Abstract
Appropriate production of mucous plays a critical role in host innate mucosal defenses against infection. However, if uncontrolled, excessive mucous may be detrimental to the host. Mucous production thus must be tightly regulated. In the present study, we investigate the role of RIP-2 in regulating nontypeable Haemophilus influenzae (NTHi)-induced up-regulation of mucin MUC5AC expression. We show that RIP-2 is a negative regulator of MUC5AC, whereas MAP kinase JNK acts as a positive regulator for MUC5AC induction by NTHi. Our studies unveil a novel role for RIP-2 in controlling mucous production in upper respiratory disease and may shed light on the identification of new therapeutic targets.
Key words
nontypeable Haemophilus influenzae, MUC5AC, negative regulation, RIP-2, JNK
Introduction
Mucosal barriers in the upper respiratory tract, including the middle ear, typically aide in trapping and clearance of infectious agents as a protective mechanism for the host [1]. Mucins are a major component of mucous in these regions. Mucins are a family of high molecular weight glycoproteins secreted by goblet cells in mucosal tissues. There are more than 20 known mucins, twelve of which have been found in the respiratory tract [2]. Mucin 5AC (MUC5AC) is known to be one of the major mucins produced during upper respiratory infections, including otitis media (OM). While appropriate mucous production is protective for the host, excessive mucous production can be deleterious [1-4]. In fact, thick mucous and neutrophil nets can create a biofilm-like environment for nontypeable Haemophilus influenzae (NTHi), one of the major bacterial pathogens of OM and chronic obstructive pulmonary disease (COPD). Overproduction of mucin MUC5AC can lead to persistent, chronic infections or can worsen acute tissue injury [1, 5]. Therefore, like inflammation, mucous production must be tightly controlled.
NTHi-caused upper respiratory infections, including COPD and OM, remain major health problems worldwide. COPD is the third leading cause of death in the U.S. with numbers rising each year. Hassett et al. reported the annual number of Americans affected by COPD in 2014 at over 14 million [6]. OM is the most common pediatric infectious disease in humans accounting for over 20 million physician visits and costing the U.S. upwards of 5 billion dollars annually [7, 8]. OM is characterized by mucous overproduction and inflammation in the middle ear cavity due to infection. Pediatric patients are highly susceptible to OM pathogens due to the shortened length of the Eustachian tube during development [7]. Currently, the standard treatment for both COPD and OM is systemically administered broad-spectrum antibiotics, which, leads to increased antibiotic resistance. Surgical intervention is also frequently utilized in pediatric OM patients by placing tubes to aid in drainage of the middle ear cavity. This commonplace surgical intervention and frequent use of antibiotics warrants concern for the development of drug-resistant strains of bacteria as well as the rising healthcare costs associated with both inflammatory diseases. Therefore, development of non-antibiotic therapeutic strategies is urgently needed based on a full understanding of the molecular pathogenesis of these inflammatory diseases.
Receptor interacting protein-2 (RIP-2) plays a key role in activation and regulation of multiple inflammatory signaling cascades, including nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK). Nucleotide-binding oligomerization domain 2 (NOD2) is a cytosolic receptor that detects bacterial peptidoglycans and other inflammatory stimuli [9]. Upon NOD2 detection of pathogen-associated molecular patterns (PAMPs), RIP-2 is recruited and interacts via CARD-CARD interactions. RIP-2 is a dual-specificity kinase downstream of NOD-like receptors, which undergoes autophosphorylation and subsequent polyubiquitination to drive transcription of inflammatory cytokines and chemokines [9]. RIP-2 is an essential signaling adaptor, and previous studies have shown NF-κB activation is sub-optimal in its absence [10]. Previous studies have also shown that RIP-2 acts as an important adaptor molecule for ovalbumin-induced lung inflammation. However, the role of RIP-2 in bacteria-induced expression of mucin MUC5AC, a major contributor of OM and COPD pathology, remains largely unknown.
