transplant ineligible (NCT02038933). The primary endpoint was ORR and secondary endpoints include further anti-tumor effects. Nivolumab was administered at 3 mg/kg every 2 weeks until progression of disease. The primary endpoint is ORR and secondary endpoints include duration of response and other anti-tumor effects [35].
Adverse effects and toxicities: consensus recommendations: As reported by Puzanov et al, consensus recommendations have been released from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. Increasing use of immune-based therapies, including immune checkpoint inhibitors, has exposed a discrete group of immune-related adverse events (irAEs) [36]. Many of these are driven by the same immunologic mechanisms responsible for the drugs’ therapeutic effects, namely blockade of inhibitory mechanisms that suppress the immune system and protect body tissues from an unconstrained acute or chronic immune response. Skin, gut, endocrine, lung and musculoskeletal irAEs are relatively common, whereas cardiovascular, hematologic, renal, neurologic and ophthalmologic irAEs occur much less frequently [36].
Overall, irAEs are broken down into two major categories, based on the opinions of the workshop organizers regarding the frequency with which they are seen in clinical practice: frequently reported (dermatologic, gastroenterological, endocrine, respiratory, and rheumatologic/musculoskeletal) and uncommon (cardiovascular, hematologic, renal, neurologic and ophthalmologic) [35]. Infusion reactions, which are more common with mAbs based on a wildtype IgG1 backbone and less common with IgG4 antibodies, are also addressed. Within each body system, information is divided into three sections: clinical presentation and epidemiology, diagnostic evaluation, and guidance on when to refer to a disease specialist. Among the SITC’s consensus recommendations are prednisone for abdominal pain and hepatitis, and thyroid replacement therapy for hypothyroidism. Due to incidences of relapse and remission and side effects observed for immune checkpoint inhibitors, the development of CAR T cells paved the way for further cancer immunotherapeutic approaches.
The development of adoptive cell therapies: Because tumor-reactive T cells cannot be reliably cultured from most human tumors, methods have been developed to engineer T cells to express genes encoding tumor-antigen specific T cell therapies. Another approach to adoptive T cell therapy is to engineer T cells to express chimeric antigen receptors (CARs). CARs are made of an antigen-recognizing receptor coupled to signaling molecules that can activate T cells expressing the CAR. The antigen receptors most commonly incorporated into CARs are single chain variable regions of a monoclonal antibody joined by a peptide linker. CD19 is promising target for antigen-specific T cell therapies because malignant B cells, normal B cells and follicular dendritic cells express it [37,38].
CARs are designed with domains derived from different origins, including an extracellular ligand binding domain, a transmembrane domain, and intracellular ligand domains. The extracellular ligand binding domain confers target specificity (Figure 2). The intracellular signaling domains drive CAR T cell effector functions. For durable T-cell activation, co- stimulatory signaling is also required. CARs allow the expressing T cell to effectively kill target tumor cells and ideally persist and provide ongoing immune surveillance. While some normal B cells die after receiving therapies targeting CD19, it is well-tolerated (Figure 3) [37].
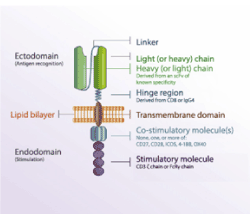
Figure 2. Anatomy of a CAR. All of the components except of the lipid bilayer which part of the host cell membrane are encoded by the plasmid. [Adapted from Gill, et al.]
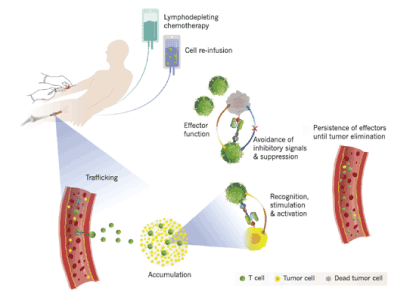
Figure 3. Components of a successful adoptive cellular immunotherapy. The patient undergoes chemotherapy that depletes the lymphocytes. CAR T cells circulate after infusion and circulate to the tumor environment, where they produce their effector functions. [Adapted from Gill, et al.]
