Abstract
Restrictive cardiomyopathy is a myocardial disorder characterized by restrictive ventricular filling. It represents one of the three phenotypes originally classified by the World Health Organization besides dilated and hypertrophic cardiomyopathies. Of the three, it is the least common phenotype but has an ominous prognosis with the highest recorded rates of sudden cardiac death. Clinical trials investigating RCM have had varied findings. This is attributable to the current classification systems of cardiomyopathy based on morphological and structural alterations rather than etiologic-based. The classification has created etiological heterogeneity requiring a combination of methods for a confirmatory diagnosis as well as complicating clinical management methods, which lacks standardized guidelines and largely relies on the standard heart failure therapy. Thus, the present review discussed current research evidence to produce a comprehensive understanding of the clinical status of RCM, including its diagnosis and clinical management.
key words
restrictive cardiomyopathy, myocardial disorder, restrictive ventricular filling
Restrictive cardiomyopathy (RCM) is the least common phenotype of cardiomyopathies with controversy in its exact definition, epidemiology and diagnostic criteria. If left untreated, the disorder could ultimately lead to heart failure. This review article documents available research-based evidence to provide a comprehensive clinical understanding of RCM, its epidemiology, diagnosis and clinical management. The article is organized into ten main sections, namely description, classification, epidemiology, clinical manifestation, prognosis, causes of death, pathophysiology, etiology, differential diagnosis, and clinical management. The article concludes with a concise summary of the main points discussed.
Up to early 1990s, the classification of primary cardiomyopathies relied on structural and functional abnormalities, which identified three major categories: dilated, hypertrophic and restrictive cardiomyopathies [1-3]. The World Health Organization (WHO) and the International Society and Federation of Cardiology (ISFC) Task Force added two more categories in 1996: arrhythmogenic right ventricular cardiomyopathy and unclassified cardiomyopathies as well as distinguished RCM into two types, primary and secondary based on the underling etiology [4]. Unlike other forms of cardiomyopathies – dilated, hypertrophic and right ventricular – that are defined using specific morphological criteria, classification of RCM is largely based on functional criteria [5]. However, since many anatomical abnormalities could lead to restrictive ventricular filling, definitive diagnosis for RCM using functional criteria remains challenging [6]. The World Health Organization further proposed dilated atria without ventricular dilation or hypertrophy as a characteristic morphological feature for the identification of RCM but these features are non-specific. The pathophysiology of endomyocardial changes causing elevated filling pressures and consequently the enlargement of atria remains unclear and the subject of ongoing research [6].
A uniform clinical definition of RCM remains a challenge due to the lack of definitive abnormal morphological characteristics. The two most common definition of primary cardiomyopathies rely on the morphology of the left ventricle (LV). Dilated cardiomyopathy defined as stretched and thinned (dilated) LV with normal wall function and impaired LV function, and hypertrophic cardiomyopathy defined as thickened (hypertrophied) LV wall with reduced LV capacity [6]. The definition of RCM as suggested by the WHO and ISFC Task Force rely on altered hemodynamics (restrictive ventricular filling) rather than abnormal morphological features. They define RCM as a disorder of the myocardium characterized by impaired ventricular filling but with normal or decreased diastolic volume in one or both ventricles. In the early phases of the disorder, systolic function is normal and depending on the underlying etiology, and wall thickness may be normal or increased [4].
Similarly, other clinical definitions of RCM proposed in literature on cardiac disorders have also relied on altered hemodynamics mainly based on echocardiographic findings. Ammash et al. [6] define primary RCM as a disorder of the heart muscle marked by echocardiographic-defined non-dilated, non-hypertrophied ventricles with bi-atrial enlargement not explained by ischemic, valvular, hypertensive, inflammatory or infiltrative cardiac disorders [6]. RCM has also been defined as myocardium disorder characterized by stiffened ventricles resulting in impaired diastolic filling but with preserved systolic function [7].
The classification of RCM is based entirely on the underlying etiology. The major classifications are primary and secondary RCM.
The primary form of RCM variously referred to as classical or non-infiltrative is idiopathic RCM, defined by the presence of hemodynamic abnormalities in the absence of discernible histological features indicating myocardial abnormalities [8,9]. Patients with idiopathic RCM may present with or without hypertrophied LV walls, but in a majority of cases (57%), they present with the familial (hereditary) form of RCM [10]. Idiopathic RCM with hypertrophied LV walls has microscopic characteristics similar to that of hypertrophic cardiomyopathy but they present with different phenotypic expression of the same genetic disorder [10]. On the other hand, the familial form of idiopathic RCM suggest etiology of a genetic abnormality usually associated with distal skeletal myopathy [11,12]. Fitzpatrick et al. reported autosomal dominant form of RCM with atrioventricular block and skeletal myopathy in five generations of an Italian family with symptoms manifesting in the forties [13]. Patients with idiopathic RCM have mild to moderate increase in cardiac weight, bi-atrial enlargement, thrombi in atrial appendages, normal ventricular cavity size and wall thickness, reduced/normal systolic function and patchy endocardial fibrosis [9].
Underlying cardiac or systemic disorder affecting the heart may result into restrictive hemodynamics inducing RCM. Systemic disorders encountered in clinical practice that may cause RCM are categorized under myocardial and endomyocardial disorders, which give rise to a variety of phenotypes collectively referred to as restrictive cardiomyopathies [5,9] (Table 1).
Table 1: Classification of restrictive cardiomyopathies [5,7,9].
Main Category |
Sub-Category |
Specific Disorder |
Myocardial |
Infiltrative |
Cardiac Amyloidosis |
Sarcoidosis |
Haemosiderosis |
Storage Diseases |
Hemochromatosis |
Fabry Disease |
Glycogen Storage Disease |
Endomyocardial |
Endomyocardial fibrosis |
Radiation |
Toxic effects of anthracylines |
However, since etiologic factors and specific pathologic processes underlying cardiac amyloidosis and endomyocardial disorders have been established, the resulting RCM is classified as secondary RCM. Secondary RCM may also develop in later stages of dilated cardiomyopathy, hypertrophic cardiomyopathy, hypertension, valvular and ischemic heart disorders [6].
Infiltrative cardiac abnormalities are one of the common causes of secondary RCM. Frequently encountered infiltrative cardiac disorders in clinical practice are cardiac amyloidosis, cardiac sarcoidosis and haemosiderosis. Other rare cardiac disorders are Gaucher’s disease, Hurler’s disease and Fatty infiltration [9].
Cardiac amyloidosis: Cardiac amyloidosis is the most common cause of RCM encountered in clinical practice and frequently referenced in many studies describing RCM. Cardiac involvement in amyloidosis causes diastolic LV dysfunction, and in mildly dilated cardiomyopathies, causes both diastolic and systolic LV dysfunction. In later stages, the interstitial infiltration of the atria causes the myocardium to become firm, rubbery and non-compliant [5]. Systemic amyloidosis on the other hand, is a dysfunction of protein metabolism where abnormal extracellular proteins are deposited in organs and tissues potentially leading to heart failure and a major cause of morbidity and mortality [14]. However, there are different forms of amyloidosis in humans and not all of them lead to restrictive ventricular filling. These are primary, secondary, hereditary and age-related amyloidosis (Table 2).
Table 2: Forms of cardiac amyloidosis [5].
Forms of Amyloidosis |
Main Characteristic |
Primary amyloidosis |
Common cardiac involvement |
Secondary amyloidosis |
Rare cardiac involvement |
Hereditary amyloidosis |
Usually autosomal dominant pattern of inheritance |
Age-related amyloidosis |
Senile/atrial |
In primary amyloidosis, cardiac involvement is more common. It is a disorder caused by plasma cells producing immunoglobulin light chains usually due to multiple myeloma. Secondary amyloidosis on the other hand is due to the production of abnormal extracellular proteins other than immunoglobulin. Secondary amyloidosis can be familial, senile or the result of chronic inflammatory processes. In primary and secondary amyloidosis, restrictive hemodynamics results from tissue injury caused by interstitial infiltrative deposits replacing normal myocardial contractile elements [15]. Amyloidosis can also be hereditary, defined as genetic variations of plasma protein (prealbumin) usually inherited in an autosomal dominant fashion. Hereditary amyloidosis causes abnormalities in ventricular filling but without clinical evidence [16,17]. The final form of amyloidosis is age-related, classified as senile in 25% of individuals aged >80 years, and atrial in 90% of individuals aged >90 years [5]. It is caused by amyloid depositions on the atrial. The deposits contain atrial natriuretic peptide and amyloid P components. It is common among the elderly and patients with chronic heart diseases such as congenital and rheumatic heart diseases [18].
There is an increasing recognition of cardiac involvement in patients with sarcoidosis. The disorder is systemic causing interstitial inflammation leading to impaired diastolic function but with an initial preserved systolic function [19]. In later stages, injury and fibrosis may lead to systolic dysfunction, and the involvement of myocardium in systemic myocardium, may cause subclinical cardiac dysfunction [20,21]. Although the disorder has a variable course, progressing rapidly to death in some asymptomatic patients or causing atrioventricular block [9]. The etiology of cardiac sarcoidosis remains unclear but many infective agents such as microbial bio-aerosols and environments exposure such as insecticides have been suggested [22]. Genetic risk factors have also been proposed for the development of the disorder because of immune response to genetic risk factors [23].
Haemosiderosis is an infiltrative cardiac disorder that may lead to restrictive ventricular filling caused by iron overload. Two clinical scenarios have been suggested that may result into iron overload. The first scenario is hereditary hemochromatosis caused by high plasma iron content exceeding transferrin iron-building capacity and normal erythropoiesis. Iron deposition occurs in parenchymal hepatocytes, cardiac muscle cells and other endocrine tissues [5]. The second clinical scenario is iron overload due to increased catabolism of erythrocytes such as in the case of transfusional iron overload. Initially, an accumulation of iron in reticuloendothelial macrophages occurs then iron spills over to parenchymal cells of the liver and heart. Iron deposition on tissues leads to tissue damage and fibrosis. Deposition on cardiac muscle cells causes myocardial fibrosis and impaired ventricular function leading to restrictive or dilated cardiomyopathy [5].
Secondary RCM may also be classified as RCM developing from storage disorders such as hemochromatosis, Fabry disease and glycogen storage disease [5,7].