In the present study, we sought to investigate the role of RIP-2 in NTHi-induced MUC5AC gene expression. Here we show that NTHi induces RIP-2 expression at mRNA and protein levels, and RIP-2 is a negative regulator of MUC5AC expression, whereas c-Jun N-terminal kinase (JNK) is a positive regulator for MUC5AC induction. Thus, our study provides the direct evidence for the first time of the negative regulation of MUC5AC induction by NTHi via RIP-2 signaling pathway and may lead to the development of new therapeutic strategies for suppressing MUC5AC overproduction.
Materials and methods
Reagents and antibodies
SP600125 was purchased from Calbiochem. Antibodies against β -actin (sc-8432), α-tubulin (sc-69969) and total JNK1/2 (sc-7345) were purchased from Santa Cruz Biotechnology. Antibodies for phospho-JNK1/2 (#9251), anti-rabbit HRP-linked antibody (#7074) and anti-mouse HRP-linked antibody (#7076) were purchased from Cell Signaling. Antibody against RIP-2 (ab8428) was purchased from Abcam.
Bacterial strains and culture conditions
Clinical isolate of NTHi 12 was used in this study. NTHi were grown as previously described [12-16]. For in vitro experiments, NTHi was resuspended in PBS and used at a multiplicity of infection (MOI) of 50. For in vivo experiments, NTHi was resuspended in isotonic saline and used at a concentration of 1 x 107 CFU per mouse.
Cell culture
Cell culture protocols were previously described [2-4, 12-17]. All media described below were supplemented with 10% by volume FBS (Sigma-Aldrich) and Pen/Strep (100 U/ml penicillin and 0.1 mg/ml streptomycin; Life Technologies). Human middle ear epithelial HMEEC-1 cells were maintained in DMEM (Cellgro) supplemented with BEGM SingleQuots (Lonza). Human bronchial epithelial cells BEAS-2B cells (ATCC) were maintained in RPMI 1640 medium (Gibco). All cells were cultured at 37°C in 5% CO2.
Real-time quantitative RT-PCR analysis
Total RNA extraction and RT-qPCR were performed as previously described [2-4, 12-17]. The relative quantities of mRNAs were determined by using the comparative Ct method and were normalized by using human cyclophilin for in vitro or mouse glyceraldehyde-3-poosphate dehydrogenase (GAPDH) for in vivo as endogenous controls. The primer sequences of human MUC5AC, mouse MUC5AC, human cyclophilin and mouse GAPDH were described previously [2, 18]. The primer sequences of RIP-2 are as follows: human RIP-2 (Forward 5’-CAA GCA CGA TAT ATA TAG CTA TGC AG-3’ and Reverse 5’-GCA AAG GAT TGG TGA CAT CCT C-3’); mouse RIP-2 (Forward 5’-GGG AAT TTG CAA TGA GCC TG-3’ and Reverse 5’-GCA GGA TGC GGA ATC TCA AT-3’).
Plasmids and transfections
RIP-2 siRNA (SR305775) and control siRNA (SR30004) were purchased from Origene. All transient transfections were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturers’ instructions. For experiments with inhibitors, the transfected cells were pretreated with or without chemical inhibitors for 1 h followed by 5 h of incubation with NTHi.
Western blot
Western blot procedures were previously described [2-4, 12-17]. Western blots were performed using whole-cell extracts, separated on 8-10 % SDS-PAGE gels, and transferred to polyvinylidene difluoride (PVDF) membranes. The membrane was blocked with a solution of Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) and 5% non-fat dry milk. The membrane was then incubated in a 1:2,000 dilution of a primary antibody in 5% bovine serum albumin-TBS-T. After three washes in TBS-T, the membrane was incubated with 1:5,000 dilution of the corresponding secondary antibody in 5% non-fat dry milk-TBS-T. Respective proteins were visualized by using Amersham ECL Prime Reagent (GE Healthcare Life Science).
Mice and animal experiments
C57BL/6 and RIP-2-/- mice (7-8 weeks old) were purchased from the Jackson Laboratory. Anesthetized mice were inoculated with NTHi at a concentration of 1 × 107 CFU per mouse or saline as control [2]. The inoculated mice were sacrificed at 6 and 12 h post-inoculation. Eardrums of mice were inspected for signs of middle ear inflammation. Dissected mouse middle ears were then subjected to total RNA extraction and histologic analyses. All animal experiments were approved by the Institutional Animal Care and Use Committee at Georgia State University.