Clinical trials for recent FDA-approved CAR-T cell agents
Kochenderfer, et al. reported in 2009 eradication of B-lineage cells and regression of lymphoma in a patient treated with T cells genetically engineered to recognize CD19. Prolonged elimination of CD19+ cells in the patient indicated in vivo antigen specific activity of anti-CD19-CAR-expressing T cells [39].
Investigators at Memorial Sloan Kettering Cancer Center (New York, NY) were the first to publish clinical trial results describing CD19-targeted CAR T-cell therapy of patients with relapsed B-ALL. In this study, all five patients treated with CAR T cells achieved minimal residual disease negative complete remission. Updated results describing the treatment of 16 patients with relapsed or refractory B-ALL treated with CAR T cells were recently published: the overall CR rate in this trial was 88% and 12 of 14 patients were classified as minimal residual disease negative. 44% of these patients went on to standard-of- care allogeneic hematopoietic stem cell transplant [40].
Initial studies from University of Pennsylvania (Philadelphia, PA) reported potent anti-leukemic effects of CD19 CAR T cell therapy in three patients with refractory chronic lymphocytic leukemia where two of the three patients achieved MRD-CR. Infused CAR T cells proliferated up to 10,000-fold and persisted in recipients for at least 6 months and shown to retain antitumor activity after six months [41].
Novartis received its first ever FDA approval for a CAR-T cell therapy, tisagenlecleucel, for children and young adults with B-cell ALL that is refractory or has relapsed at least twice. First-in-class therapy showed an 83% (52/63) overall remission rate in this patient population with limited treatment options and historically poor outcomes [42]. Tisagenlecleucel’s approval was followed by FDA approval of asicabtagene ciloleucel, which employs similar mechanisms [20].
A recent report released by the American Society of Clinical Oncology (ASCO) showed that CAR-T therapy trials show longer term benefits [21]. In addition to being named the 2018 Advance of the Year by ASCO, CAR-T cell therapy, unlike checkpoint inhibitors, boosts the ability of the body’s immune system to fight cancer using the patient’s own genetically re-engineered cells
ZUMA-6 (NCT02926833), a phase II trial of a second type of anti-CD19 CAR-T therapy, called axicabtagene ciloleucel (Yescarta), enrolled 111 adults with refractory large B-cell lymphoma [21]. After a median time of 15.4 months, 82% of patients had decreased signs and symptoms of disease and 54% experienced a complete regression of disease [21]. Furthermore, 52% of patients were alive 18 months after treatment. There were some significant adverse effects, including myelosuppression, cytokine release syndrome, and neurologic events. The FDA approved the drug for use in this population in October 2017 [21].
There are a number of toxicities and limitations associated with CAR T-cell therapy. Cytokine Release Syndrome (CRS) is a condition hallmarked by increased cytokine production and associated inflammation. CRS generally correlates with increased IFN, GM-CSF, IL-10, and IL-6, as well as CAR T cell expansion [20,43]. Cytokine release syndrome clinical presentation includes fever, nausea, anorexia, tachycardia and/or hypotension, cardiac dysfunction, renal impairment, and hepatic failure among other conditions. Higher disease burdens correlated with greater CRS severity. Anti-tumor activity of CAR T therapies, however, was not predicated by disease severity, as patients who did not experience CRS may still responded to therapy. Clinical trial results from the University of Pennsylvania/ Children’s Hospital of Philadelphia, as well as the ELIANA trial indicated that CRS severity specifically correlated with patient IL-6 levels. As a result, the FDA concurrently approved tocilizumab with tisagenlecleucel for the treatment of CAR T-associated CRS. Subsequent studies have confirmed tocilizumab efficacy in reversing CRS symptoms, as well as having little to no inhibitory effect upon CAR T therapeutic efficacy [20,44].
Patients who receive CAR T-cells can also develop reversible neurotoxicity independently of CRS. While global encephalopathy is the most common toxicity, other symptoms have been reported (seizures, aphasia, hallucinations). The symptoms are usually brief and self-limited. It is of unknown etiology. NTX, which can present as CAR T-related encephalopathy (CRES), includes a variety of neurological symptoms, including confusion, delirium, expressive aphasia, obtundation, myoclonus, and seizure. White matter degradation has also been observed in some severe cases [20]. To date, causality of CRES is unknown, but studies suggest that cytokine secretion and subsequent breakdown of the blood-brain barrier may play a role. of note, patients treated with blinatumomab – a bi-specific anti-CD19/CD3 BiTE antibody – also have increased incidence of neurological toxicities similar to those observed in patients treated with CAR T [45].