Haemochromatosis: Haemochromatosis is an uncommon disorder marked by cirrhosis, diabetes, skin discoloration, endocrine and heart failure and anthropathy. The hereditary form of the disease results from inherited mutations in at least four genes associated with iron metabolism in autosomal recessive inheritance in the Mendelian fashion [5]. The disorder causes abnormal regulation in iron absorption leading to iron-overload in the entire body and secondary damage to the tissue in multiple organs including the heart. Clinical symptoms of the disorder results from abnormal iron production by enterocytes and macrophages causing progressive iron deposits on parenchymal cells of the heart and subsequently restrictive ventricular filling with normal erythropoiesis [24].
Fabry disease: Fabry disease is an X-linked lysosomal storage disorders (LSD) due to mutations in the gene that encodes the lysosomal enzyme alpha-galactosidase A (GLA). Deficiency in alpha-galactosidase disrupts glycosphingolipid metabolism resulting in intra-lysosomal accumulation and deposition of glycosphingolipids in various organs including the heart with significant morbidity and mortality [25]. Patients with Fabry disease may present with restrictive cardiomyopathy with full expression in males and incomplete expression in females presenting late in life [7,26]. Echocardiographic findings or RCM in the context of Fabry disease are LV hypertrophy, normal modest diastolic filling abnormalities and valve thickening [27-29] but ventricular wall thickness is very rare [30].
Glycogen storage disease: Glycogen storage disease refers to a group of congenital inherited disorders marked by deposition of glycogen on tissues [31,32]. It is caused by abnormality or deficiency in enzymes such as liver glycogen phosphorylase used for breaking down glycogen leading to its accumulation and deposition in tissues [33]. The disorder causes neuromuscular deficits, abnormal liver/kidney function and central nervous system and restrictive cardiomyopathy [31].
Endomyocardial fibrosis is a secondary cause of RCM. The disorder produces patchy fibrosis on the endocardium surface penetrating into the myocardium. Endomyocardial fibrosis increases stiffness of the heart leading to a reduction in compliance and ultimately restrictive hemodynamics. It causes a reduction in conduction velocity, impaired activate pattern and provision of substrate inducing wave break and re-entry [5]. The disorder principally affects right and left ventricular apex and sometimes the atrioventricular valves through the involvement of the papillary muscles causing tricuspid and mitral regurgitation. Ventricular stiffness together with atrioventricular valvular regurgitation causes the atria to enlarge [32].
The exact prevalence and incidence of RCM in the general population remains undefined because of the lack of population-based epidemiology studies. However, small studies on defined samples in different populations suggest RCM is the least common form of cardiomyopathies [8]. In Western countries, RCM accounts for less than 5% of cardiomyopathic disorders but the prevalence varies in certain populations [34]. The disorder could occur sporadically across the world but RCM in the context of endomyocardial fibrosis is endemic in equatorial Africa, and parts of India, Asia and South and Central America. In equatorial Africa, it is a frequent cause of heart failure and accounts for 15-25% of cardiac deaths [35]. In children, RCM is the least common form of cardiomyopathy accounting for 2.5-5.0% of diagnosed cardiomyopathic disorders. Two small studies reported a prevalence rate of less than one per one million children with more incidents in girls than in boys [36,37]. Reported mean age at diagnosis is between five and six years. Children in a family with a history of cardiomyopathy are thrice more likely to develop the disorder [8]. Undefined prevalence and incidence of RCM in the general population warrants a population-based epidemiology study to determine the exact distribution of the disorder globally and in different patient populations
Since RCM is a rare form of cardiomyopathy and lacks early screening indicators, there is a lack of complete largescale studies to survey and determine the clinical course of RCM. However, small-scale studies suggest, in a majority of cases, clinical representation of patients with RCM is heart failure symptoms with normal systolic function [38-40]. Patients may experience dyspnea, fatigue, a third discernible heart sound, pulmonary edema and cardiac murmurs from valve incompetence. Their heart size may be normal or enlarged with predominating features of right ventricular failure such as elevated JVP, prominent x/y descent, hepatomelogy, edema and ascites [38]. About three quarters of RCM patients may develop atrial fibrillation [40]. Clinical presentation of RCM phenotype has a striking similarity with that of constrictive pericarditis posing a diagnostic challenge. Constrictive pericarditis is due to a thickened and calcified pericardium and clinically managed through surgical procedures [39].
Clinical manifestations of RCM are heterogeneous and may vary with the age of the patient. At advanced stage of the disease, adult patients may present with cardiopulmonary symptoms [7]. They may complain of paroxysmal nocturnal dyspnea, fatigue and exercise intolerance and orthopnea. RCM patients suffering from heart failure may suffer from bilateral lower extremity edema and right upper quadrant pain and occasional abdominal discomfort or liver tenderness. RCM in the context of cardiac amyloidosis rarely causes angina-like chest pain mimicking myocardial ischemia because of the compression of small vessels by the myocardium. Some patients may experience palpitations due to RCM-induced atrial fibrillation or supraventricular arrhythmias [41]. About a third of RCM patients may suffer from thromboembolic complications, particularly pulmonary emboli caused by blood clots in the limbs. In the context of atrial fibrillation, patients may exhibit symptoms of left atrial clots and systemic emboli [7].
In children, initial signs of RCM are unrelated cardiac problems. They frequently present with lung-related problems such as repeated infections or asthma but referred to a cardiologist when chest x-ray reveals a large heart [7]. In other physical findings such as ascites and hepatomegaly and edema, gastroenterologist first examines them to exclude gastrointestinal etiologies before making a referral to a cardiologist based on additional cardiac signs such as abnormal heart sounds, signs of heart failure or fainting. In some patients, sudden death is an unfortunate clinical end-point and a symptom of RCM [39]. Typical RCM signs in pediatrics are not being lively, fatigue and syncope [42,43]. However, studies on clinical presentation of RCM in children are mainly case studies or case series that do not provide an accurate picture of signs and symptoms general to pediatric populations [44].
RCM has the poorest prognosis in all cardiomyopathic disorders. Resultant restrictive hemodynamics often always leads to the development of heart failure and pulmonary hypertension [45]. RCM has the highest recorded rates of sudden cardiac death with reported mortality rates at the second and fifth year at 50% and 70% respectively [45]. Prognosis varies depending on the underlying cause with RCM in the context of cardiac amyloidosis showing worse prognosis [45]. RCM is associated with poor tolerance of medication for cardiovascular conditions and sometimes heart transplantation, which has otherwise had favorable long-term prognosis for other forms of cardiomyopathies such as dilated and hypertrophic cardiomyopathy [46]. However, recent advances made on RCM diagnosis and prompt clinical management have promised a favorable prognosis [47].
In pediatric patients, long-term prognosis varies based on the symptoms at the time of diagnosis or in the setting of pulmonary hypertension [38,45]. About 20% of children diagnosed with RCM including those who have undergone heart transplantation, develop thromboembolic events with a two-year survival rate of 25% to 70% [47]. Irreversible pulmonary hypertension is a major risk factor associated with poorer prognosis in pediatrics. For favorable prognosis, early consideration for heart transplantation is recommended to optimize survival because of uncertainty in the time to acquire a donor heart. In addition, close monitoring of pediatric patients by a cardiologist specializing in pediatric heart failure, arrhythmias, cardiomyopathy and heart transplantation to ensure responsiveness to poor prognosis [48].
Predictors of prognosis refer clinical indicators used to assess disease progression or the efficacy of medical therapy. Age is an important predictor of RCM prognosis. Pediatric patients report poorer prognosis with 50% survival rate in the second year [38]. Ascites, abnormal heart rhythm, thromboembolic events or progressive pulmonary hypertension suggest unfavorable prognosis in pediatrics [44,49]. Prognosis in adult RCM patients is also poor because of progressive deterioration of cardiac function. They usually experience prolonged heart failure and complications associated with cardiac cirrhosis and thromboembolic events. Refractory to pharmacotherapy is another indicator of poor prognosis in adult patients leading to death due to low-cardiac output [47]. Generally, syncope is predictor of prognosis in all RCM patients suggesting disease progression or poor clinical outcomes with reported deaths within four months [50]. RCM in the context of infiltrative and fibrotic cardiac disorders have a poorer prognosis than idiopathic RCM [51]. However, systemic and pulmonary venous congestion, atrial fibrillation at presentation in patients with idiopathic RCM suggest poor prognosis especially for men aged 70 years or above and with higher NYHA class [6,36]. Hemodynamic features especially the extent of elevation of left atrial to aortic root dimension ratio defined by echocardiographic findings at presentation is a strong predictor of poor prognosis. Myocardial contraction fraction, defines as the ratio of stroke volume to left ventricular myocardial volume, is a new prognosis marker with better clinical significance than ventricular ejection fraction in RCM caused by infiltrative disorders [36]. Others predictors of prognosis are the extent of elevation in end-diastolic and pulmonary capillary wedge pressures [42].
RCM is a significant cause of mortality. Its hallmark is restrictive physiology, which has been associated with death or cardiac transplantation in 71.2% of patients, 3.5 times increase in the rate of hospitalization, 3.8 increased in risk of death or aborted sudden cardiac death and 5.7 times increase in the risk of death or heart failure [52]. In most cases, death in RCM patients occurs as sudden cardiac death or congestive heart failure in the setting of chronic cardiac disorders and associated complications but the underlying cause remains unclear, warranting further exploration. However, ischemia, arrhythmias and heart failure have been suspected as possible underlying cause of death in RCM patients [53].
Myocardial ischemia, defined as inadequate blood and oxygen supply to heart muscles, is one of the suspected underlying causes of sudden cardiac death in RCM patients. A study of five sudden cardiac death pediatric patients suggested a correlation between myocardial ischemia and sudden cardiac death. All the five showed signs of syncope, angina and ischemic ECG showing ST depression but without signs of heart failure. Further, autopsy analysis of four hearts revealed acute/chronic ischemia affecting papillary muscles and sub-endocardium [44]. Similar findings were reported in another study [54], which found signs of myocardial ischemia without signs of heart failure in 15 sudden cardiac death patients. However, two other RCM studies [55,56] observed syncope, chest pain and ST segment depression in SCD patients but did not find any histological evidence of ischemia to suggest it is the underlying cause of death [55,56]. Although myocardial ischemia may occur in RCM patients, current evidence is inconclusive as to whether it is an underlying cause of sudden cardiac death in these patients suggesting the need of further autopsy studies in SCD patients or studies on coronary microcirculation to determine the role of ischemia in RCM progression and death.