Histology
As previously described [2], formalin-fixed paraffin-embedded mouse middle ear tissues were sectioned (4 µm), and then stained with hematoxylin and eosin (H&E) to visualize inflammatory responses and pathological changes in the middle ear. Images of stained cells and tissue sections were recorded with light microscopy system (Carl Zeiss).
Statistical analyses
All experiments were repeated in at least three independent experiments. Data are shown as mean ± SD of n determinations. Statistical analysis was assessed with two-tailed unpaired Student t-test; p < 0.05 was considered statistically significant.
Results and discussion
NTHi induces RIP-2 in human epithelial cells in vitro and in mouse middle ear in vivo
RIP-2 is a dual specificity kinase known to modulate inflammatory signaling pathways. Previous studies have shown the role of RIP-2 to be complex in various diseases. For example, RIP-2 deficient mice were shown to be resistant to developing arthritis and autoimmune-mediated cephalomyelitis associated with a multiple sclerosis model [9,19,20]. Paradoxically, dysregulation of the NOD2/RIP-2 pathway has been implicated in dysregulation of cell signaling pathways during viral infection [9, 21]. To investigate the role of RIP-2 in NTHi-induced mucin expression in our experimental models, we first examined whether NTHi induces RIP-2 in vitro. We treated human epithelial cells with NTHi and measured RIP-2 mRNA expression by performing quantitative PCR (Q-PCR) analysis. NTHi induced RIP-2 expression at mRNA level in human middle ear epithelial HMEEC-1 cells (Figure 1A) and human bronchial epithelial BEAS-2B cells (Figure 1B). We also confirmed a time-dependent induction of RIP-2 at the protein level (Figure 1C). Consistent with in vitro findings, NTHi-induced RIP-2 mRNA expression was also confirmed in the middle ear of mice by q-PCR analysis (Figure 1D). Collectively, these results that NTHi induces expression of RIP-2 in vitro and in vivo.
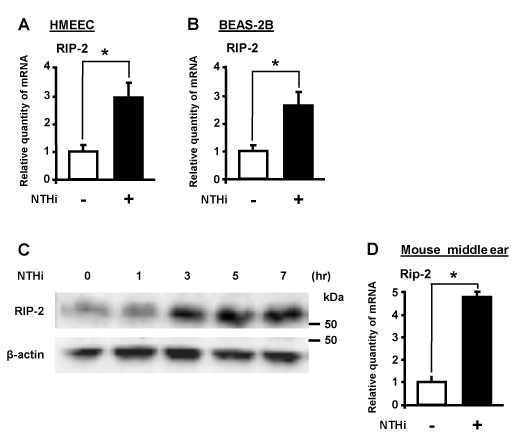
Figure 1. NTHi induces RIP-2 in human epithelial cells in vitro and in mouse middle ear in vivo. HMEEC-1 (A) and BEAS-2B (B) cells were stimulated with NTHi for 5h, and RIP-2 mRNA expression was analyzed by Q-PCR analysis. (C) Cells were stimulated with NTHi for various time periods (1, 3, 5, or 7 h) and RIP-2 protein expression was analyzed by Western blot analysis. (D) C57BL/6 mice were transtympanically inoculated with NTHi or control for 6h, and RIP-2 mRNA expression was analyzed by Q-PCR analysis. Data in A-B, D are means ± SD (n = 3); *P<0.05.