CRES treatment includes supportive care and consistent neurological evaluation. CRES also requires a neurological evaluation, as well as tocilizumab administration if symptoms are associated with CRS. Unlike CRS where tocilizumab has a clear role, the efficacy of tocilizumab in reversing CRES is not well established. CRES requires ICU transfer, and some centers utilize tocilizumab administration if associated with CRS. Corticosteroids - including dexamethasone or methylprednisolone - are also recommended for grade 3/4 CRES.
Combination of immune checkpoint inhibitors with t-cell based therapies: Adoptive T cell therapy in combination with immune checkpoint inhibitors may elicit promising treatments. The antitumor effect of combinational therapy with CAR T-cells and PD-1 inhibitor was investigated preclinically using transgenic Her2 mice treated with Her2-specific CAR T-cells. Tumor Her2 antigen triggered PD-1 upregulation in CAR T-cells, and PD-1 blockade enhanced Her2-specific T-cell functions and decreased MDSC in the tumor microenvironment, leading to enhanced anti-tumor effect [43]. A clinical trial to study the combination of CD19-CAR T-cells and ipilimumab in patients with B-cell lymphoid malignancies including pediatric patients has been initiated (NCT00586391).
When non-clonal T-cells are gene-modified with CAR or armed with bispecific antibodies, they mediate potent anti-tumor cytotoxicity, leading to strong T-cell activation and production of proinflammatory cytokines [26]. However, despite promising clinical responses (e.g. CD19-directed T-cell based immunotherapy), tumor recurrence was observed, partly because of genomic instability and the effects of cancer immune editing [20]. Additional resistance mechanisms include downregulation or loss of target antigen expression, tumor-associated dendritic cell dysfunction, increased T-regulatory cells, immunosuppressive cytokines, activation of alternative signaling pathways, and anti-antibody formation [26].
Use of hematopoietic cell transplantation in conjunction with immune checkpoint inhibitors and car t-cell therapies:
As described earlier, hematopoietic cell transplantation has been considered the standard curative therapy for many patients with hematological malignancies. In addition to the cytotoxic effects of the chemotherapy and radiation used in the conditioning regimen, the benefits of hematopoietic cell transplantation are derived from a reset of the immune system and harnessing the ability of donor T cells to eliminate malignant cells. With the dawn of the era of immunotherapies in the form of checkpoint inhibitors and chimeric antigen receptor T cells, the role of hematopoietic cell transplantation has evolved [46,47].
Ghosh et al. found that immunotherapy with checkpoint inhibitors is increasingly being used for relapsed Hodgkin and non-Hodgkin lymphoma after auto-HCT. Checkpoint inhibitors are also being tested after allo-HCT with observable benefits in treating hematological malignancies, but with a potential risk of increased GVHD and transplant-related mortality. Immunotherapy with CD19 CAR T cells are powerful options with aggressive B cell malignancies both for therapy and as induction leading to allo-HCT [48,49]. While immunotherapies with checkpoint inhibition and CAR T cells are increasing being used to treat hematological malignancies, HCT remains a standard of care for most of the diseases with the best chance of cure. Combination of these therapies with HCT has the potential to more effectively treat hematological malignancies [50,51].
Summary
Clinical data reveal safety and efficacy in Phase I and II trials for both approaches that have subsequently achieved Phase III approvals. Relapsed and refractory B cell malignancies, particularly classical Hodgkin lymphoma in adults and B cell acute lymphoblastic leukemia in pediatric patients, can now be considered to be amenable for immune checkpoint inhibitors and CAR T-cell treatments. Associated immune related adverse events are treatable, and in some cases reversible, but may affect patients’ quality of lives adversely. Future studies may concentrate on developing lines of treatment that avoid immune-related toxicities and benefit adult and pediatric patient populations.