Ventricular arrhythmias, defined as an abnormal heart rhythm or rate, which can be too fast (tachycardia), too slow (bradycardia) or irregular, originating from the bottom chambers of the heart, is an extensively studied mechanism in cardiac disorders associated with sudden cardiac death [57,58]. Elevated myofilament calcium sensitivity has been suggested as an independent risk factor inducing fatal ventricular arrhythmias [59,60]. Mutations causing RCM have been linked with elevated myocardial calcium sensitivity, increasing susceptibility of inherited/familial RCM patients to fatal ventricular arrhythmias [61]. However, six studies [6,44,49,62,63] observe abnormal electrocardiography (ECG) patterns in almost all (99%) RCM patients but only 15% to 30% had arrhythmias. The six studies suggest atrial fibrillation/flutter are the most common causes of ECG abnormalities encountered in clinical practice while heart block, bradycardia and fatal ventricular arrhythmias are rare. Current research-based evidence is unclear of the role of ventricular arrhythmias in SCD death in RCM patients since ischemia is one of the possible causes of arrhythmias [53,58,59]
Death due to heart failure is a common outcome in RCM patients, which is more severe in familial RCM due to mutation in the genes encoding sarcomere proteins than in on other forms of RCM in the setting of cardiac disorders or systemic diseases [9,43,49,64]. However, the exact role of heart failure in the clinical progression of RCM is not well understood due to the lack of large-scale studies [53]. Current evidence comes from several case reports in pediatric and adult RCM patients. The onset of RCM is often always asymptomatic RCM-related ventricular refilling pattern but with a mild enlargement of the atria, which progresses with symptoms of dyspnea, exercise intolerance and syncope [65-68]. Over time, the symptoms worsen, which includes aggravated impairment of diastolic function and enlargement of the atria ultimately causing fatal heart failure or sudden cardiac death. However, there is inconclusive evidence on whether diastolic dysfunction alone with preserved systolic function throughout RCM progression can develop congestive heart failure. Additional studies are thus warranted to explore the underlying causes of systolic dysfunction in end-stage RCM [37,69].
RCM is a disorder of the myocardium resulting from increased stiffness of the myocardium causing increased ventricular pressure with small increases in ventricular volume. Ventricular filling is accentuated during early diastole terminating at the end of rapid filling phase with ventricular pressure tracings revealing a characteristic diastole dip/plateau pattern mimicking that of constrictive pericarditis [70]. RCM patients present with increased diastolic stiffness leading to reduced ventricular filling and reduced cardiac output. In the early clinical phase, systolic function is preserved in RCM patients and in the case of RCM in the setting of infiltrative cardiac disorders such as amyloidosis and sarcoidosis there may be an increase in wall thickness but not as pronounced as the extent observed in patients with hypertrophic RCM [71].
As RCM progresses, there may be a variable systolic dysfunction accompanied by symptoms of reduced cardiac output including fatigue and lethargy. Increases in ventricular pressure may induce pulmonary and systemic congestion. In the case of RCM affecting both the left ventricle (LV) and/or the right ventricle (RV), symptoms of left or right sided heart failure may manifest. Complete heart block in some patients results from endomyocardial fibrosis on the sino-atrial or atrioventricular nodes [5,6]. Pathology findings further classify RCM into two forms. The first is obliterative RCM, defined as ventricles filled with thrombus. It is a very rare disorder occurring in the end-stage of RCM in the setting of endomyocardial fibrosis extending into atrioventricular valves causing regurgitation [6]. The second RCM form is non-obliterative, defined as progressive myocardial fibrosis in the absence of thrombus, which lacks specific histopathologic changes [71].
Etiologies of RCM may be categorized into two: genetic and non-genetic. Forms of RCM derived their names based on the underlying etiologies [72].
Genetic etiologies inherited in the autosomal dominant pattern with complete penetrance has been demonstrated as one of the causes of idiopathic RCM in 30% of all reported RCM incidents [45]. Genetic etiologies are mutations in the genes encoding sarcomere contractile proteins: troponin T, α-cardiac actin and β-myosin heavy chain [65,73]. Components of sarcomere proteins involved in heart muscle contraction are actin (the thin filament) and myosin (the thick filament). Actin and myosin slide over each other to cause contraction and force development. The movement is regulated in a calcium environment, and activities of troponin complex and tropomyosin located on actin [74]. Also involved in the cardiac contractile regulation are myosin light chains and myosin binding protein. Mutations in genes encoding various sarcomere proteins destroys the integrity of these proteins especially myofibril proteins to induce some forms of cardiomyopathies – restrictive, dilated and hypertrophic cardiomyopathies [75,76].
Mutation screening of individuals and families with RCM have identified several mutations in genes encoding sarcomere proteins, cytoskeletal and Z-line-associated proteins [77]. These mutations include missense mutation in the gene cardiac actin, two different missense mutation in β-myosin gene heavy chain, seven missense mutations and two deletions causing frame-shift mutations and deletion in cardiac troponin T [65-67,78,79]. Mutation screening for genetic mutations especially in the myofibril protein is widely accepted as a method for diagnosis of RCM and stratification of family members at risk of developing idiopathic RCM [53].
Mutations in the DES gene have also been linked with genetic etiology of familial RCM [80,81]. Desmin is an intermediate filament protein connecting myofibril and anchoring them to the sarcomere protein [80]. Desmin-associated myopathies are rare and cause intracytoplasmic accumulation of desmin. Four mutations in the desmin (DES) gene or beta-crystallin (CRYAB) causes accumulation and disorganization of desmin leading to the development of RCM characterized by abnormalities in atrioventricular conduction and skeletal myopathy [81].
Non-genetic etiologies refer to RCM occurring in the setting of local or systemic disorders. Main etiologies for secondary RCM are infiltrative, storage and inflammatory disorders (Figure 1).
Figure 1: Non-Genetic Etiologies of RCM [7].
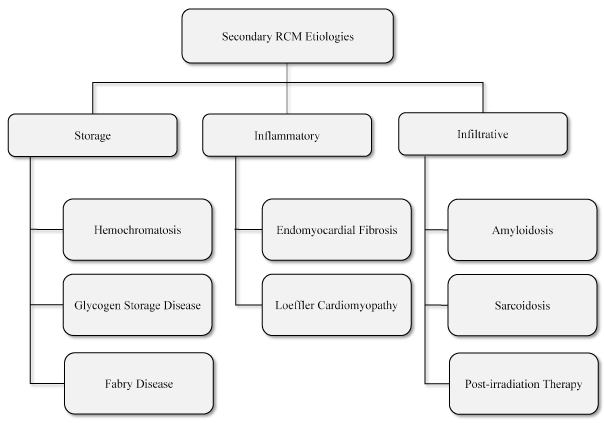
The main non-genetic etiologies of RCM are classified into storage diseases, inflammatory disorders and infiltrative disorders as discussed in text.
Secondary or non-genetic RCM develops in the setting of systemic cardiac disorders. Unlike primary RCM, which affects only the myocardium, secondary RCM affects myocardium and endocardium. Systemic cardiac disorders cause accumulation and deposition of protein substances on cardiac tissues impairing cardia contractility [53]. Storage disorders, hemochromatosis, causes iron accumulation and deposition, lysosomal storage disorders or Fabry disease causes accumulation of glycosphingolipid while glycogen storage disease causes accumulation of glycogen on cardiac tissues. Infiltrative cardiac disorders, amyloidosis, causes deposition of amyloid proteins while sarcoidosis induces RCM through rhythm disturbance by non-caseating granulomas. Radiation, endomyocardial fibrosis and Loeffler disease also cause RCM through fibrotic tissues or endothelial cell damage and subsequent microvascular dysfunction [7,53].
Patients with RCM usually present with heterogonous clinical signs and symptoms that make definitive diagnosis challenging [53]. Some patients are asymptomatic and unaware of any cardiac anomalies until they undergo medical examination based on family history of RCM or symptoms that raise suspicion of RCM [51]. According the ECS and AHA, the diagnostic work-up of RCM should involve multiple methods to arrive at a confirmatory diagnosis. The common diagnostic methods include physical examination, echocardiography, electrocardiography (ECG), cardiac catheterization and endomyocardial biopsy. Differential diagnosis aims to distinguish RCM from other cardiac disorders presenting with similar symptoms especially constrictive pericarditis. Other cardiac conditions are metastases, carcinoid, radiation and constrictive cardiac tamponade, which suggest or raise suspicion of RCM [9] (Table 3).
Table 3: Diagnostic methods and their outcomes raising suspicion of RCM [9].
Diagnosis Method |
Outcomes Raising Suspicion of RCM |
Physical Examination |
Kussmaul’s sign, prominent apical impulse, S3 present, S4 rare, regurgitation murmurs. |
Electrocardiography |
Low voltage (amyloidosis), pseudo-infarction, left-axis deviation, atrial fibrillation, conduction disturbances. |
Echocardiography |
Increased wall thickness, thickened cardiac valves, granular sparkling (common in amyloidosis) |
Doppler Echocardiography |
Reduced RV/LV velocities, augmentation of hepatic vein diastole flow, mitral and tricuspid regurgitation |
Cardiac catheterization |
LVEDP > 5 mm Hg either greater than RVEDP or identical |
Endomyocardial biopsy |
Reveals specific cause of RCM |
CT/MRI |
Reveals normal pericardium to distinguish RCM from constrictive pericarditis (enlarged pericardium). |
CT: Computed Tomography, LV: Left Ventricular, LVEDP: Left Ventricular End-Diastolic Pressure, MRI: Magnetic Resonance Imaging, RV: Right Ventricular, RVEDP: Right Ventricular End-Diastolic
Physical examination looks for extra-cardiac signs, symptoms and manifestations of systemic disorders such as amyloidosis, sarcoidosis or hemochromatosis, which suggest secondary causes of RCM. Physical examination directs particular attention towards assessing signs and symptoms of cardiovascular or respiratory systems [72,82]. General signs and symptoms of RCM include ascites, edema in lower extremities, and enlarged and pulsatile liver, which may be painful. Some patients may experience cardiac cachexia (weight loss due to heart disease). Systemic findings such as periorbital purpura and macroglossia suggests RCM in the setting of amyloidosis [82]. Common physical signs for adult patients with RCM include dyspnea, orthopnea, edema, palpitation, fatigue and chest pains. For pediatric patients, common physical signs for referral to a cardiologist are pulmonary-related, regurgitation murmurs, gallop rhythm, loud P2, ascites, edema and hepatomegaly while syncope, palpitation and chest pains are rare [6,50].