RIP-2 acts as a negative regulator for NTHi-induced MUC5AC expression
On the basis that mucous overproduction is a hallmark of NTHi-infections and NTHi markedly induce RIP-2 expression, we hypothesized that RIP-2 may play a critical role in regulating mucin production [22]. To determine the role of RIP-2 in MUC5AC induction, we utilized small interfering RNA (siRNA) specific for RIP-2 in human epithelial cells. We first evaluated the efficiency of RIP-2 knockdown. As shown in Figure 2A, RIP-2 siRNA efficiently depleted RIP-2 expression. Next, we evaluated the effect of siRNA RIP-2 on MUC5AC induction. Interestingly, siRIP-2 markedly enhanced NTHi-induced MUC5AC up-regulation in HMEEC-1 cells (Figure 2B). Moreover, we determined if NTHi also induces effusion in the middle ear of the mouse otitis media models. As shown in Figure 2C, increased effusion was observed in RIP-2 deficient mice compared with WT control mice as early as 12 hours post-infection. Together, our data indicate that RIP-2 is a negative regulator for NTHi-induced MUC5AC expression.
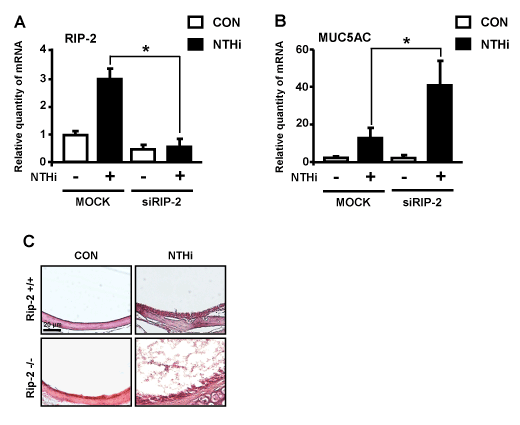
Figure 2. RIP-2 acts as a negative regulator for NTHi-induced MUC5AC expression. (A & B) HMEEC-1 cells were transfected with siRNA for RIP-2 or control for 40 h and were then stimulated with NTHi for 5 h and analyzed for RIP-2 (A) and MUC5AC (B) mRNA expression by Q-PCR analysis. (C) Middle ear tissue sections from C57BL/6 or RIP-2-/- mice transtympanically inoculated with NTHi (1 x 107 CFU) or control were stained with hematoxylin and eosin stain, magnification x 200, visualized with Carl Zeiss light microscope. Scale bar: 20 µm. Data in A-B are means ± SD (n = 3); *P<0.05.
JNK acts as a positive regulator for NTHi-induced MUC5AC up-regulation
MAP kinase JNK has been shown to play a critical role in mediating bacteria-induced inflammatory responses [4, 23]. Thus, we sought to determine the role of JNK in NTHi-induced MUC5AC expression. We first confirmed the activation of JNK by NTHi. NTHi induced potent activation of JNK in a time-dependent manner in human epithelial cells (Figure 3A). We next evaluated the effects of JNK specific inhibitor (SP600125) on NTHi-induced MUC5AC expression. As shown in Figure 3B, inhibition of JNK suppressed NTHi-induced MUC5AC expression, thereby indicating a critical role of JNK as a positive regulator for NTHi-induced MUC5AC expression. The role of JNK in regulating bacteria-induced host mucosal immune responses is highly complex and may act differently depending on the pathogen as well as on the host genes. The positive regulation of MUC5AC expression by JNK may be due to its role in mediating the activation of the signaling pathways including the transcription factors leading to the transcriptional up-regulation of MUC5AC expression. We previously reported that JNK negatively regulates MUC5AC expression via targeting negative activator protein-1 (AP-1) site in Streptococcus pneumoniae infection [4]. Thus, it is possible that JNK positively regulates NTHi-induced MUC5AC expression through the AP-1 site in the promoter region of MUC5AC gene. Future studies are needed to confirm this hypothesis by using AP-1 site mutants of MUC5AC-Luc luciferase reporter promoter constructs.
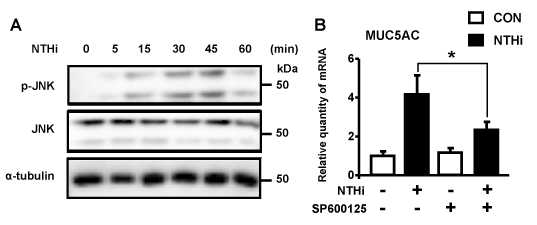
Figure 3. JNK acts as a positive regulator for NTHi-induced MUC5AC expression. (A) HMEEC-1 cells were treated with NTHi for indicated time periods, and cell lysates were analyzed by Western blot analysis with indicated antibodies. (B) HMEEC-1 cells were pretreated with 5 µM JNK inhibitor (SP600125) for 1 h followed by stimulation with NTHi for 5 h, and MUC5AC mRNA expression was then analyzed by Q-PCR analysis. Data in B are means ± SD (n=3); *P<0.05.