Conflicts of Interest:
No conflicts of interest to disclose.
- Armand P (2015) Immune checkpoint blockade in hematologic malignancies. Blood 125: 3393-3400 .
- La-Beck NM, Jean GW, Huynh C, Alzghari S, Lowe D (2015) Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacotherapy 35: 963-976. [Crossref]
- Bröckelmann, P, Engert A (2017) Checkpoint Inhibition in Hodgkin Lymphoma – A Review. Oncol Res Treat 40: 654-660. [Crossref]
- Gill S, Maus MV, Porter DL (2016) Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev 30: 157-167. [Crossref]
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168: 707-723. [Crossref]
- Allard B, Aspeslagh S, Garaud S, Dupont FA, Solinas C, et al. (2018) Immuno-oncology 101: overview of major concepts and translational perspectives. Semin Cancer Biol 52: 1-11. [Crossref]
- Xu-Monette ZY, Zhou J, Young KH (2018) PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 131: 68-83. [Crossref]
- Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, et al. (2014) CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev 257: 91-106. [Crossref]
- Leukemia & Lymphoma Society (2019) Facts and Statistics.
- Ok C, Young K (2017) Checkpoint inhibitors in hematological malignancies. J Hematol Oncol 10:103-119. [Crossref]
- Dada R (2018) Program death inhibitors in classical Hodgkin’s lymphoma: a comprehensive review. Ann Hematol 97:555-561. [Crossref]
- Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39: 1-10. [Crossref]
- Goodman A, Patel SP, Kurzrock R (2017) PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol 14: 203-220. [Crossref]
- Maleki Vareki S, Garrigós C, Duran I (2017) Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncol Hematol 116: 116-124. [Crossref]
- Turtle CJ, Hay KA, Hanafi, L-A, Li D, Cherian S, et al. (2017) Durable Molecular Remissions in Chronic Lymphocytic Leukemia with CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol 35: 3010-3020. [Crossref]
- Herrera AF (2018) Where does PD-1 blockade fit in HL therapy? Hematology Am Soc Hematol Educ Program 2018: 213-220. [Crossref]
- Tang C, Jiang W, Yap T (2018) Efficacy and Toxic Effects of Cancer Immunotherapy Combinations-A Double-edged Sword. JAMA Oncol 4: 1116-1117. [Crossref]
- Matsuki E, Younes A (2016) Checkpoint Inhibitors and Other Immune Therapies for Hodgkin and Non-Hodgkin Lymphoma. Curr Treat Options Oncol 17: 31-57. [Crossref]
- Vick E, Mahadevan D (2016) Programming the immune checkpoint to treat hematologic malignancies. Expert Opin Investig Drugs 25: 755-770. [Crossref]
- Boyiadzis MM, Dhodapkar MV, Brentjens RJ, Kochenderfer JN, Neelapu SS, et al. (2018) Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective J Immunother Cancer 6: 1-12. [Crossref]
- Pal SK, Miller MJ, Agarwal N, Chang SM, Chavez-MacGregor M, et al. (2019) Clinical Cancer Advances 2019: Annual Report on Progress Against Cancer from the American Society of Clinical Oncology. J Clin Oncol 37: 834-849. [Crossref]
- Burugu S, Dancsok AR, Nielsen TO (2018) Emerging targets in cancer immunotherapy. Semin Cancer Biol 52: 39-52. [Crossref]
- Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, et al. (2016) Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol 34: 2699-2704. [Crossref]
- Kitano S (2017) Development of immune checkpoint inhibitors. Rinsho Ketsueki 58: 966-976. [Crossref]
- Thallinger C, Füreder T, Preusser M, Heller G, Müllauer L, et al. (2018) Review of cancer treatment with immune checkpoint inhibitors : Current concepts, expectations, limitations and pitfalls. Wien Klin Wochenschr 130: 85-91. [Crossref]
- Park J, Cheung NV (2017) Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat Rev 58: 22-33. [Crossref]
- Tsirigotis P, Savani BN, Nagler A (2016) Programmed death-1 immune checkpoint blockade in the treatment of hematological malignancies. Ann Med 48: 428-439. [Crossref]