Laboratory tests further assists in detecting secondary causes of RCM or in differentiating RCM from constrictive pericarditis. Tests for complete blood cell count using peripheral smear detects eosinophilia as possible underlying cause [9]. In selected cases, laboratory tests for blood gas analysis (to measure and monitor hypoxia), serum electrolyte, creatinine levels, blood urea nitrogen and liver functional tests. In hemochromatosis-induced RCM, serum iron concentrations, saturation of total iron-binding capacity and levels of ferritin are increased [5]. Assessing the levels of serum brain natriuretic peptide (BNP) should be considered to differentiate RCM from constrictive pericarditis. In RCM, BNP levels are elevated while in constrictive pericarditis, the levels are almost normal [83].
Electrocardiography (ECG) assess electrical/conduction abnormalities. Its findings depend on the stage of the RCM or specific diagnostic tests performed. Findings of ECG are usually abnormal in 99% of RCM patients, especially in patients with idiopathic RCM but the pattern varies greatly [53]. The most frequently encountered ECG abnormality is tall bi-phasic P wave, ST-T wave abnormalities, left/right atrial enlargement but are non-specific. In few RCM patients (less than 10%), especially RCM secondary to amyloidosis-induced, ECG findings may be normal or showing nonspecific changes in ST-T waves [84]. A pseudoinfarct pattern is common in RCM in the setting of myocardial infiltration or ischemia or infarction [41]. Rhythm disorders are frequently diagnosed in ECG with 74% of RCM patients presenting with atrial fibrillation. Further, case reports find Torsades depointe and significant ST depression similar that of ischemia in idiopathic RCM patients [85].
Typical echocardiography findings in RCM patients include enlarged atria. Normal/reduced LVEDD/LVED, normal ventricular wall thickness, restrictive filling pattern and normal systolic function [53] (Figure 2).
Figure 2: Typical Echo Findings in RCM Patients [53].
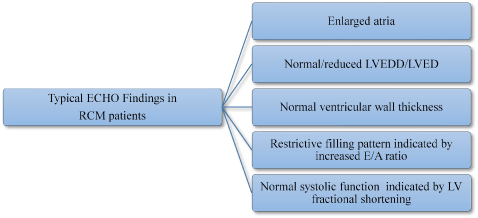
In HCM patients, typical echocardiography findings are enlarge atrial, normal or reduced LVEDD/LVED, normal LV thickness, E/A ratio defined restrictive filling pattern and normal systolic function defines by LV fractional shortening.
The availability and utility of standard echocardiography underlies the recommendation as the primary diagnostic imaging modality for RCM [86]. Echo findings in non-infiltrative RCM (idiopathic or RCM secondary to inflammatory cardiac disorders) show non-dilated, non-hypertrophied and normal contracting LV and large atrial dilation. In infiltrative RCM (secondary to amyloidosis, sarcoidosis and glycogen storage diseases), echo findings reveal increased LV wall thickness, intra-atrial thickening, and normal/reduced ventricular cavity caused by amyloid protein deposits on the myocardium. In RCM secondary to amyloidosis, thrombi may occur in left atrium [87]. Other frequently encountered echo findings include dilation of both atria, thickening of cardiac valves, pericardial effusion and preserved ejection fraction until end-stage RCM [86].
Doppler echocardiography findings reveal restriction in diastolic dysfunction, which in infiltrative RCM precedes reduced LV ejection fraction. Diastolic dysfunction has a correlation with the degree of wall thickening in RCM secondary to amyloidosis. Initial accentuated ventricular diastolic filling (E), reduced deceleration time and atrial filling (A) causes high E/A ratio on mitral inflow velocities. Differences in diastolic blood flow and respiration assist in differential diagnosis of RCM with constrictive pericarditis [86,87]. Figure 3 illustrates echo findings of RCM patients (A) 2-dimensional apical 4-chamber view showing moderate right atria and severely enlarged left atria and (B) pulsed Doppler mitral inflow pattern showing high E/A wave velocity ratio [44].
Figure 3: Echo Findings of RCM Patient [44].
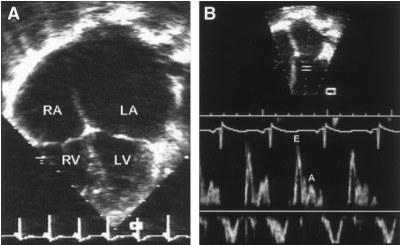
Echo features of RCM. (A) 2-D image (apical 4-chmaber view) demonstrating moderate right and severe left atrial enlargement relative to normal bi-ventricular size and thickness. (B) Pulsed Doppler Mitral inflow pattern consistent with restrictive physiology demonstrating rapid E-wave deceleration phase, high E/A wave velocity ratio and reduced isovolumic relaxation time. LA indicates left atrium; LV, left ventricle; RA, right atrium; and RV, right ventricle.
In addition to standard Doppler, pulsed-wave tissue Doppler provides information about myocardial velocity gradient to measure myocardial contraction and relaxation, and specifically to distinguish RCM from constrictive pericarditis [88]. Novel Echo methods for strain imaging assesses myocardial deformation and changes in contractility revealing initial stages of RCM and especially amyloidosis-induced RCM. Speckle tracking echo assesses global and local ventricular stain to reveal contractile direction enabling differentiation of amyloid-related wall thickness from that of hypertrophic cardiomyopathy [87].
Cardiac catheterization in RCM diagnosis is helpful in tracing ventricular pressure. The method measures increases in right heart pressure indicated by a typical venous wave pattern and dip/plateau contour of ventricular diastolic pressure, which is similar to findings of constrictive pericarditis. These patterns are seen in atrial pressure tracing marked by prominent descent followed a rapid rise to plateau. The differences in RV and LV diastolic pressures is greater than 5 mm Hg, RVSP higher than 50 mm Hg, which is more than two-thirds of RVEDP [88].
Nuclear imaging is rarely used in RCM diagnosis. In RCM secondary to cardiac amyloidosis, nuclear imaging shows increased diffuse uptake of technetium-99m pyrophosphate and indium-111 [88].
Cardiac magnetic resonance imaging (MRI) assess myocardial structure and function to provide useful information – LV mass, volume and contractility, myocardial strain analysis, tissue mapping and extracellular volumes – for the diagnosis of RCM [89]. Cardiac MRI is frequently used to assess myocardial interstitium and has high accuracy in detecting abnormal morphological factors in cardiac amyloidosis [87]. Recent findings suggest contrast (gadolinium) enhanced cardiac MRI reveals a characteristic pattern in sub-endocardium and abnormal myocardial kinetics. For instance, Figure 4 shows late gadolinium enhanced cardiac MRI image of a patient with RCM, short-axis (left) and apical 4-chamber view (right). The arrows indicate regions of local intra-myocardial brightness indicating intra-myocardial fibrosis.
Figure 4: Cardiac MRI with Delayed Gadolinium Enhancement [5].
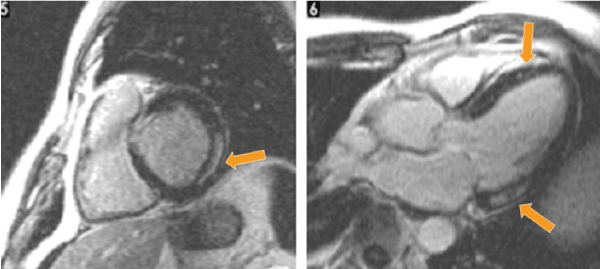
Cardiac MRI with delayed gadolinium enhancement in short axis (left) and apical 4-chamber (right) revealing localized intramyocardial brightness (arrows) suggesting intramyocardial fibrosis.
Novel cardiac MRI techniques using dynamic cine imaging provides clear visibility of all heart chamber enabling tracking of changes in endocardium and epicardium as well as valvular diseases. Same to Echo, cardiac MRI strain analysis is a promising novel method to improve the detection of infiltrative disease causing RCM [88].
Cardiac computed tomography (CT), is a modality for investigating coronary artery disease (CAD) in selected patients with RCM and symptoms of angina-like chest pains. However, the role of cardiac CT in RCM diagnostic remain limited. Same to cardiac MRI, cardiac CT is able to track the distribution of contrast agent in both healthy and diseased cardiac tissues. Cardiac CT thus is an alternative imaging modality for cardiac MRI if it is not available or contra-indicated [88].
Biopsy from RV or LV has been used to determine the presence of myocardial or endocardial disorders in RCM. It has a high diagnostic yield in systemic disorders that cause restrictive hemodynamics. Biopsy is invasive and often always used when non-invasive methods have failed to confirm RCM diagnosis. Myocardial biopsy or fine-needle aspiration of abdominal fat provides useful information for confirmatory diagnosis of primary RCM and amyloidosis-induced RCM using plasma cell disorders. Liver biopsy is used for assessment and diagnosis of hemochromatosis (iron-overload) [88]. Figure 5 illustrates images from endocardial biopsy of specimens from two patients (A and B) with idiopathic RCM.
Restrictive cardiomyopathy (RCM) lacks definitive morphological features. Its diagnosis largely relied on functional and hemodynamic characteristics of the LV. The use of various functional characteristics has seriously undermined the development of standardized diagnostic features because they are not distinctive from constrictive pericarditis [9,53]. The objective of this literature-based meta-analysis was to combine research findings on different methods and the specific LV parameters used in the diagnosis of RCM to improve diagnostic accuracy.
For inclusion in this literature-based meta-analysis, studies were required to meet the following inclusion criteria. (a) Enrolled patients either prospectively or retrospectively; (b) enlisted patients with RCM; (c) used at least one diagnostic method to detect RCM; and (d) reported the findings of the diagnosis. Studies were identified through searching several online medical databases including Biological Abstracts, PubMed, EMBASE, Medline and other non-indexed citations such as Google Scholar. Additional studies not included in the electronic search outcomes were retrieved from screening of bibliographies of the included studies, reviews and unpublished data. The search terms used were restrictive cardiomyopathy, diagnosis, echocardiography, biopsy, cardiac magnetic resonance and histological findings. There was no restriction on publication date, publication language or age of the patients. All patients, adults and children were eligible for inclusion. Data extracted from the included studies were first author, publication year, number of patients recruited, diagnostic method used, LV parameter assessed and a summary of the findings (Table 4).
Table 4: Summary of studies on RCM diagnosis.