JNK acts as a negative regulator for NTHi-induced RIP-2 expression
Having demonstrating the positive role of JNK in mediating the up-regulation of mucin MUC5AC by NTHi, we next sought to explore if JNK may also mediate up-regulation of MUC5AC by suppressing the expression of RIP-2, the negative regulator for NTHi-induced MUC5AC expression. Interestingly, inhibition of JNK using JNK specific inhibitor enhanced NTHi-induced expression of RIP-2 (Figure 4). This exciting result provides direct evidence to unveil the complex molecular mechanism by which JNK mediates NTHi-induced mucin MUC5AC expression likely via up-regulation of MUC5AC transcription and also via inhibition of the induction of negative regulator RIP-2 expression (Figure 5).
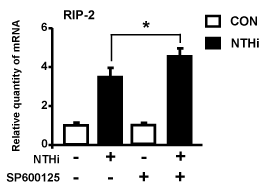
Figure 4. JNK acts as a negative regulator for NTHi-induced up-regulation of RIP-2 expression. HMEEC-1 cells were pretreated with 5 µM JNK inhibitor (SP600125) for 1 h followed by stimulation with NTHi for 5 h, and RIP-2 mRNA expression was then analyzed by Q-PCR analysis. Data are means ± SD (n = 3); *P<0.05.
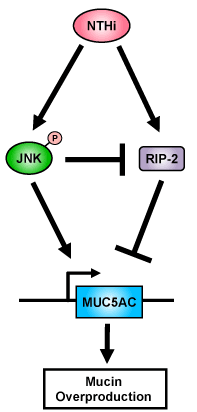
Figure 5. A Schematic diagram illustrating that JNK acts as a positive regulator for NTHi-induced up-regulation of mucin MUC5AC expression. In contrast, RIP-2 acts as a negative regulator for MUC5AC induction.
Taken together, our studies bring new insights into the complex regulatory mechanism underlying bacteria-induced up-regulation of mucous and may be helpful for developing new therapeutic strategies for suppressing mucous overproduction in upper respiratory infectious diseases. Future studies are needed for further investigating the precise molecular mechanisms underlying tight regulation of mucous overproduction.
Acknowledgement
This work was supporte2021 Copyright OAT. All rights reservants DC005843, DC013833 and GM107529 (to J.-D.L.). J.-D.L. is a Georgia Research Alliance Eminent Scholar in Inflammation and Immunity.
Conflicts of interest
The authors declare no conflicts of interest.