- Shin DS, Ribas A (2015) The evolution of checkpoint blockade as a cancer therapy: what's here, what's next? Curr Opin Immunol 33: 23-35. [Crossref]
- Davis KL, Agarwal AM, Verma AR (2017) Checkpoint inhibition in pediatric hematologic malignancies. Pediatr Hematol Oncol 34: 379-394. [Crossref]
- Postow MA, Callahan MK, Wolchok JD (2015) Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 33: 1974-1982. [Crossref]
- Janakiram M, Pareek V, Cheng H, Narasimhulu DM, Zang X (2016) Immune checkpoint blockade in human cancer therapy: lung cancer and hematologic malignancies. Immunotherapy 8: 809-819. [Crossref]
- Tie Y, Ma X, Zhu C, Mao Y, Shen K, et al. (2017) Safety and efficacy of nivolumab in the treatment of cancers: A meta-analysis of 27 prospective clinical trials. Int J Cancer 140: 948-958. [Crossref]
- Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, et al. (2016) Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N Engl J Med 375: 143-153. [Crossref]
- Xia Y, Medeiros L, Young K (2016) Immune checkpoint blockade: Releasing the brake towards hematological malignancies. Blood Rev 30: 189-200. [Crossref]
- Bryan LJ, Gordon LI (2015) Blocking tumor escape in hematologic malignances: The anti-PD-1 strategy. Blood Rev 29: 25-32. [Crossref]
- Puzanov I, Diab A, Abdallah K, Bingham CO 3rd, Brogdon C, et al. (2017) Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer 5: 1-28. [Crossref]
- Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, et al. (2016) Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016;127:1117–1127. [Crossref]
- Fesnak AD, June CH, Levine BL (2016) Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 16: 566-581. [Crossref]
- Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, et al. (2009) Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother 32: 689-702. [Crossref]
- Pegram H, Smith E, Rafq, S, Brentjens, R (2015) CAR therapy for hematological cancers: can success seen in the treatment of B-cell acute lymphoblastic leukemia be applied to other malignances? Immunotherapy 7: 545-561. [Crossref]
- Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, et al. (2015) Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7: 1-25. [Crossref]
- FDA Approves Personalized Cellular Therapy for Advanced Leukemia Developed by University of Pennsylvania and Children’s Hospital of Philadelphia.
- Anwer F, Shaukat AA, Zahid U, Husnain M, McBride A, et al. (2017) Donor origin CAR T cells: graft versus malignancy effect without GVHD: a systematic review. Immunotherapy 9: 123-130. [Crossref]
- Frey N (2017) The what, when and how of CAR T cell therapy for ALL. Best Pract Res Clin Haematol 30: 275-281. [Crossref]
- Liu J, Zhong JF, Zhang X, Xhang C (2017) Allogeneic CD19–CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. J Hematol Oncol 10: 35. [Crossref]
- Valton J, Guyot V, Marechal A, Filhol JM, Juillerat A, et al. (2015) A Multidrug-resistant Engineered CAR T cell for Allogeneic Combination Immunotherapy. Mol Ther 23: 1507-1518. [Crossref]
- Ghosh A, Politikos I, Perales MA (2017) Stop and go: hematopoietic cell transplantation in the era of chimeric antigen receptor T cells and checkpoint inhibitors. Curr Opin Oncol 29: 474-483. [Crossref]
- Freeman CL, Gribben JG (2016) Immunotherapy in Chronic Lymphocytic Leukaemia (CLL). Curr Hematol Malig Rep 11: 29-36. [Crossref]
- Herrera AF, Moskowitz AJ, Bartlett NL, Vose JM, Ramchandren R, et al. (2017) Interim results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Hematol Oncol 35: 85-86. [Crossref]
- Hay KA, Turtle CJ (2018) CD19-specific chimeric antigen receptor-modified (CAR)-T cell therapy for the treatment of chronic lymphocytic leukemia in the ibrutinib era. Immunotherapy 10: 251-254. [Crossref]
- Jain N, O’Brien S (2016) Targeted therapies for CLL: Practical issues with the changing treatment paradigm. Blood Rev 30: 233-244. [Crossref]