1st Author [Ref. No.] |
Year |
Sample |
Method |
Measurement |
Findings |
Palka [54] |
2000 |
15 |
Doppler Echo |
MVG measured from LV posterior wall in early diastole |
MVG measured during rapid ventricular filling/isovolumic relaxation differentiates RCM from CP |
Hatle [90] |
1998 |
12 |
Doppler Echo |
LV isovolumic, relaxation time and in early mitral and tricuspid flow velocities at onset of inspiration |
Respiratory variation in transvalvular flow patterns differentiates CP from RCM. |
Garcia [91] |
1996 |
7 |
Doppler Echo |
Peak early velocity of longitudinal axis expansion (EA) |
RCM has reduced EA compared to CP and normal patients |
Ha [92] |
2004 |
52 |
Doppler Echo |
Early diastolic mitral annular velocity (E`) |
E` velocity higher in CP than in RCM patients useful to distinguish CP from RCM. |
Schoenfeld [93] |
1987 |
38 |
Endomyocardial Biopsy |
Biopsy abnormalities (amyloid/myocarditis) in 39% of patients |
Biopsy is useful to identify patients that thoracotomy should be avoided |
Leya [94] |
2005 |
11 |
Lab tests |
Brain Natriuretic Peptide (BNP) higher in RCM than in CP |
BNP is a useful noninvasive marker for differentiating CP from RCM. |
Parakh [95] |
2015 |
49 |
Lab tests |
NT-pro-BNP significantly elevated in RCM than CP |
NT-pro-BNP could be a useful non-invasive marker for discriminating RCM from CP |
Masui [96] |
1992 |
17 |
Spin-echo MRI |
RV pericardial thickening (low signal intensity) in CP but not in RCM |
Spin eco MRI can be useful in discriminating CP from RCM |
Francone [97] |
2006 |
15 |
Cine-MRI |
Ventricular coupling. Decreased excursion in RCM compared to CP |
2021 Copyright OAT. All rights reserv
Cine-MRI helpful to distinguish CP from RCM especially in normal/mildly thickened pericardium |
Amaki [98] |
2014 |
92 |
Cardiac MRI/Echo |
Pericardial thickness, LV cavity size and enlarged atria |
Cardiac MRI and Echo provide comparable diagnostic information for differentiating CP from RCM |
Cheng [99] |
2011 |
70 |
LGE-MRI |
Pericardial thickening, relative atrial volume and LGE |
Cardiac MRI with LGE and RAR provide additional information to distinguish CP from RCM |
Isner [100] |
1983 |
30 |
Contrast-enhanced Cardiac CT |
Pericardial thickening (over 2mm) in 3 CP patients, none in RCM |
Cardiac CT measurement of pericardial thickening can differentiate CP from RCM |
Weiss [101] |
1995 |
12 |
Cine Cardiac CT |
Thickened pericardium in CP patients |
Cine cardiac CT is a promising imaging tool to differentiate CP from RCM |
CP: Constrictive Pericarditis, MVG: Myocardial Velocity Gradient, RV: Right Ventricular, BNP: Brain Natriuretic Peptide, MRI: Magnetic Resonance Imaging, LGE: Late Gadolinium Enhancement
After screening title, abstract and full article, against the inclusion criteria, thirteen (13) studies were included in the present meta-analysis [54,90-101]. The studies were published between 1983 and 2014 with a combined population of 420 RCM patients. They assessed various methods used in the differential diagnosis of RCM and the focus was largely discriminating RCM from constrictive pericarditis because the two disorders have characteristically indistinguishable hemodynamic abnormalities. The majority of the 13 included studies examined Doppler echocardiography [54,90-92] and cardiac MRI [96-99], while fewer studies investigated laboratory tests [94,95], cardiac CT [100,101], and endomyocardial biopsy [93].
Echocardiography studies assessed hemodynamic properties such as myocardial velocity gradient [54], LV isovolumic and tricuspid flow velocities [90], early velocity of longitudinal axis expansion [91] and early diastolic mitral annular velocity [92]. While theses echo studies suggest differences in hemodynamics between RCM and constrictive pericarditis, the differences are not sufficient for a definitive diagnosis. Advanced imaging techniques such as Cardiac CT and MRI studies assess pericardial thickness providing additional information for differential diagnosis of RCM and constrictive pericarditis as well as aid in the detection of other infiltrative conditions resulting in restrictive hemodynamics [96-101]. In addition, CT provides information in pericardial calcification but it is not definitive since in about 50% of patients with constrictive pericarditis do not develop calcified pericardium [101]. Analysis of myocardial biopsy may provide information for underlying causes and definitive diagnosis of RCM [93]. Finally, laboratory tests on brain natriuretic peptide (BNP) and NT-pro-BNP is proving an important non-invasive marker for discriminating RCM from constrictive pericarditis [94,95]. The high amounts of the two hormones in RCM is due to restrictive effect in the ventricular myocardium causes the atrial to stretch while in constrictive pericarditis there is no stretching of the myocardium [95].
Diagnosis of RCM is challenging due to the lack of definitive morphological features and striking similarity of functional alterations with constrictive pericarditis as well as other cardiac conditions such as metastases, carcinoid, radiation and constrictive cardiac tamponade [9]. In the present meta-analysis, echocardiography imaging is the most common initial imaging modality for assessing alterations in cardiac hemodynamics (functional characteristics) mainly myocardial velocity gradient, LV isovolumic and tricuspid flow velocities, early velocity of longitudinal axis expansion and early diastolic mitral annular velocity [90-92]. However, echocardiography characterization of functional alterations alone does not provide sufficient clues to distinguish RCM from constrictive pericarditis.
Cardiac MRI is an additional imaging modalities used in the visualization and characterization of pericardial thickness to provide complementary diagnostic clue for differentiating RCM from constrictive pericarditis. Cardiac MRI is also useful for excluding other underlying infiltrative disorders that could explain the constrictive hemodynamics [96-101]. Although cardiac CT is not in common use in the diagnostic of RCM, it may be considered to provide additional information on pericardial calcification, which is a feature of constrictive pericarditis but absent in RCM. However, since about half of constrictive pericarditis cases do not exhibit calcified pericardium, it has a limited value in the differential diagnosis of RCM from constrictive pericarditis [95]. To differentiated RCM from constrictive pericarditis, assessment of the levels of BNP and NT-pro-BNP, with RCM having higher levels and consequent stretching of the atrium. Finally, endomyocardial biopsy may also help in differentiating RCM from constrictive pericarditis but has limited use today due to its invasive nature [95].
The present findings confirm that the definitive diagnosis of RCM requires multiple methods, especially in the exclusion of other underlying cardiac disorders and infiltrative diseases that could lead to restrictive hemodynamics. The present findings support the proposals by Kushwaha et al. [9] that diagnosis of RCM lacks a gold standard method. No single method could be used to confirm RCM. Instead, diagnosis requires a combination of diagnostic methods: physical examination, ECG, cardiac imaging methods, cardiac catheterization and endomyocardial biopsy. Physical examination assessed for regurgitation murmurs, ECG for conduction disturbances, echocardiography for alterations in RV/LV velocities, cardiac MRI/CT assessing normal pericardium and endomyocardial biopsy for identifying specific etiologies for RCM [9]. With demonstrable changes in RV/LV hemodynamics and normal pericardium, elevated levels of brain serum – BNP and pro-NT BNP – could exclude constrictive pericarditis and confirm the diagnosis RCM. However, although it is of limited use, endomyocardial biopsy has a high diagnostic yield, and recommended for diagnosis of RCM when imaging modalities provide inconclusive findings [88].
In summary, the mainstay of the diagnosis of RCM is the characterization of RV/LV functional alterations. However, because RCM has striking similarity of restrictive hemodynamics with that caused by constrictive pericarditis and other infiltrative cardiac disorders, its diagnosis is achieved through using a multi-method diagnosis work-up. Imaging methods may reveal changes in RV/LV hemodynamics (echocardiography) and normal pericardium (cardiac MRI/CT), while laboratory examination may reveal elevated levels of BNP and pro-NT BNP for a confirmatory diagnosis. If non-invasive methods do not confirm diagnosis, endomyocardial biopsy may be considered.
Idiopathic RCM has no specific therapy or standardized guidelines. Clinical management targets underlying cardiac disorders and related symptoms such as venous pressure, control heart rate, increase ventricular filling time, maintaining atrial contraction, and correcting atrioventricular disturbances [3]. In many cases, management is difficult because the underlying causes may not respond effectively to therapy. To improve clinical outcomes, the choice of therapy usually depends on clinical condition, symptoms and clinical outcomes of current therapy. The frequently used therapies in the management of RCM are pharmacotherapy, surgery and heart transplantation [90].
Some patients with RCM may develop signs and symptoms of heart failure caused by abnormal contractility of the heart. Such patients usually receive drugs used to treat heart failure such as diuretics, beta-blockers, angiotensin-converting enzyme (ACE) inhibitors and anticoagulants [43].
Diuretics, also referred to as “water pill” are drugs that work by reducing excess fluids in the lungs and other organs by increasing the amount water and salt (urine) removed from the body [38,43]. In the management of RCM, diuretics are used as therapies for venous congestion in pulmonary and systemic circulation [102]. The drugs reduce venous filling pressures causing a decrease in cardiac output and associated signs of fatigue, hypotension and hypoperfusion. In patients with RCM secondary to amyloidosis, diuretics such as Digoxin should be used with caution because they are potentially arrhythmogenic [9].
Beta-adrenergic blocking agents or beta-blockers are cardiac medications used to control heart rates. They slow the heart rate and increase cardiac relaxation time allowing the heart to fill with blood better before each heartbeat [38]. Beta-blockers manage symptoms associated with restrictive hemodynamics and abnormal cardiac contractility [43]. Common beta-blockers include metoprolol, carvedilol and propranolol. Side effects frequently encountered include reduced heart rate, dizziness, low blood pressure and sometimes edema, and fatigue [38]. In RCM patients with atrial fibrillation, beta-blockers in combination with non-dihydropyridine calcium-channel blockers are used to control heart rate [103].
Anticoagulants are drugs that reduce the natural ability of blood to clot. In RCM patients, abnormal contractility especially in pediatrics increases the likelihood of thrombus (blood clot) formation and the risk of a stroke. Thrombus formation could occur in in the atrial appendage increasing the risk of thromboembolic complications. RCM patients with atrial fibrillation and low cardiac output or valvular regurgitation have an increased risk of thrombus formation [38]. In these patients, anticoagulants are prescribed to prevent thrombus formation. Common side effects of anticoagulants in RCM patients include bleeding or bruising from minor skin injuries and possible drug-drug-interaction [43].