References
- Koeppen M, McNamee EN, Brodsky KS, Aherne CM, Faigle M, et al. (2013) Detrimental role of the airway mucin Muc5ac during ventilator-induced lung injury. Mucosal Immunol 6: 762-775. [Crossref]
- Lee JY, Komatsu K, Lee BC, Miyata M, O'Neill Bohn A, et al. (2015) Vinpocetine inhibits Streptococcus pneumoniae-induced upregulation of mucin MUC5AC expression via induction of MKP-1 phosphatase in the pathogenesis of otitis media. J Immunol 194: 5990-5998. [Crossref]
- Lee J, Komatsu K, Lee BC, Lim JH, Jono H, et al. (2012) Phosphodiesterase 4B mediates extracellular signal-regulated kinase-dependent up-regulation of mucin MUC5AC protein by Streptococcus pneumoniae by inhibiting cAMP-protein kinase A-dependent MKP-1 phosphatase pathway. J Biol Chem 287: 22799-22811. [Crossref]
- Lim JH, Kim HJ, Komatsu K, Ha U, Huang Y, et al. (2009) Differential regulation of Streptococcus pneumoniae-induced human MUC5AC mucin expression through distinct MAPK pathways. Am J Transl Res 1: 300-311. [Crossref]
- Juneau RA, Pang B, Weimer KE, Armbruster CE, Swords WE (2011) Nontypeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infect Immun 79: 431-8. [Crossref]
- Hassett DJ, Borchers MT, Panos RJ (2014) Chronic obstructive pulmonary disease (COPD): evaluation from clinical, immunological and bacterial pathogenesis perspectives. J Microbiol 52: 211-226. [Crossref]
- Vergison A (2008) Microbiology of otitis media: a moving target. Vaccine 26 Suppl 7: G5-10. [Crossref]
- Allen EK, Manichaikul A, Sale MM (2014) Genetic contributors to otitis media: agnostic discovery approaches. Curr Allergy Asthma Rep14: 411. [Crossref]
- Jun JC, Cominelli F, Abbott DW (2013) RIP2 activity in inflammatory disease and implications for novel therapeutics. J Leukoc Biol 94: 927-932. [Crossref]
- Kobayashi K, Inohara N, Hernandez LD, Galán JE, Núñez G, et al. (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416: 194-199. [Crossref]
- Goh FY, Cook KL, Upton N, Tao L, Lah LC, et al., (2013) Receptor-interacting protein 2 gene silencing attenuates allergic airway inflammation. J Immunol 191: 2691-2699. [Crossref]
- Andrews CS, Matsuyama S, Lee BC, Li JD (2016) Resveratrol suppresses NTHi-induced inflammation via up-regulation of the negative regulator MyD88 short. Sci Rep 6: 34445. [Crossref]
- Andrews CS, Miyata M, Susuki-Miyata S, Byung-Cheol L, Komatsu K, et al., (2015) Nontypeable Haemophilus influenzae-Induced MyD88 Short Expression Is Regulated by Positive IKKbeta and CREB Pathways and Negative ERK1/2 Pathway. PLoS One 10: e0144840. [Crossref]
- Konduru AS, Lee BC, Li JD (2016) Curcumin suppresses NTHi-induced CXCL5 expression via inhibition of positive IKKβ pathway and up-regulation of negative MKP-1 pathway. Sci Rep 6: 31695. [Crossref]
- Konduru AS, Matsuyama S, Lee BC, Komatsu K, Li JD (2017) Curcumin Inhibits NTHi-Induced MUC5AC Mucin Overproduction in Otitis Media via Upregulation of MAPK Phosphatase MKP-1. Int J Inflam 2017: 4525309. [Crossref]
- Tasaki Y, Wang YW, Konduru AS, Komatsu K, Matsuyama S, et al. (2016) cAMP-dependent protein kinase A acts as a negative regulator for nontypeable Haemophilus influenzae-induced GM-CSF expression via inhibition of MEK-ERK signaling pathway. Int Mol Med 3: 1-5.
- Komatsu K, Lee JY, Miyata M, Hyang Lim J, Jono H, et al. (2013) Inhibition of PDE4B suppresses inflammation by increasing expression of the deubiquitinase CYLD. Nat Commun 4: 1684. [Crossref]
- Susuki-Miyata S, Miyata M, Lee BC, Xu H, Kai H, et al. (2015) Cross-talk between PKA-Cβ and p65 mediates synergistic induction of PDE4B by roflumilast and NTHi. Proc Natl Acad Sci U S A 112: E1800-1809. [Crossref]
- Vieira SM, Cunha TM, França RF, Pinto LG, Talbot J, et al. (2012) Joint NOD2/RIPK2 signaling regulates IL-17 axis and contributes to the development of experimental arthritis. J Immunol 188: 5116-5122. [Crossref]
- Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, et al. (2011) Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity 34: 75-84. [Crossref]
- Kim YG, Park JH, Reimer T, Baker DP, Kawai T, et al. (2011) Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe 9: 496-507. [Crossref]
- Yang S, Wang B, Humphries F, Jackson R, Healy ME, et al. (2013) Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol 14: 927-936. [Crossref]
- Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9: 537-549. [Crossref]