Cardiac transplantation is considered for symptomatic RCM pediatric patients with reactive pulmonary hypertension [38] and in patients with idiopathic or familial RCM with refractory symptoms. Cardiac transplantation has also been considered in RCM secondary to sarcoidosis but cases of recurrent sarcoid granulomata have been reported in the transplanted heart. Cardiac transplantation is often not indicated for RCM secondary to systemic disorders such as amyloidosis [104]. The disadvantage with cardiac transplantation is donor shortage, a long waiting time and development of irreversible pulmonary hypertension [105]. Others are infection risks during operation, long-term complications, risk of organ rejection and coronary artery disease [38].
The use of left ventricular assist device (LVAD) the treatment and management of end-stage dilated and ischemic cardiomyopathy has been increasingly but its application in RCM has not received sufficient research support. However, one small study recruiting 83 patients observed axial flow LVAD therapy benefits RCM patients with end stage heart failure similar to patients with hypertrophic cardiomyopathy [105]. Prospective clinical trials are warranted to recommend LVAD therapy for end stage RCM.
Clinical management of RCM lacks standardized guidelines and current treatment protocols target correction of cardiac hemodynamics and/or its symptoms. The present literature-based meta-analysis combines research findings on clinical management methods of RCM to advance understanding on the common methods and their efficacy in the prevention or treatment of RCM.
Studies were included in the present literature-based meta-analysis if they: (a) were prospective or retrospective trials; (b) enrolled patients diagnosed with RCM; (c) used at least one clinical management method for RCM; and (d) recorded clinical outcomes of the clinical management methods used. Studies were searched on several online electronic databases: PubMed, EMBASE, Medline and Google Scholar. The search terms used were restrictive cardiomyopathy, treatment, medication, cardiac/heart transplantation and device therapy. Additional studies were retrieved from screening of bibliographies of included studies and review articles. All articles irrespective of publication date, language and age of patients were included. Studies with only abstract with insufficient data for extraction, provided, case series recruiting one or very few patients, and conference papers because they are not final (subject to changes) were excluded. Data extracted included name of first author, publication year, clinical management method, survival or improvement rate, and a summary of findings (Table 5).
Table 5: Summary of studies on HCM clinical management.
1st Author [Ref. No.] |
Year |
Sample |
Method |
Clinical Endpoint |
Survival/ Improvement |
Main Findings |
Kimberling [105] |
2002 |
8 |
CTx |
Elevated Pulmonary vascular resistance (PVR) |
37.5% had elevated PVR successfully reversed by nitric oxide |
Cardiac transplantation can be successfully done in pediatric patients showing reversibility of PVR |
|
Al-Kindi [106] |
2016 |
45,041 |
CTx |
LoS, early mortality, survival rates |
Radiation induced RCM had LoS and survival rates that other RCM and other etiologies |
End-stage radiation induced RCM have higher early post-operative mortality than other RCM and heart etiologies. |
|
DePasquale [107] |
2012 |
38,190 |
CTx |
Mortality, survival rate |
Mortality and survival rates of RCM comparable to non-RCM except radiation/amyloid RCM |
CTX has sub-optimal outcomes for RCM secondary to amyloid disease or radiation. |
|
Oliveira [108] |
2012 |
232 |
CTx |
Short/long-term post-transplant survival |
Higher infection rates/malignancies but comparable survival rates RCM and non-ischemic cardiomyopathy |
CTx improves survival rates for chemotherapy-induced RCM |
|
Roig [109] |
2009 |
25 |
CTx |
Post-transplant survival |
60% mortality rate at 1.3 years. |
Poor CTx outcomes for amyloid-induced RCM. |
|
Davis [110] |
2014 |
19 |
CTx |
Post-transplant survival |
89.5% survival rate at 1.3 years |
Patient selected and aggressive control of amyloid light chains, CTx could have better outcomes for amyloid induced RCM. |
|
Oliveira [111] |
2014 |
3,812 |
MCS |
Survival rate |
Similar survival rates with transplantation or RV assist device support |
Patients with radiation induced RCM have similar survival rate with other MCS patients. |
|
Topilsky [112] |
2011 |
83 |
LVAD |
1-year actuarial survival rates and Los |
No difference with HCM/DCM on survival/hospitalization |
Continuous axial flow LVAD has beneficial outcomes in treating end stage heart failure due to RCM |
|
LoS: Length of Hospital Stay, CTx: Cardiac Transplantation, MCS: Mechanical Circulatory Support, LVAD: Left Ventricular Assist
After screening, titles, abstracts and full article against the inclusion criteria, eight (8) studies were included [105-112] published between 2002 and 2016. In total, the eight studies recruited 87,410 patients. The main clinical endpoints assessed were elevated pulmonary vascular resistance [105], length of hospital stay [106], survival rates [105,111], mortality rates [105], and post-transplant survival rates [108-110]. A majority of the studies investigated long-term outcomes of surgical procedures particularly cardiac transplantation (CTx) [105-110] and mechanical circulatory support, and right or left ventricular assist device [111,112]. Cardiac transplantation has been demonstrated to have beneficial effect on 1-5 year survival rates of 40-89.5% on patients with RCM. However, patients with RCM due to radiation [106,108,111] or amyloidosis [107,109] have poorer outcomes suggesting the need to consider genetic or idiopathic RCM for cardiac transplantation due to shortages and long wait times for donors. However, patient selection and aggressive therapies to reduce amyloid depositions on myocardial tissues suggest improvement of short-term (380 days) survival rates [110]. In addition, left ventricular assist devices has been shown to improve survival rates of RCM similar to that of dilated and hypertrophic cardiomyopathy. However, due to the small population studied, there is need for clinical trials recruiting larger samples to demonstrate chronic therapeutic value of left ventricular assist devices in the long-term survival or RCM patients [112].
Pharmacotherapy of RCM is an important initial clinical management approach of RCM targeting the treatment of venous congestion (diuretics) [102], control heart rate (beta-adrenergic blocking agents) [38,43], and prevention of blood clotting and the risk for stroke due to restrictive hemodynamics (anticoagulants) [43]. However, due to poor tolerance for medication [45], the present meta-analysis focused on chronic treatment of RCM using non-pharmacological support. Non-medical therapy are usually indicated for symptomatic RMC patients or symptoms refractory to medical therapy. Cardiac transplantation is an effective chronic RCM therapy with favorable post-transplant survival rates of up to 89.5% at 1.3 years. It reverses elevated pulmonary vascular resistance and improved survival on radiation-induced RCM. However, cardiac transplantation is not universal and mainly contra-indicated in some cohorts of RCM patients such as amyloid-induced RCM, and suffers the disadvantage of donor shortage, high infection rates and malignancies [107,108]. Besides cardiac transplantation, device therapy such as LV assist devices and mechanical circulatory support have been indicated for RCM patients [111,112]. Device therapy have comparable survival rates with cardiac transplantation [111], and has no differences on survival rates compared to HCM and DCM phenotypes. The main indication of continuous axial flow LV assist devices is RCM patients with end-stage heart failure [112].
The present findings support earlier studies [45,46] reporting that although pharmacotherapy is readily available and a preferred first-line of RCM treatment, RCM has a general ominous prognosis due to restrictive hemodynamics leading to pulmonary hypertension and progressive heart failure, and a demonstrated poor tolerance to medical therapy [45,46]. Moreover, RCM has one of the leading recorded rates of sudden cardiac death of up to 70% in the fifth year in diagnosed cardiomyopathies [45]. The development of surgical treatment approaches using either device therapy or cardiac transplantation could improve long-term survival rates for RCM patients [105-107]. However, surgical treatment has important limitations such as contra-indication RCM secondary to systemic diseases such as amyloidosis, and has procedural complication, reducing their effectiveness in the long-term. To improve clinical management, there is need for improvement in diagnosis to enable early diagnosis and to support etiologic-specific treatment. In summary, clinical management of RCM targets on relieving restrictive filling and associated symptoms. The main treatment options are medical therapy and surgical through LV assist devices and cardiac transplantation. However, prognosis of RCM remains ominous warranting additional studies to investigate diagnosis and support development of standardized guidelines targeting the underlying etiology.
Restrictive cardiomyopathy (RCM) represents one of the three phenotypes of cardiomyopathy originally recognized by the World Health Organization (WHO) and the International Society and Federation of Cardiology (ISFC) Task Force on classification of cardiomyopathies in 1994. Of the three phenotypes, it is the least common accounting for less than 5% of the total diagnosed cases of cardiomyopathies. Its two main classifications are primary RCM occurring in the setting of definitive genetic mutations and secondary (non-genetic) RCM occurring in the setting of cardiac or systemic disorders affecting the heart. Its clinical manifestation varies with age with adults presenting with cardiopulmonary symptoms (paroxysmal nocturnal dyspnea, fatigue and exercise intolerance and orthopnea) and children with presenting with symptoms unrelated to cardiac problems such as lung-related problems such as repeated infections or asthma. Its prognosis is ominous with the highest recorded rates of sudden cardiac death due to myocardial ischemia, lethal ventricular arrhythmias and heart failure. Its main etiologies are mutations in the genes encoding sarcomere proteins that cause severe fibrosis or gene defects from secondary cardiac disorders leading to extracellular accumulation of substrates such as iron, amyloid, glycolipid and desmin. Its principal pathophysiologic mechanisms is the stiffening of ventricular walls caused by extracellular accumulation of substrates or infiltrative diseases such as myocardial fibrosis. The mainstay of diagnosis is characterizing RV/LV dysfunction obtained through a combination of laboratory tests, electrocardiography, echocardiography and cardiac imaging. RCM has no specific therapy but the principal clinical management approaches include pharmacotherapy and surgical interventions, which focus on managing symptoms of heart failure, arrhythmias and hypertension.
References
- Goodwin JF, Gordon H, Hollman A, Bishop MB (1961) Clinical aspects of cardiomyopathy. Br Med J 1: 69-79. [Crossref]
- Brandenburg RO, Chazov E, Cherian G, Falase AO, Grosgogeat Y, Kawai C (1981) Report of the WHO/ISFC task force on definition and classification of cardiomyopathies. Circulation 64: 437A-438A.
- Abelmann WH (1984) Classification and natural history of primary myocardial disease. Prog Cardiovasc Dis 27: 73-94. [Crossref]
- McKenna RP, Bristow M, Maisch B, Mautner B (1996) Report of the 1995 World Health organization/International Society and Federation of cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Nihoyannopoulos P, Dawson D (2009) Restrictive cardiomyopathies. Eur J Echocardiogr 10: iii23-33. [Crossref]
- Ammash NM, Seward JB, Bailey KR, Edwards WD, Tajik, AJ (2000) Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation 101: 2490-2496. [Crossref]
- Mogensen J, Arbustini E (2009) Restrictive cardiomyopathy. Curr Opin Cardiol 24: 214-220. [Crossref]
- Rapezzi C, Ortolani P, Binetti G, Picchio FM, Magnani B (1990) Idiopathic restrictive cardiomyopathy in the young: report of two cases. Int J Cardiol 29: 121-126. [Crossref]
- Kushwaha SS, Fallon JT, Fuster V (1997) Restrictive cardiomyopathy. N Engl J Med 336: 267-276. [Crossref]
- Angelini A, Calzolari V, Thiene G, Boffa GM, Valente M, et al. (1997) Morphologic spectrum of primary restrictive cardiomyopathy. Am J Cardiol 80: 1046-1050. [Crossref]
- Katritsis D, Wilmshurst PT, Wendon JA, Davies MJ, Webb-Peploe MM (1991) Primary restrictive cardiomyopathy: clinical and pathologic characteristics. J Am Coll Cardiol 18: 1230-1235. [Crossref]
- Ishiwata S, Nishiyama S, Seki A, Kojima S (1993) Restrictive cardiomyopathy with complete atrioventricular block and distal myopathy with rimmed vacuoles. Japanese Circulation Journ 57: 28-933.
- Fitzpatrick AP, Shapiro LM, Rickards AF, Poole-Wilson, PA (1990) Familial restrictive cardiomyopathy with atrioventricular block and skeletal myopathy. Heart 63: 114-118.
- Pepys MB (1995) Amyloidosis. In: Weatherall DJ, Ledingham JGG, Warrell DA (Eds) Oxford Textbook of Medicine. Oxford: University Press, 2: 1512-1524.
- Smith TJ, Kyle RA, Lie JT (1984) Clinical significance of histopathologic patterns of cardiac amyloidosis. Mayo Clin Proc 59: 547-555. [Crossref]
- Kinoshita OS, Hongo MI, Yamada HI, Misawa TA, Kono J, et al. (1989) Impaired left ventricular diastolic filling in patients with familial amyloid polyneuropathy: a pulsed Doppler echocardiographic study. Heart 61: 198-203.
- Hongo M, Fujii T, Hirayama J, Kinoshita O, Tanaka M, et al. (1989) Radionuclide angiographic assessment of left ventricular diastolic filling in amyloid heart disease: a study of patients with familial amyloid polyneuropathy. J Am Coll Cardiol 13: 48-53. [Crossref]
- Lool LM (1993) Isolated atrial amyloidosis: a clinicopathologic study indicating increased prevalence in chronic heart disease. Human Pathology 24: 602-607.
- Angomachalelis N1, Hourzamanis A, Vamvalis C, Gavrielides A (1992) Doppler echocardiographic evaluation of left ventricular diastolic function in patients with systemic sarcoidosis. Postgrad Med J 68 Suppl 1: S52-56. [Crossref]
- Perry A, Vuitch, F (1995) Causes of death in patients with sarcoidosis. A morphologic study of 38 autopsies with clinicopathologic correlations. Arch Pathol Lab Med 119: 167-172. [Crossref]
- Gibbons WJ, Levy RD, Nava S, Malcolm I, Marin JM, et al. (1991) Subclinical cardiac dysfunction in sarcoidosis. Chest 100: 44-50. [Crossref]
- Dubrey SW, Bell A, Mittal TK (2007) Sarcoid heart disease. Postgrad Med J 83: 618-623. [Crossref]
- Doughan AR, Williams BR (2006) Cardiac sarcoidosis. Heart 92: 282-288. [Crossref]
- Pietrangelo A (2004) Hereditary hemochromatosis--a new look at an old disease. N Engl J Med 350: 2383-2397. [Crossref]
- Linhart A, Elliott PM (2007) The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 93: 528-535. [Crossref]
- Furth PA, Futterweit W, Gorlin, R (1985) Case report: Refractory biventricular heart failure in secondary hemochromatosis. Am J Med Sci 290: 209-213.
- Zarate YA, Hopkin RJ (2008) Fabry's disease. Lancet 372: 1427-1435. [Crossref]
- Clarke JT (2007) Narrative review: Fabry disease. Ann Intern Med 146: 425-433. [Crossref]
- Linhart A, Lubanda JC, Palecek T, Bultas J, Karetova D, et al. (2001) Cardiac manifestations in Fabry disease. J Inherit Metabol Dis 24: 75-83.
- Cantor WJ, Butany J, Iwanochko M, Liu P (1998) Restrictive cardiomyopathy secondary to Fabry's disease. Circulation 98: 1457-1459. [Crossref]
- Patel P, Wan SH, Sinak L (2015) Glycogen storage disease induced restrictive cardiomyopathy. J Am Coll Cardiol 65: A609.
- Sisakian H (2014) Cardiomyopathies: Evolution of pathogenesis concepts and potential for new therapies. World J Cardiol 6: 478-494. [Crossref]
- Hers, HG (1963) a-Glucosidase deficiency in generalized glycogen-storage disease (Pompe's disease). Biochem J 86: 11. [Crossref]
- Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, et al. (2000) Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. New Eng J Med 342: 1077-1084.
- Goodwin JF (1992) Cardiomyopathies and specific heart muscle diseases. Definitions, terminology, classifications and new and old approaches. Postgrad Med J 68: S3-6. [Crossref]
- Lewis AB (1992) Clinical profile and outcome of restrictive cardiomyopathy in children. Am J Heart123: 1589-1593.
- Cetta F, O'leary PW, Seward JB, Driscoll DJ (1995) Idiopathic restrictive cardiomyopathy in childhood: diagnostic features and clinical course. Mayo Clin Proc 70: 634-640.
- American Heart Association (2017) Restrictive cardiomyopathy. Retrieved from https://www.heart.org/idc/groups/heart-public/@wcm/@hcm/documents/downloadable/ucm_312227.pdf
- Zwas DR, Gotsman I, Admon D, Keren A (2012) Advances in the differentiation of constrictive pericarditis and restrictive cardiomyopathy. Herz 37: 664-673. [Crossref]
- Mohmand-Borkowski A, Tang, WW (2014) Atrial fibrillation as manifestation and consequence of underlying cardiomyopathies: from common conditions to genetic diseases. Heart Failure Reviews 19: 295-304.
- Niemann, JT (2016) Cardiomyopathies and pericardial disease. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Ed. New York, NY: McGraw-Hill, 380-387.
- Russo LM, Webber SA (2005) Idiopathic restrictive cardiomyopathy in children. Heart 91: 1199-1202. [Crossref]
- Chen SC, Balfour IC, Jureidini S (2001) Clinical spectrum of restrictive cardiomyopathy in children. J Heart Lung Transplant 20: 90-92. [Crossref]
- Rivenes SM, Kearney DL, Smith EB, Towbin JA, Denfield, SW (2000) Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 102: 876-882.
- Huby AC, Mendsaikhan U, Takagi K, Martherus R, Wansapura J, et al. (2014) Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J Am Coll Cardiol 6: 2765-2776.
- Esplin BL, Gertz MA (2013) Current trends in diagnosis and management of cardiac amyloidosis. Curr Probl Cardiol 38: 53-96. [Crossref]
- Sharma N, Howlett J (2013) Current state of cardiac amyloidosis. Curr Opin Cardiol 28: 242-248. [Crossref]
- Chaulagain CP, Comenzo RL (2013) New insights and modern treatment of AL amyloidosis. Curr Hematol Malig Rep 8: 291-298.
- Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, et al. (2008) Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 94: 1478-1484.
- Denfield SW, Rosenthal G, Gajarski RJ, Bricker JT, Schowengerdt KO, et al. (1997) Restrictive cardiomyopathies in childhood. Etiologies and natural history. Texas Heart Institute Journal, 24: 38.
- Benotti JR, Grossman W, Cohn PF (1980) Clinical profile of restrictive cardiomyopathy. Circulation 61: 1206-1212. [Crossref]
- Maskatia SA, Decker JA, Spinner JA, Kim JJ, Price JF, et al. (2012) Restrictive physiology is associated with poor outcomes in children with hypertrophic cardiomyopathy. Pediatr cardiol 33: 141-149.
- Jean-Charles PY, Li YJ, Nan CL, Huang XP (2011) Insights into restrictive cardiomyopathy from clinical and animal studies. Journal of Geriatric Cardiology, 8: 168-183.
- Palka P, Lange A, Donnelly JE, Nihoyannopoulos P (2000) Differentiation between restrictive cardiomyopathy and constrictive pericarditis by early diastolic Doppler myocardial velocity gradient at the posterior wall. Circulation 102: 655-662.
- Rai TS, Ahmad S, Ahluwalia TS, Ahuja M, Bahl A, et al. (2009) Genetic and clinical profile of Indian patients of idiopathic restrictive cardiomyopathy with and without hypertrophy. Mol Cell Biochem 331: 187-192. [Crossref]
- Hayashi T, Tsuda E, Kurosaki K, Ueda H, Yamada O, Echigo S (2007) Electrocardiographic and clinical characteristics of idiopathic restrictive cardiomyopathy in children. Circulation 71: 1534-1539.
- National Heart, Lung and Blood Institute (2011) What is arrhythmias? Available at https://www.nhlbi.nih.gov/health/health-topics/topics/arr
- Zipes DP, Wellens HJ (1998) Sudden cardiac death. Circulation 98: 2334-2351. [Crossref]
- Baudenbacher F1, Schober T, Pinto JR, Sidorov VY, Hilliard F, et al. (2008) Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118: 3893-3903. [Crossref]
- Huke S, Knollmann BC (2010) Increased myofilament Ca 2+-sensitivity and arrhythmia susceptibility. J Mol Cel Cardiol 48: 824-833.
- Yumoto F, Lu QW, Morimoto S, Tanaka H, Kono N, et al. (2005) Drastic Ca 2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem Biophys Res Comm 338: 1519-1526.
- Denfield SW (2002) Sudden death in children with restrictive cardiomyopathy. Cardiac Electrophysiology Review 6: 163-167.
- Weller RJ, Weintraub R, Addonizio LJ, Chrisant MR, Gersony WM, et al. (2002) Outcome of idiopathic restrictive cardiomyopathy in children. Am J Cardiol 90: 501-506. [Crossref]
- Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, et al. (2007) Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol 49: 2419-2426.
- Mogensen J1, Kubo T, Duque M, Uribe W, Shaw A, et al. (2003) Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 111: 209-216. [Crossref]
- Ware SM, Quinn ME, Ballard ET, Miller E, Uzark K, Spicer RL (2008) Pediatric restrictive cardiomyopathy associated with a mutation in ß-myosin heavy chain. Clin gen 73: 165-170.
- Peddy SB, Vricella LA, Crosson JE, Oswald GL, Cohn RD, et al. (2006) Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatr 117: 1830-1833.
- Weller RJ, Weintraub R, Addonizio LJ, Chrisant MR, Gersony WM, et al. (2002) Outcome of idiopathic restrictive cardiomyopathy in children. Am J Cardiol 90: 501-506. [Crossref]
- Menon SC, Michels VV, Pellikka PA, Ballew JD, Karst ML, et al. (2008) Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin gen 74: 445-454.
- Higano ST, Azrak E, Tahirkheli NK, Kern MJ (1999) Hemodynamic rounds series II: hemodynamics of constrictive physiology: influence of respiratory dynamics on ventricular pressures. Catheter Cardiovasc Interv 46: 473-486. [Crossref]
- Katritsis D, Wilmshurst PT, Wendon JA, Davies MJ, Webb-Peploe MM (1991) Primary restrictive cardiomyopathy: clinical and pathologic characteristics. J Am Coll Cardiol 18: 1230-1235. [Crossref]
- Davies MJ, Mann JM (1995) Systemic pathology. The Cardiovasc Syst 10: 1409-1416.
- Garcia MJ (2016) Constrictive pericarditis versus restrictive cardiomyopathy? J Am Coll Cardiol 67: 2061-2076.
- Towbin JA (1998) The role of cytoskeletal proteins in cardiomyopathies. Curr Opin Cell Biol 10: 131-139. [Crossref]
- Chang AN, Potter JD (2005) Sarcomeric protein mutations in dilated cardiomyopathy. Heart Failure Rev 10: 225-235. [Crossref]
- Xu Q, Dewey S, Nguyen S, Gomes AV (2010) Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J Mol Cell Cardiol 48: 899-909. [Crossref]
- Kostareva A, Kiselev A, Gudkova A, et al. (2016) Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS One 11: e0163362. [Crossref]
- Karam S, Raboisson MJ, Ducreux C, Chalabreysse L, Millat G, Bozio A, Bouvagnet P (2008) A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congen Heart Dis 3:138-143.
- Pinto JR, Parvatiyar MS, Jones MA, Liang J, Potter JD (2008) A troponin T mutation that causes infantile restrictive cardiomyopathy increases Ca2+ sensitivity of force development and impairs the inhibitory properties of troponin. J Biol Chem 283: 2156-2166. [Crossref]
- Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, et al. (2006) Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Euro Jour Heart Failure 8: 477-483. [Crossref]
- Arbustini E, Morbini P, Grasso M, Fasani R, Verga L, et al. (1998) Restrictive cardiomyopathy, atrioventricular block and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J Am Coll Cardiol 31: 645-653. [Crossref]
- Wald DS, Gray HH (2003) Restrictive cardiomyopathy in systemic amyloidosis. QJM 96: 380-382. [Crossref]
- Leya FS, Arab D, Joyal D, Shioura KM, Lewis BE, Steen LH, Cho L (2005) The efficacy of brain natriuretic peptide levels in differentiating constrictive pericarditis from restrictive cardiomyopathy. J Am Coll Cardiol 45: 1900-1902. [Crossref]
- Hayashi T, Tsuda E, Kurosaki K, Ueda H, Yamada O, Echigo S (2007) Electrocardiographic and clinical characteristics of idiopathic restrictive cardiomyopathy in children. Circulation 71: 1534-1539. [Crossref]
- Selvaganesh M, Arul AS, Balasubramanian S, Ganesan N, Naina Mohammed S, et al. (2015) An unusual ECG pattern in restrictive cardimyopathy. Indian Heart J 67: 362-367. [Crossref]
- White JA, Fine NM (2016) Recent advances in cardiovascular imaging relevant to the management of patients with suspected cardiac amyloidosis. Curr Cardiol Rep 18: 1-11.
- Falk RH, Quarta CC (2015) Echocardiography in cardiac amyloidosis. Heart Fail Rev 20: 125-131. [Crossref]
- Reardon L (2017) Restrictive Cardiomyopathy Workup. Medscape. Available at http://emedicine.medscape.com/article/153062-workup#showall
- Saeed M, Liu H, Liang CH, Wilson MW (2017) Magnetic resonance imaging for characterizing myocardial diseases. Int J Cardiovasc Imaging 1-20. [Crossref]
- Hatle LK, Appleton CP, Popp RL (1989) Differentiation of constrictive pericarditis and restrictive cardiomyopathy by Doppler echocardiography. Circulation 79:357-370. [Crossref]
- Garcia MJ, Rodriguez L, Ares M, Griffin BP, Thomas JD, Klein AL (1996) Differentiation of constrictive pericarditis from restrictive cardiomyopathy: assessment of left ventricular diastolic velocities in longitudinal axis by Doppler tissue imaging. J Am Coll Cardiol 27: 108-114. [Crossref]
- Ha JW, Ommen SR, Tajik AJ, Barnes ME, Ammash NM, et al. (2004) Differentiation of constrictive pericarditis from restrictive cardiomyopathy using mitral annular velocity by tissue Doppler echocardiography. Am J Cardiol 94: 316-319. [Crossref]
- Schoenfeld MH, Supple EW, Dec GW, Fallon JT, Palacios IF (1987) Restrictive cardiomyopathy versus constrictive pericarditis: Role of endomyocardial biopsy in avoiding unnecessary thoracotomy. Circulation 75: 1012-1017. [Crossref]
- Leya FS, Arab D, Joyal D, Shioura KM, Lewis BE, Steen LH, Cho L (2005) The efficacy of brain natriuretic peptide levels in differentiating constrictive pericarditis from restrictive cardiomyopathy. J Am Coll Cardiol 45: 1900-1902. [Crossref]
- Parakh N, Mehrotra S, Seth S, Ramakrishnan S, Kothari SS, et al. (2015) NT pro B type natriuretic peptide levels in constrictive pericarditis and restrictive cardiomyopathy. Ind Heart Jour 67: 40-44. [Crossref]
- Masui T, Finck S, Higgins CB (1992) Constrictive pericarditis and restrictive cardiomyopathy: evaluation with MR imaging. Radiol 182: 369-373. [Crossref]
- Francone M, Dymarkowski S, Kalantzi M, Rademakers FE, Bogaert J (2006) Assessment of ventricular coupling with real-time cine MRI and its value to differentiate constrictive pericarditis from restrictive cardiomyopathy. Europ Radiol 16: 944-951. [Crossref]
- Amaki M, Savino J, Ain DL, Sanz J, Pedrizzetti G, et al. (2014) Diagnostic Concordance of Echocardiography and Cardiac Magnetic Resonance–Based Tissue Tracking for Differentiating Constrictive Pericarditis from Restrictive Cardiomyopathy. Circ Cardiovasc Imaging 7: 819-827. [Crossref]
- Cheng H1, Zhao S, Jiang S, Lu M, Yan C, et al. (2011) The relative atrial volume ratio and late gadolinium enhancement provide additive information to differentiate constrictive pericarditis from restrictive cardiomyopathy. J Cardiovasc Magn Reson 13: 15. [Crossref]
- Isner JM, Carter BL, Bankoff MS, Pastore JO, Ramaswamy K, et al. (1983) Differentiation of constrictive pericarditis from restrictive cardiomyopathy by computed tomographic imaging. Am Heart Jour 105: 1019-1025.
- Weiss RM, Otoadese T, Oren RM (1995) Differentiation of constrictive pericarditis from restrictive cardiomyopathy: the case for high-resolution dynamic tomographic imaging. Med Imaging 2433: 329-336.
- Leviner DB, Hochhauser E, Arad M (2015) Inherited cardiomyopathies--Novel therapies. Pharmacol Ther 155: 36-48. [Crossref]
- Mohmand-Borkowski A, Tang WW (2014) Atrial fibrillation as manifestation and consequence of underlying cardiomyopathies: from common conditions to genetic diseases. Heart Failure Reviews 19: 295-304.
- Oni AA, Hershberger RE, Norman DJ, (1992) Recurrence of sarcoidosis in a cardiac allograft: Control with augmented corticosteroids. J Heart Lung Transplant 11: 367-369.
- Kimberling MT, Balzer DT, Hirsch R, Mendeloff E, Huddleston CB, Canter CE (2002) Cardiac transplantation for pediatric restrictive cardiomyopathy: presentation, evaluation, and short-term outcome. J Heart Lung Transplant 21: 455-459.
- Al-Kindi SG, Oliveira GH (2016) Heart transplantation outcomes in radiation-induced restrictive cardiomyopathy. J Cardiac Failure 22: 475-478. [Crossref]
- DePasquale EC, Nasir K, Jacoby DL (2012) Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant 31: 1269-1275. [Crossref]
- Oliveira GH, Hardaway BW, Kucheryavaya AY, Stehlik J, Edwards LB, Taylor DO (2012) Characteristics and survival of patients with chemotherapy-induced cardiomyopathy undergoing heart transplantation. J Heart Lung Transplant 31: 805-810. [Crossref]
- Roig E, Almenar L, González-Vílchez F, Rabago G, Delgado J, et al. (2009) Outcomes of heart transplantation for cardiac amyloidosis: subanalysis of the spanish registry for heart transplantation. Am J Transplant 9: 1414-1419. [Crossref]
- Davis M, Kale P, Witteles RM (2014) Outcomes After Heart Transplantation for Amyloid Cardiomyopathy in the Modern Era. J Heart Lung Transplant 33: S51. [Crossref]
- Oliveira GH, Dupont M, Naftel D, Myers SL, Yuan Y, et al. (2014) Increased need for right ventricular support in patients with chemotherapy-induced cardiomyopathy undergoing mechanical circulatory support: outcomes from the INTERMACS Registry (Interagency Registry for Mechanically Assisted Circulatory Support) J Am Coll Cardiol 63: 240-248. [Crossref]
- Topilsky Y, Pereira NL, Shah DK, Boilson B, Schirger JA, et al. (2011) LVAD therapy in patients with restrictive and hypertrophic cardiomyopathy. Circulation: Heart Failure, CIRCHEARTFAILURE-110. [Crossref]